
Disentangling Crocus Series Verni and Its Polyploids

 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and Sanger Sequencing
2.3. Genotyping-by-Sequencing
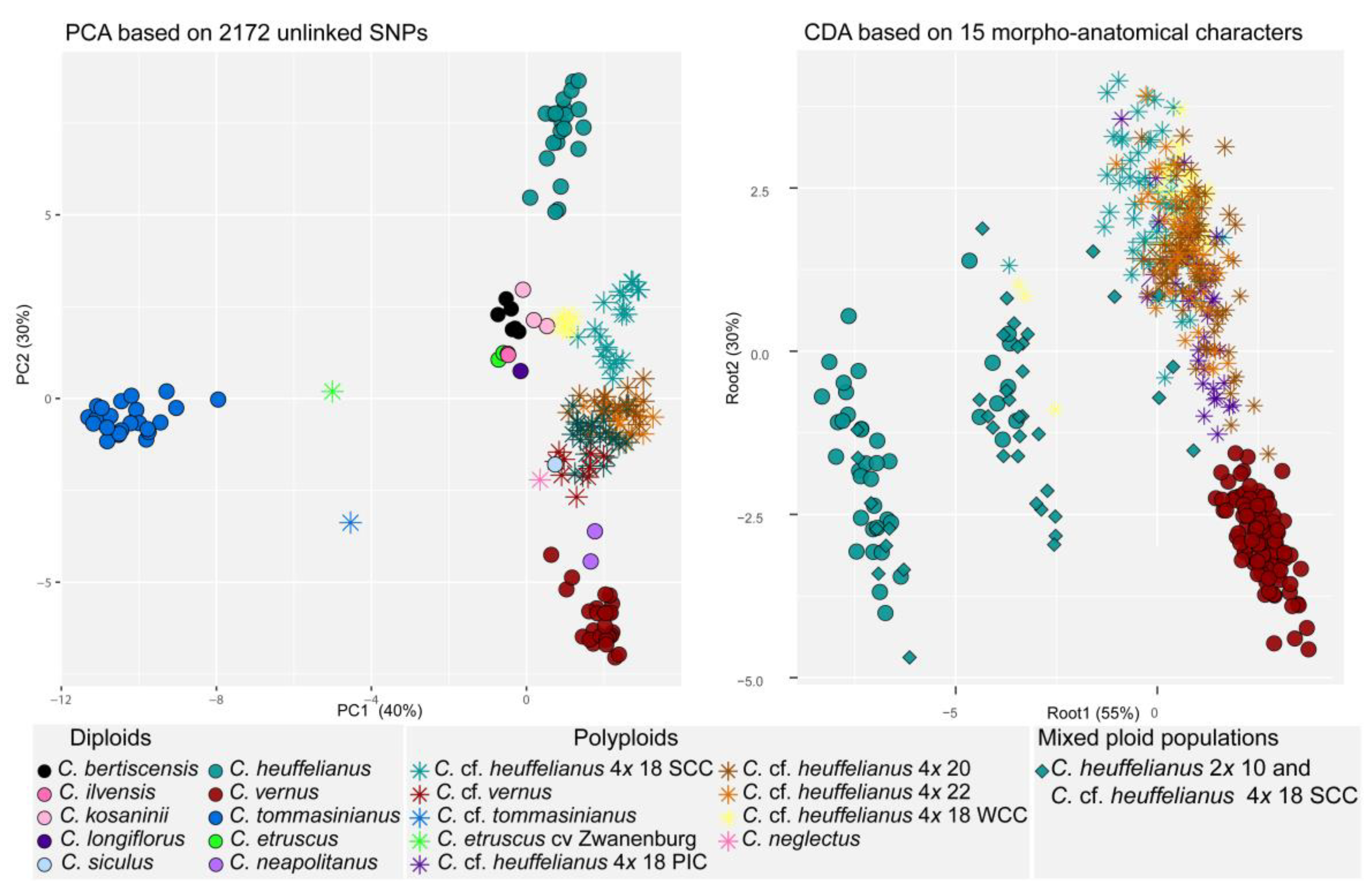
2.4. Analyses of Population Structure
2.5. Heterozygosity and Fst Determination
2.6. Next-Generation Sequencing of Nuclear Markers
2.7. Chromosome Counts
2.8. Genome Size Measurements
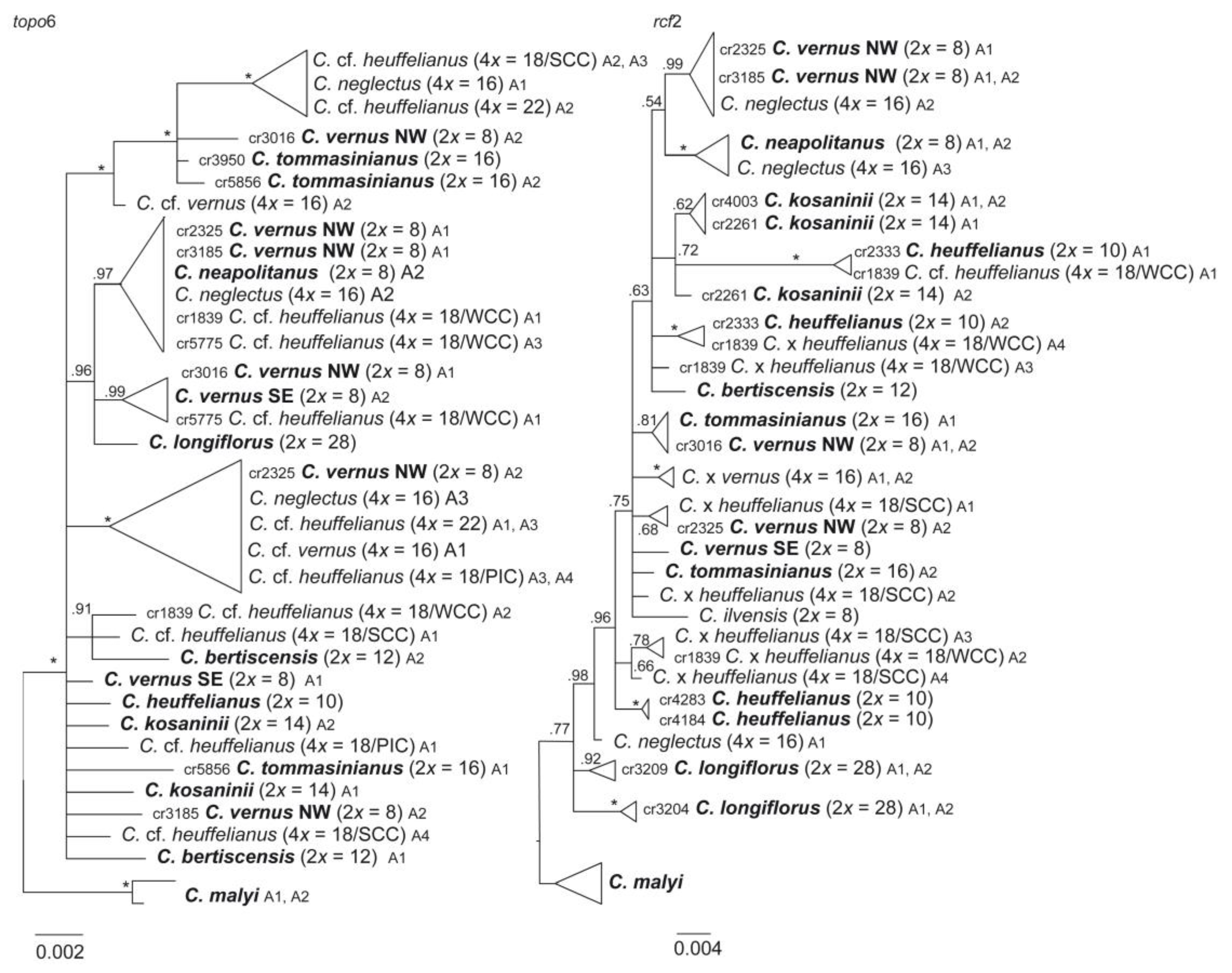
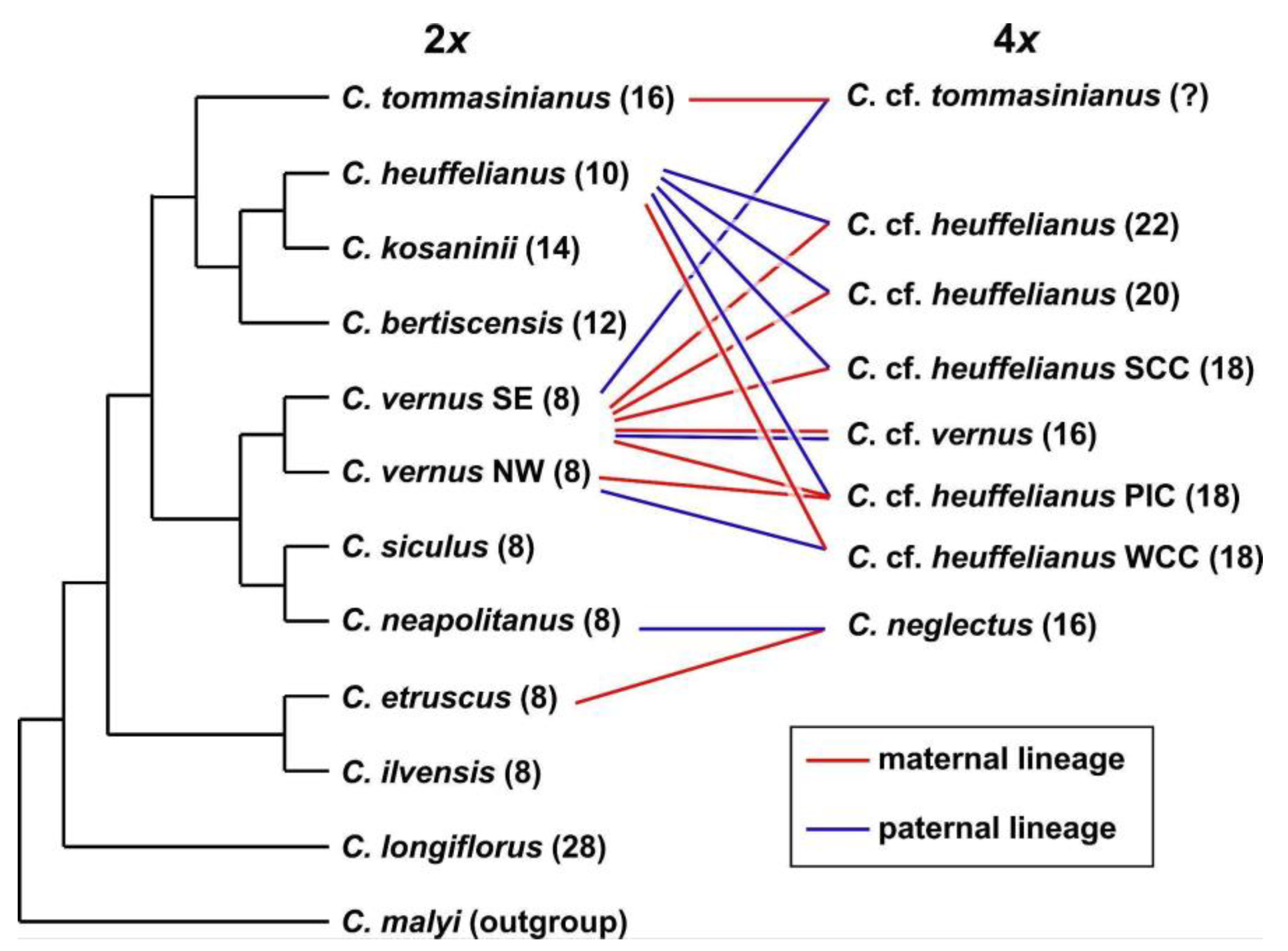
2.9. Phylogenetic Inferences and Origin of Allopolyploids
2.10. Morpho-Anatomical Analyses
3. Results
3.1. Determination of Ploidy Levels
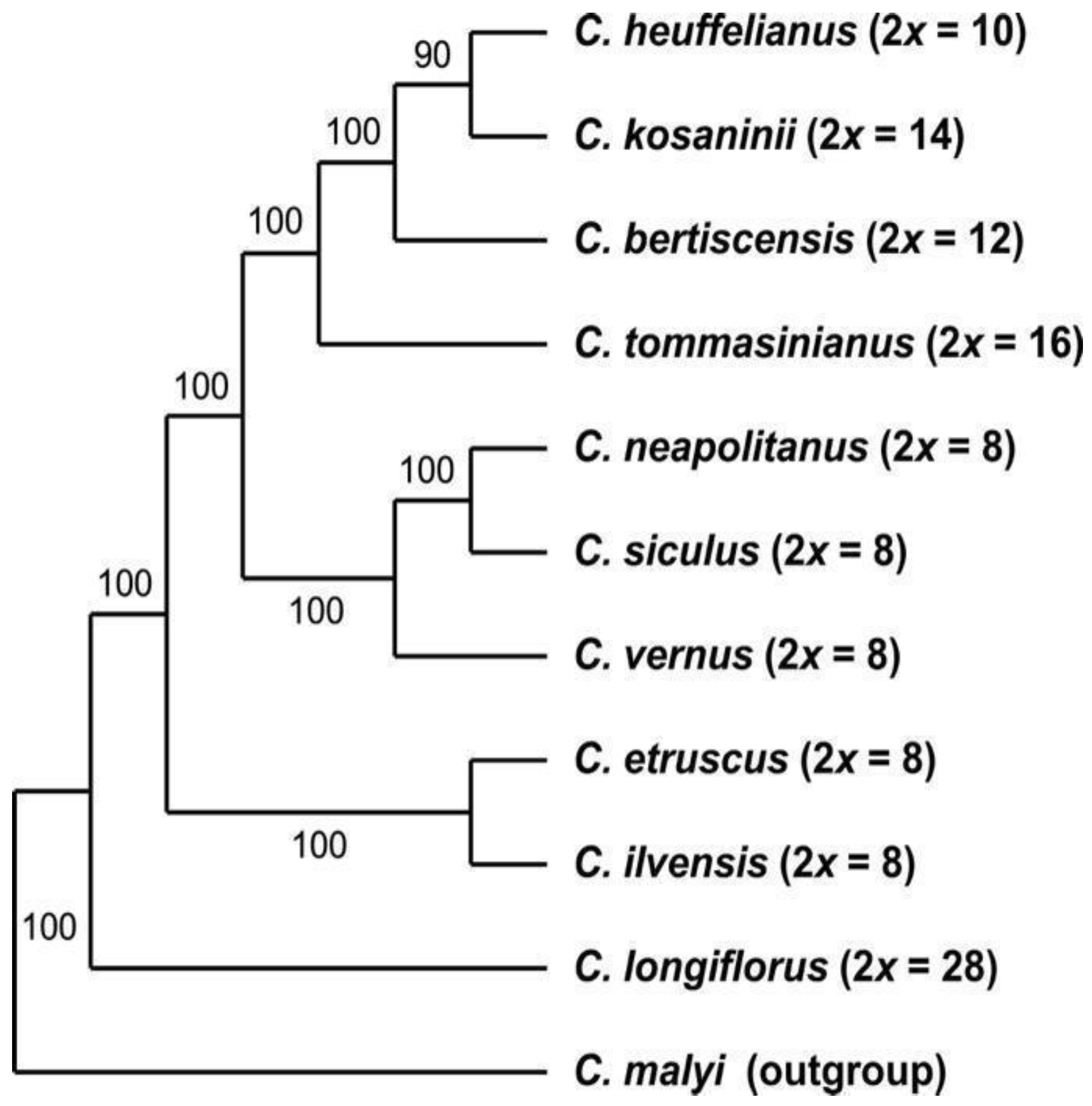
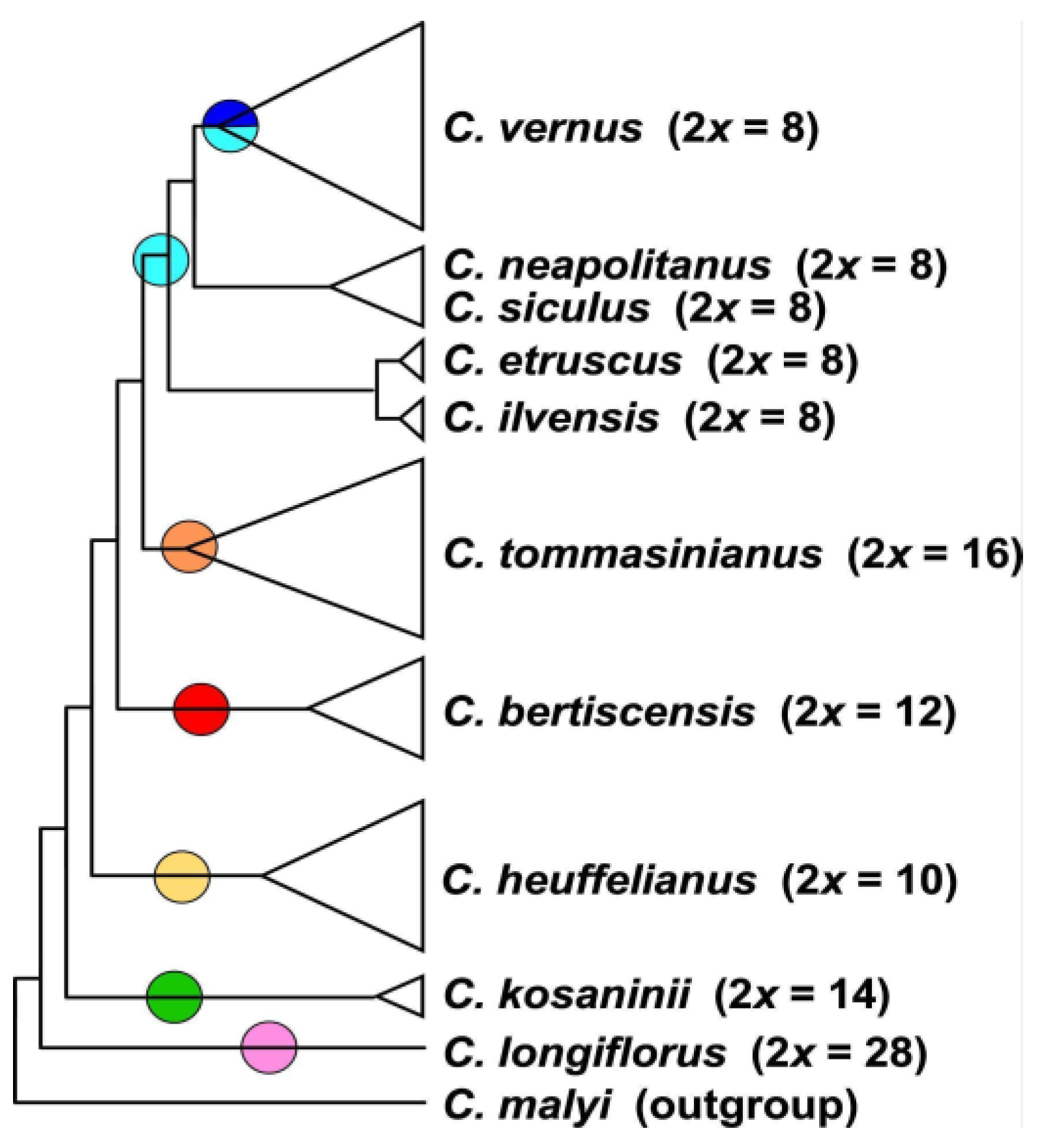
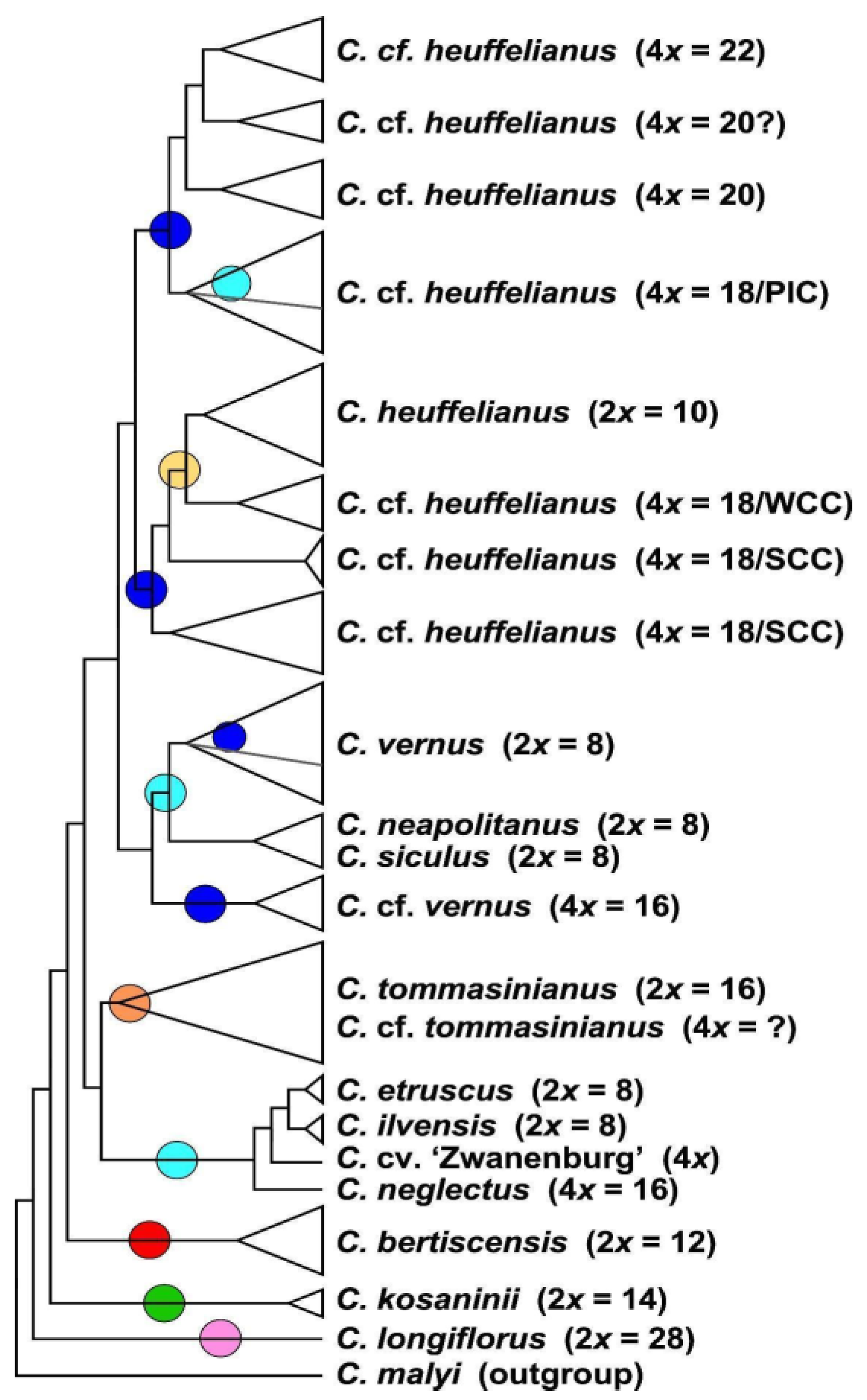
3.2. Phylogenetic Inference and Origin of Allopolyploids
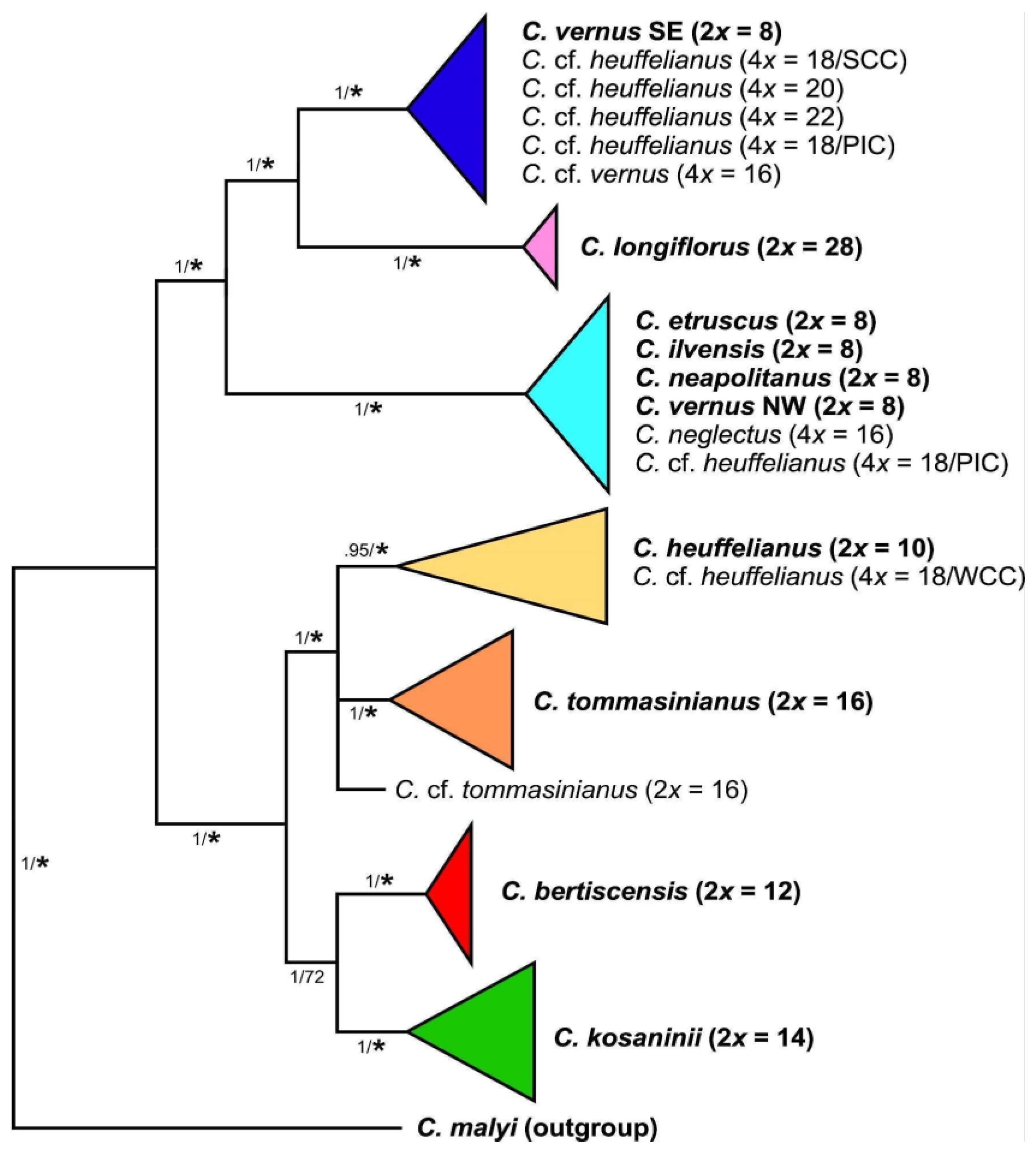
3.3. Phylogenomic Analysis
3.4. Morpho-Anatomical Analyses
4. Discussion
4.1. Recognition of Recent Polyploids and Their Parents
4.2. General Results Regarding Phylogeny and Systematics
4.3. Chromosome and Genome Size Evolution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soltis, D.E.; Visger, C.J.; Marchant, D.B.; Soltis, P.S. Polyploidy: Pitfalls and paths to a paradigm. Am. J. Bot. 2016, 103, 1146–1166. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Fawcett, J.A.; Proost, S.; Sterck, L.; Vandepoele, K. The flowering world: A tale of duplications. Trends Plant Sci. 2009, 14, 680–688. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Ashman, T.L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An evolutionary and ecological force in stressful times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Vu, G.T.H.; Schmutzer, T.; Bull, F.; Cao, H.X.; Fuchs, J.; Tran, T.D.; Jovtchev, G.; Pistrick, K.; Stein, N.; Pecinka, A.; et al. Comparative genome analysis reveals divergent genome size evolution in a carnivorous plant genus. Plant Genome 2015, 8, plantgenome2015-04. [Google Scholar] [CrossRef]
- Brassac, J.; Blattner, F.R. Species-level phylogeny and polyploid relationships in Hordeum (Poaceae) inferred by next-generation sequencing and in silico cloning of multiple nuclear loci. Syst. Biol. 2015, 64, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Jakob, S.S.; Meister, A.; Blattner, F.R. The considerable genome size variation of Hordeum species (Poaceae) is linked to phylogeny, life form, ecology, and speciation rates. Mol. Biol. Evol. 2004, 21, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Albach, D.C.; Greilhuber, J. Genome size variation and evolution in Veronica. Ann. Bot. 2004, 94, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Leitch, I.J.; Bennett, M.D. Genome downsizing in polyploid plants. Biol. J. Linn. Soc. 2004, 82, 651–663. [Google Scholar] [CrossRef]
- Leitch, I.J.; Fay, M. Plant genome horizons: Michael Bennett’s contribution to genome research. Ann. Bot. 2008, 101, 737–746. [Google Scholar] [CrossRef]
- Meudt, H.M.; Rojas-Andrés, B.M.; Prebble, J.M.; Low, E.; Garnock-Jones, P.J.; Albach, D.C. Is genome downsizing associated with diversification in polyploid lineages of Veronica? Bot. J. Linn. Soc. 2015, 178, 243–266. [Google Scholar] [CrossRef]
- Petrov, D.A. Evolution of genome size: New approaches to an old problem. Trends Genet. 2001, 17, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Vu, G.T.H.; Cao, H.X.; Reiss, B.; Schubert, I. Deletion-bias in DNA double-strand break repair differentially contributes to plant genome shrinkage. New Phytol. 2017, 214, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.; Seberg, O.; Thorsøe, S.; Jørgensen, T.; Mathew, B. A phylogeny of the genus Crocus (Iridaceae) based on sequence data from five plastid regions. Taxon 2008, 57, 487–499. [Google Scholar] [CrossRef]
- Harpke, D.; Meng, S.; Rutten, T.; Kerndorff, H.; Blattner, F.R. Phylogeny of Crocus (Iridaceae) based on one chloroplast and two nuclear loci: Ancient hybridization and chromosome number evolution. Mol. Phylogenet. Evol. 2013, 66, 617–627. [Google Scholar] [CrossRef]
- Harpke, D.; Carta, A.; Tomović, G.; Ranđelović, V.; Ranđelović, N.; Blattner, F.R.; Peruzzi, L. Phylogeny, karyotype evolution and taxonomy of Crocus series Verni (Iridaceae). Plant Syst. Evol. 2015, 301, 309–325. [Google Scholar] [CrossRef]
- Raca, I.; Harpke, D.; Shuka, L.; Ranđelović, V. A new species of Crocus ser. Verni (Iridaceae) with 2n = 12 chromosomes from the Balkans. Plant Biosyst. 2022, 156, 36–42. [Google Scholar] [CrossRef]
- Karasawa, K. Karyological studies in Crocus. III. Jap. J. Bot. 1943, 12, 475–503. [Google Scholar]
- Brighton, C.A. Cytological problems in the genus Crocus (Iridaceae): I. Crocus vernus aggregate. Kew Bull. 1976, 31, 33–46. [Google Scholar] [CrossRef]
- Baldini, R.M. Karyological observations on two Crocus species (Iridaceae) from Tuscany (Italy). Caryologia 1990, 43, 341–345. [Google Scholar] [CrossRef]
- Micevska, D. Caryological Analysis of the Species of the Genus Crocus. Master’s Thesis, University of Skopje, Skopje, North Macedonia, 2000. [Google Scholar]
- Mitic, D. Caryological Analysis of Some Species of the Genus Crocus L. Master’s Thesis, University of Skopje, Skopje, North Macedonia, 2001. [Google Scholar]
- Peruzzi, L.; Carta, A. Crocus ilvensis sp. nov. (sect. Crocus, Iridaceae), endemic to Elba Island (Tuscan Archipelago, Italy). Nord. J. Bot. 2011, 29, 6–13. [Google Scholar] [CrossRef]
- Peruzzi, L.; Carta, A.; Garbari, F. Lectotypification of the name Crocus sativus var. vernus L. (Iridaceae) and its consequences within Crocus ser. Verni. Taxon 2013, 62, 1037–1040. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Kerndorff, H.; Harpke, D. Crocus cobbii Kerndorff, Pasche & Harpke species nova (Liliiflorae, Iridaceae) and its relatives. Stapfia 2022, 113, 5–32. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Wendler, N.; Mascher, M.; Nöh, C.; Himmelbach, A.; Scholz, U.; Ruge-Wehling, B.; Stein, N. Unlocking the secondary gene-pool of barley with next-generation sequencing. Plant Biotechnol. J. 2014, 12, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Overcast, I. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 2020, 36, 2592–2594. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Wickham, H.; Wickham, M.H. Package ‘Tidyverse’, 2019; pp. 1–5. Available online: http://tidyverse.tidyverse.org (accessed on 20 July 2022).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; pp. 1–189. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R. Direct amplification of the entire ITS region from poorly preserved plant material using recombinant PCR. Biotechniques 1999, 27, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R. Phylogenetic analysis of Hordeum (Poaceae) as inferred by nuclear rDNA ITS sequences. Mol. Phylogenet. Evol. 2004, 33, 289–299. [Google Scholar] [CrossRef]
- Waminal, N.E.; Park, H.M.; Ryu, K.B.; Kim, J.H.; Yang, T.J.; Kim, H.H. Karyotype analysis of Panax ginseng C.A.Meyer, 1843 (Araliaceae) based on rDNA loci and DAPI band distribution. Comp. Cytogenet. 2012, 6, 425–441. [Google Scholar] [CrossRef]
- Rodríguez-Domínguez, J.M.; Ríos-Lara, L.L.; Tapia-Campos, E.; Barba-Gonzalez, R. An improved technique for obtaining well-spread metaphases from plants with numerous large chromosomes. Biotech. Histochem. 2017, 92, 159–166. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (* and Other Methods); Sinauer Assoc.: Sunderland, MA, USA, 2002. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef]
- Malinsky, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and fineRADstructure: Population inference from RADseq data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Gligorijević, S.; Pejčinović, D. Contribution to the methodology of anatomical sections preparation. Acta Biol. Med. Exp. 1983, 8, 43–45. [Google Scholar]
- Raca, I.; Ljubisavljević, I.; Jušković, M.; Ranđelović, N.; Ranđelović, V. Comparative anatomical study of the taxa from series Verni Mathew (Crocus L.) in Serbia. Biol. Nyssana 2017, 8, 15–22. [Google Scholar] [CrossRef]
- Raca, I.; Jovanovic, M.; Ljubisavljevic, I.; Juskovic, M.; Randelovic, V. Morphological and leaf anatomical variability of Crocus cf. heuffelianus Herb. (Iridaceae) populations from the different habitats of the Balkan Peninsula. Turk. J. Bot. 2019, 43, 645–658. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Mosolygo-L, A.; Sramko, G.; Barabas, S.; Czegledi, L.; Javor, A.; Molnar, V.A.; Suranyi, G. Molecular genetic evidence for allotetraploid hybrid speciation in the genus Crocus L. (Iridaceae). Phytotaxa 2016, 258, 121–136. [Google Scholar] [CrossRef]
- Raca, I. Taxonomy and Phylogeny of Series Verni Mathew (Crocus L.) in Southeastern Europe—Morpho-Anatomical, Cytological and Molecular Approach. Ph.D. Thesis, University of Niš, Niš, Serbia, 2021. [Google Scholar]
- Statsoft Inc. STATISTICA (Data Analysis Software System), Version 7; Statsoft Inc.: Tulsa, OK, USA, 2004.
- Herbert, W.; Lindley, J. A history of the species of Crocus. J. R. Hortic. Soc. 1847, 2, 249–293. [Google Scholar]
- Meusel, H.; Jäger, E.J.; Weinert, E. Vergleichende Chorologie der zentraleuropäischen Flora; VEB Fischer: Jena, Germany, 1965.
- Malynovsky, K.A. Roslynnisty vysokogirja Ukrainskykh Karpat; Naukova dumka: Kyiv, Ukraine, 1980. [Google Scholar]
- Beck-Mannagetta, G. Flora von Bosnien, der Herzegowina und des Sandzaks Novipazar; C. Gerold: Vienna, Austria, 1904. [Google Scholar]
- Peruzzi, L. Crocus heuffelianus (Iridaceae), a new record for the Italian flora. Phytotaxa 2016, 261, 291–294. [Google Scholar] [CrossRef]
- Carta, A.; Probert, R.; Moretti, M.; Peruzzi, L.; Bedini, G. Seed dormancy and germination in three Crocus ser. Verni species (Iridaceae): Implications for evolution of dormancy within the genus. Plant Biol. 2014, 16, 1065–1074. [Google Scholar] [CrossRef]
- Carta, A.; Flamini, G.; Cioni, P.L.; Pistelli, L.; Peruzzi, L. Flower bouquet variation in four species of Crocus ser. Verni. J. Chem. Ecol. 2015, 41, 105–110. [Google Scholar] [CrossRef]
- Molloy, E.K.; Warnow, T. To include or not to include: The impact of gene filtering on species tree estimation methods. Syst. Biol. 2017, 67, 285–303. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef]
- Murat, F.; Armero, A.; Pont, C.; Klopp, C.; Salse, J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 2017, 49, 490–496. [Google Scholar] [CrossRef]
- Mandakova, T.; Lysak, M.A. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant Biol. 2018, 42, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Glombik, M.; Bačovský, V.; Hobza, R.; Kopecký, D. Competition of parental genomes in plant hybrids. Front. Plant Sci. 2020, 11, 200. [Google Scholar] [CrossRef]
- Lysak, M.A.; Cheung, K.; Kitschke, M.; Bures, P. Ancestral chromosomal blocks are triplicated in Brassiceae species with varying chromosome number and genome size. Plant Physiol. 2007, 145, 402–410. [Google Scholar] [CrossRef] [PubMed]
- El-Shehawi, A.M.; Elseehy, M.M. Genome size and chromosome number relationship contradicts the principle of Darwinian evolution from common ancestor. J. Phylogenet. Evol. Biol. 2017, 5, 179. [Google Scholar] [CrossRef]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M. Agmatoploidy and symploidy: A critical review. Genet. Mol. Biol. 2016, 39, 492–496. [Google Scholar] [CrossRef]
- Schubert, I.; Lysak, M.A. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 2011, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Tank, D.C.; Eastman, J.M.; Pennell, M.W.; Soltis, P.S.; Soltis, D.E.; Hinchliff, C.E.; Brown, J.W.; Sessa, E.B.; Harmon, L.J. Nested radiations and the pulse of angiosperm diversification: Increased diversification rates often follow whole genome duplications. New Phytol. 2015, 207, 454–467. [Google Scholar] [CrossRef]
- Murat, F.; Xu, J.-H.; Tannier, E.; Abrouk, M.; Guilhot, N.; Pont, C.; Messing, J.; Salse, J. Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Res. 2010, 20, 1545–1557. [Google Scholar] [CrossRef]
- Kelly, L.J.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Novák, P.; Neumann, P.; Lysak, M.A.; Day, P.D.; Berger, M.; Fay, M.F.; et al. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytol. 2015, 208, 596–607. [Google Scholar] [CrossRef]
- Macas, J.; Novak, P.; Pellicer, J.; Cizkova, J.; Koblizkova, A.; Neumann, P.; Fukova, I.; Doležel, J.; Kelly, L.J.; Leitch, I.J. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef] [PubMed]
- Brighton, C.A.; Mathew, B.; Marchant, C.J. Chromosome counts in the genus Crocus (Iridaceae). Kew Bull. 1973, 28, 451–464. [Google Scholar] [CrossRef]
- Thiv, M.; Wörz, A. Die neue Identität des Zavelsteiner Krokus als Crocus neglectus nach DNA-Untersuchungen. Jahresh. Ges. Nat. Württemberg 2015, 171, 163–172. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Individuals in Chloroplast/ GBS/NSCG 1/Morpho-Anatomical Analyses | Origin | x/Chromosome Number/Average 2C Genome Size |
|---|---|---|---|
| Crocus bertiscensis Raca, Harpke, Shuka, and V.Randjel. | 4/6/1/0 | Albania (ALB), Montenegro (MNE) | 2x/12/6.66 pg |
| Crocus etruscus Parl. | 2/3/0/0 | Italy (ITA) | 2x/8/7.58 pg |
| Crocus ilvensis Peruzzi and Carta | 4/5/1/0 | Italy (ITA) | 2x/8/7.88 pg |
| Crocus kosaninii Pulević | 2/3/2/0 | Kosovo (XKX), Serbia (SRB) | 2x/14/7.95 pg |
| Crocus longiflorus Raf. | 2/1/2/0 | Italy (ITA) | 2x/28/3.21 pg |
| Crocus neglectus Peruzzi and Carta | 1/1/1/0 | Germany (GER) | 4x/16/12.24 pg |
| Crocus neapolitanus (Ker Gawl.) Loisel. | 3/2/1/0 | Italy (ITA) | 2x/8/– |
| Crocus siculus Tineo | 0/1/0/0 | Italy (ITA) | 2x/8/– |
| Crocus tommasinianus Herb. | 5/25/2/0 | Bosnia and Herzegovina (BIH), Italy (ITA), Montenegro (MNE), Serbia (SRB) | 2x/16/5.53 pg |
| Crocus vernus (L.) Hill | 14/27/4/100 | Albania (ALB), Bosnia and Herzegovina (BIH), France (FRA), Montenegro (MNE), Slovenia (SLO), Switzerland (CHE) | 2x/8/5.78 pg |
| Crocus heuffelianus Herb. | 8/22/4/70 | Romania (ROU), Slovakia (SVK), Ukraine (UKR) | 2x/10/7.73 pg |
| Crocus cf. heuffelianus (SCC) | 7/19/1/70 | Romania (ROU) | 4x/18/12.84 pg |
| Crocus cf. heuffelianus (PIC) | 5/26/1/40 | Bosnia and Herzegovina (BIH), Slovenia (SLO) | 4x/18/10.88 pg |
| Crocus cf. heuffelianus (WCC) | 3/9/2/35 | Slovakia (SVK) | 4x/18/12.75 pg |
| Crocus cf. heuffelianus | 12/23/0/80 | Montenegro (MNE), Serbia (SRB) | 4x/20/11.82 pg |
| Crocus cf. heuffelianus | 3/14/1/40 | Albania (ALB), Kosovo (XKX) | 4x/22/11.95 pg |
| Crocus cf. vernus | 5/9/1/0 | Albania (ALB) | 4x/16/12.38 pg |
| Crocus cf. tommasinianus | 0/1/0/0 | Montenegro (MNE) | 4x/-/- |
| Crocus malyi Vis. (outgroup) | 2/2/2/0 | Croatia (HRV) | 2x/30/– |
| Chloroplast (matK–trnQ + ycf1) | GBS Data 2x/2x + 4x | Nuclear Gene Regions orcp/rcf2/topo6/ITS | |
|---|---|---|---|
| Number of sequences | 81 | 93/194 | 37/42/39/22 |
| Alignment lengths | 4123 | 187,846 | 1160/568/819/646 |
| Constant characters | 3989 | 178,748/176,958 | 982/490/737/599 |
| Variable characters | 134 | 9098/10,888 | 178/78/82/47 |
| Parsimony-informative characters | 114 | 5899/6954 | 125/47/38/33 |
| Number of MP trees | 4 | 45/5400 | 142/10k/10k/10k 1 |
| MP tree length | 156 | 16,298/25,300 | 333/99/89/56 |
| Consistency index (CI) | 0.87 | 0.58/0.45 | 0.57/0.83/0.96/0.88 |
| Retention index (RI) | 0.98 | 0.83/0.77 | 0.66/0.88/0.97/0.93 |
| Model of sequence evolution (BIC) | GTR + I | GTR + I + Γ | HKY + I + Γ/HKY + I + Γ/TrN + I/HKY + I |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raca, I.; Blattner, F.R.; Waminal, N.E.; Kerndorff, H.; Ranđelović, V.; Harpke, D. Disentangling Crocus Series Verni and Its Polyploids. Biology 2023, 12, 303. https://doi.org/10.3390/biology12020303
Raca I, Blattner FR, Waminal NE, Kerndorff H, Ranđelović V, Harpke D. Disentangling Crocus Series Verni and Its Polyploids. Biology. 2023; 12(2):303. https://doi.org/10.3390/biology12020303
Chicago/Turabian StyleRaca, Irena, Frank R. Blattner, Nomar Espinosa Waminal, Helmut Kerndorff, Vladimir Ranđelović, and Dörte Harpke. 2023. "Disentangling Crocus Series Verni and Its Polyploids" Biology 12, no. 2: 303. https://doi.org/10.3390/biology12020303
APA StyleRaca, I., Blattner, F. R., Waminal, N. E., Kerndorff, H., Ranđelović, V., & Harpke, D. (2023). Disentangling Crocus Series Verni and Its Polyploids. Biology, 12(2), 303. https://doi.org/10.3390/biology12020303