CAR Based Immunotherapy of Solid Tumours—A Clinically Based Review of Target Antigens



Abstract
Simple Summary
Abstract

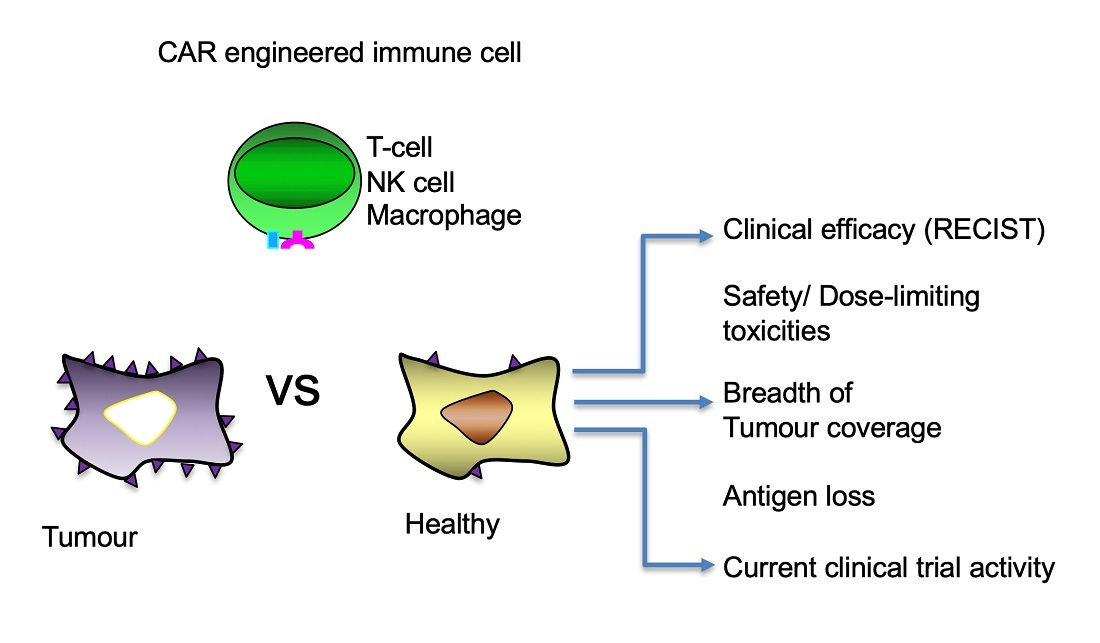
1. Introduction
2. Mesothelin
3. Receptor Tyrosine Kinases
3.1. HER2
3.2. Epidermal Growth Factor Receptor (EGFR)
3.3. Epidermal Growth Factor Receptor Variant III (EGFRvIII)
3.4. Receptor Tyrosine Kinase-like Orphan Receptor Family Member (ROR)1
3.5. c-MET (Hepatocyte Growth Factor Receptor)
3.6. Vascular Endothelial Growth Factor Receptor (VEGFR) 2
4. Mucins
4.1. MUC-1 (Mucin-1)
4.2. MUC-16 (Mucin-16)
4.3. Tumour-Associated Glycoprotein 72 (TAG-72)
5. Claudins
5.1. CLDN6 (Claudin-6)
5.2. CLDN18.2 (Claudin-18.2)
6. FR (Folate Receptor)-α
7. IL13Rα2
8. Prostate-Specific Membrane Antigen (PSMA)
9. GD2 (Disialoganglioside 2)
10. B7 Family Members
10.1. B7-H3 (CD276)
10.2. Programmed Death Receptor Ligand 1 (PD-L1)
11. Glypican 3
12. NKG2D Ligands
13. Prostate Stem Cell Antigen (PSCA)
14. Carcinoembryonic Antigen (CEA)
15. CD70
16. Carboxy Anhydrase IX (CAIX)
17. CD133
18. Erythropoietin-Producing Human Hepatocellular Carcinoma (Ephrin) Type A Receptor 2 (EphA2)
19. Fibroblast Activation Protein (FAP)
20. Adhesion Molecules
20.1. Epithelial Cell Adhesion Molecule (EpCAM)
20.2. Neuronal L1 Cell Adhesion Molecule (L1CAM; CD171)
21. Roundabout Guidance Receptor 1 (ROBO1)
22. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B-cell malignancy: The story so far. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520927164. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.; Usmani, S.Z.; Yong, K. CAR T-cell therapy for multiple myeloma: State of the art and prospects. Lancet Haematol. 2021, 8, e446–e461. [Google Scholar] [CrossRef] [PubMed]
- Glover, M.; Avraamides, S.; Maher, J. How Can We Engineer CAR T Cells to Overcome Resistance? Biologics 2021, 15, 175–198. [Google Scholar] [CrossRef] [PubMed]
- Boccalatte, F.; Mina, R.; Aroldi, A.; Leone, S.; Suryadevara, C.M.; Placantonakis, D.G.; Bruno, B. Advances and Hurdles in CAR T Cell Immune Therapy for Solid Tumors. Cancers 2022, 14, 5108. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Klaver, Y.; Gratama, J.W.; Sleijfer, S.; Debets, R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem. Soc. Trans. 2016, 44, 951–959. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- Tanyi, J.; Haas, A.; Aggarwal, C.; O’Hara, M.; Lacey, S.; Golden, R.; Hwang, W.-T.; Young, R.; Sheppard, N.; Albelda, S.; et al. Phase I study of autologous T cells bearing fully-humanized chimeric antigen receptors targeting mesothelin in mesothelin-expressing cancers (314). Gynecol. Oncol. 2022, 166, S164–S165. [Google Scholar] [CrossRef]
- Castelletti, L.; Yeo, D.; van Zandwijk, N.; Rasko, J.E.J. Anti-Mesothelin CAR T cell therapy for malignant mesothelioma. Biomark Res. 2021, 9, 11. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Riviere, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Q.J.; Yang, S.; Kochenderfer, J.N.; Zheng, Z.; Zhong, X.; Sadelain, M.; Eshhar, Z.; Rosenberg, S.A.; Morgan, R.A. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 2009, 183, 5563–5574. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Ghobadi, A.; Pennella, E.J.; Vanas, J.; Powell, C.; Pavelova, M.; Wagner, C.; Kuo, M.; Ullmann, C.D.; Hassan, R.; et al. Feasibility and preliminary safety and efficacy of first-in-human intraperitoneal delivery of MCY-M11, anti-human-mesothelin CAR mRNA transfected into peripheral blood mononuclear cells, for ovarian cancer and malignant peritoneal mesothelioma. J. Clin. Oncol. 2020, 38, 3014. [Google Scholar] [CrossRef]
- Chen, J.; Hu, J.; Gu, L.; Ji, F.; Zhang, F.; Zhang, M.; Li, J.; Chen, Z.; Jiang, L.; Zhang, Y.; et al. Anti-mesothelin CAR-T immunotherapy in patients with ovarian cancer. Cancer Immunol. Immunother. 2022, 72, 409–425. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Stancovski, I.; Schindler, D.G.; Waks, T.; Yarden, Y.; Sela, M.; Eshhar, Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J. Immunol. 1993, 151, 6577–6582. [Google Scholar] [CrossRef]
- Budi, H.S.; Ahmad, F.N.; Achmad, H.; Ansari, M.J.; Mikhailova, M.V.; Suksatan, W.; Chupradit, S.; Shomali, N.; Marofi, F. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor (CAR) for tumor immunotherapy; recent progress. Stem Cell. Res. Ther. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Heslop, H.E. Safer CARS. Mol. Ther. 2010, 18, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Navai, S.A.; Derenzo, C.; Joseph, S.; Sanber, K.; Byrd, T.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Mathew, P.R.; et al. Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; p. LB–147. [Google Scholar]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Pearson, A.D.; Perry, R.H.; Jaros, E.; Kelly, P.J. Prognostic significance of the c-erbB-2 oncogene product in childhood medulloblastoma. Br. J. Cancer 1995, 71, 473–477. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Kunkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor-Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; Dai, H.; Shi, F.; Yang, Q.; Han, W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020, 22, 573–580. [Google Scholar] [CrossRef]
- Albert, C.M.; Pinto, N.R.; Taylor, M.; Wilson, A.; Rawkings-Rhea, S.; Mgebroff, S.; Brown, C.; Lindgren, C.; Huang, W.; Seidel, K.; et al. STRIvE-01: Phase I study of EGFR806 CAR T-cell immunotherapy for recurrent/refractory solid tumors in children and young adults. J. Clin. Oncol. 2022, 40, 2541. [Google Scholar] [CrossRef]
- Davies, D.M.; Foster, J.; Van Der Stegen, S.J.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L.; et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 565–576. [Google Scholar] [CrossRef]
- Papa, S.; Adami, A.; Metoudi, M.; Achkova, D.; van Schalkwyk, M.; Parente Pereira, A.; Bosshard-Carter, L.; Whilding, L.; van der Stegen, S.; Davies, D.M.; et al. T4 immunotherapy of head and neck squamous cell carcinoma using pan-ErbB targeted CAR T-cells. Cancer Res. 2017, 77, CT118. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Tang, O.Y.; Tian, L.; Yoder, T.; Xu, R.; Kulikovskaya, I.; Gupta, M.; Melenhorst, J.J.; Lacey, S.F.; O’Rourke, D.M.; Binder, Z.A. PD1 Expression in EGFRvIII-Directed CAR T Cell Infusion Product for Glioblastoma Is Associated with Clinical Response. Front Immunol. 2022, 13, 872756. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M.; Moshfegh, A.; Daneshmanesh, A.H.; Khan, A.S.; Mikaelsson, E.; Osterborg, A.; Mellstedt, H. The receptor tyrosine kinase ROR1--an oncofetal antigen for targeted cancer therapy. Semin. Cancer Biol. 2014, 29, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J. ROR1: An orphan becomes apparent. Blood 2022, 140, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Kamizaki, K.; Minami, Y. The Ror-Family Receptors in Development, Tissue Regeneration and Age-Related Disease. Front. Cell Dev. Biol. 2022, 10, 891763. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Barrientos, J.C.; Furman, R.R.; Mei, M.; Barr, P.M.; Choi, M.Y.; de Vos, S.; Kallam, A.; Patel, K.; Kipps, T.J.; et al. Zilovertamab Vedotin Targeting of ROR1 as Therapy for Lymphoid Cancers. NEJM Evid. 2021, 1, 1–11. [Google Scholar] [CrossRef]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503. [Google Scholar] [CrossRef]
- Peng, H.; Nerreter, T.; Mestermann, K.; Wachter, J.; Chang, J.; Hudecek, M.; Rader, C. ROR1-targeting switchable CAR-T cells for cancer therapy. Oncogene 2022, 41, 4104–4114. [Google Scholar] [CrossRef]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208. [Google Scholar] [CrossRef]
- Specht, J.M.; Lee, S.; Turtle, C.; Berger, C.; Veatch, J.; Gooley, T.; Mullane, E.; Chaney, C.; Riddell, S.; Maloney, D. Phase I study of immunotherapy for advanced ROR1+ malignancies with autologous ROR1-specific chimeric antigen receptor-modified (CAR)-T cells. J. Clin. Oncol. 2018, 36, TPS79. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef]
- Burchell, J.M.; Beatson, R.; Graham, R.; Taylor-Papadimitriou, J.; Tajadura-Ortega, V. O-linked mucin-type glycosylation in breast cancer. Biochem. Soc. Trans. 2018, 46, 779–788. [Google Scholar] [CrossRef]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- You, F.; Jiang, L.; Zhang, B.; Lu, Q.; Zhou, Q.; Liao, X.; Wu, H.; Du, K.; Zhu, Y.; Meng, H.; et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci. China Life Sci. 2016, 59, 386–397. [Google Scholar] [CrossRef]
- Gutierrez, R.; Shah, P.D.; Hamid, O.; Garfall, A.L.; Posey, A.; Bishop, M.R.; Blumenschein, G.R.; Johnson, M.L.; Lee, S.; Luke, J.J.; et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J. Clin. Oncol. 2021, 39, e14513. [Google Scholar] [CrossRef]
- Specht, J.M.; Maloney, D.G.; Yeung, C.; Wu, V.; Bamdad, C. Phase I study of adoptive immunotherapy for advanced MUC1* positive breast cancer with autologous T cells engineered to express a chimeric antigen receptor, huMNC2-CAR44 specific for a cleaved form of MUC1 (MUC1*). J. Clin. Oncol. 2021, 39, TPS2663. [Google Scholar] [CrossRef]
- Zhang, Y.; Kozlowska, A.; Fritz, J.; Zhao, Y.; Torre, C.P.L.; Cranert, S.; Wang, S.; Codde, R.; Argus, E.; Ibitokou, S.; et al. 123 P-MUC1C-ALLO1: A fully allogeneic stem cell memory T cell (TSCM) CAR-T therapy with broad potential in solid tumor. J. ImmunoTherapy Cancer 2021, 9, A132. [Google Scholar] [CrossRef]
- Lee, D.H.; Choi, S.; Park, Y.; Jin, H.S. Mucin1 and Mucin16: Therapeutic Targets for Cancer Therapy. Pharmaceuticals 2021, 14, 1053. [Google Scholar] [CrossRef]
- Bast, R.C., Jr.; Klug, T.L.; St John, E.; Jenison, E.; Niloff, J.M.; Lazarus, H.; Berkowitz, R.S.; Leavitt, T.; Griffiths, C.T.; Parker, L.; et al. A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N. Engl. J. Med. 1983, 309, 883–887. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Evtimov, V.J.; Hammett, M.V.; Nguyen, N.N.; Zhuang, J.; Hudson, P.J.; Howard, M.C.; Pupovac, A.; Trounson, A.O.; Boyd, R.L. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol. Ther. Oncolytics 2021, 20, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Woll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Jin, Q.; Quan, C. CLDN6: From Traditional Barrier Function to Emerging Roles in Cancers. Int. J. Mol. Sci. 2021, 22, 13416. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.; Mackensen, A.; Koenecke, C.; Alsdorf, W.; Wagner-Drouet, E.; Heudobler, D.; Borchmann, P.; Bokemeyer, C.; Klobuch, S.; Desuki, A.; et al. BNT211: A Phase I trial to evaluate safety and efficacy of CLDN6 CAR-T cells and CARVac-mediated in vivo expansion in patients with CLDN6-positive advanced solid tumors. Cancer Res. 2022, 82, CT002. [Google Scholar] [CrossRef]
- Carvalho, T. mRNA vaccines boost BioNTech’s CAR T cell therapy. Nat. Med. 2022, 28, 1968–1969. [Google Scholar] [CrossRef]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Zhan, X.; Wang, B.; Li, Z.; Li, J.; Wang, H.; Chen, L.; Jiang, H.; Wu, M.; Xiao, J.; Peng, X.; et al. Phase I trial of Claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. J. Clin. Oncol. 2019, 37, 2509. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M.; Hulet, S.W.; Connor, J.R.; Gillespie, G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Cancer Res. 1999, 5, 985–990. [Google Scholar]
- Kioi, M.; Kawakami, M.; Shimamura, T.; Husain, S.R.; Puri, R.K. Interleukin-13 receptor alpha2 chain: A potential biomarker and molecular target for ovarian cancer therapy. Cancer 2006, 107, 1407–1418. [Google Scholar] [CrossRef]
- Fujisawa, T.; Nakashima, H.; Nakajima, A.; Joshi, B.H.; Puri, R.K. Targeting IL-13Ralpha2 in human pancreatic ductal adenocarcinoma with combination therapy of IL-13-PE and gemcitabine. Int. J. Cancer 2011, 128, 1221–1231. [Google Scholar] [CrossRef]
- Barderas, R.; Bartolome, R.A.; Fernandez-Acenero, M.J.; Torres, S.; Casal, J.I. High expression of IL-13 receptor alpha2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012, 72, 2780–2790. [Google Scholar] [CrossRef]
- Kawakami, M.; Kawakami, K.; Kasperbauer, J.L.; Hinkley, L.L.; Tsukuda, M.; Strome, S.E.; Puri, R.K. Interleukin-13 receptor alpha2 chain in human head and neck cancer serves as a unique diagnostic marker. Clin. Cancer Res. 2003, 9, 6381–6388. [Google Scholar]
- Jaen, M.; Martin-Regalado, A.; Bartolome, R.A.; Robles, J.; Casal, J.I. Interleukin 13 receptor alpha 2 (IL13Ralpha2): Expression, signaling pathways and therapeutic applications in cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188802. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Mol. Med. 2000, 6, 440–449. [Google Scholar] [CrossRef]
- Gauchat, J.F.; Schlagenhauf, E.; Feng, N.P.; Moser, R.; Yamage, M.; Jeannin, P.; Alouani, S.; Elson, G.; Notarangelo, L.D.; Wells, T.; et al. A novel 4-kb interleukin-13 receptor alpha mRNA expressed in human B, T, and endothelial cells encoding an alternate type-II interleukin-4/interleukin-13 receptor. Eur. J. Immunol. 1997, 27, 971–978. [Google Scholar] [CrossRef]
- Knudson, K.M.; Hwang, S.; McCann, M.S.; Joshi, B.H.; Husain, S.R.; Puri, R.K. Recent Advances in IL-13Ralpha2-Directed Cancer Immunotherapy. Front. Immunol. 2022, 13, 878365. [Google Scholar] [CrossRef]
- Debinski, W.; Thompson, J.P. Retargeting interleukin 13 for radioimmunodetection and radioimmunotherapy of human high-grade gliomas. Clin. Cancer Res. 1999, 5, 3143s–3147s. [Google Scholar]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 2022, 24, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Xella, A.; Hibbard, J.C.; Salvary, V.; Aguilar, B.; Wagner, J.; Dezube, B.; Niss, K.; Bayless, L.; Edinger, J.; et al. Abstract CT541A: Oncolytic viral reshaping of the tumor microenvironment to promote CAR T cell therapy for glioblastoma. Cancer Res. 2022, 82, CT541A. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Kuratsukuri, K.; Landas, S.; Imaida, K.; Rovito, P.M., Jr.; Wang, C.Y.; Haas, G.P. Expression of prostate-specific membrane antigen in normal and malignant human tissues. World J. Surg. 2006, 30, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Sacha, P.; Zamecnik, J.; Barinka, C.; Hlouchova, K.; Vicha, A.; Mlcochova, P.; Hilgert, I.; Eckschlager, T.; Konvalinka, J. Expression of glutamate carboxypeptidase II in human brain. Neuroscience 2007, 144, 1361–1372. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Sathekge, M.; de Spiegeleer, B.; De Jonghe, P.J.; Debruyne, P.R.; Borms, M.; Beels, L.; Maes, A. PSMA expression on neovasculature of solid tumors. Histol. Histopathol. 2020, 35, 919–927. [Google Scholar] [CrossRef]
- Noto, B.; Vrachimis, A.; Schafers, M.; Stegger, L.; Rahbar, K. Subacute Stroke Mimicking Cerebral Metastasis in 68Ga-PSMA-HBED-CC PET/CT. Clin. Nucl. Med. 2016, 41, e449–e451. [Google Scholar] [CrossRef]
- Oh, G.; Miles, K. Subacute Cerebellar Infarction with Uptake on 68Ga-Prostate-Specific Membrane Antigen PET/CT. Clin. Nucl. Med. 2018, 43, 134–135. [Google Scholar] [CrossRef]
- Wong, V.C.K.; Shen, L.; Nasser, E.; Adams, D.N.; Mansberg, R. 68Ga-Prostate-Specific Membrane Antigen Uptake in Cerebral Tuberculosis. Clin. Nucl. Med. 2020, 45, 238–240. [Google Scholar] [CrossRef]
- Vadi, S.K.; Kumar, R.; Singh, H.; Singh, S.K.; Mittal, B.R. 68Ga-Prostate-Specific Membrane Antigen Expression in Neurocysticercosis Lesions in a Patient with Prostate Carcinoma. Clin. Nucl. Med. 2018, 43, e122–e124. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Slovin, S.F.; Wang, X.; Hullings, M.; Arauz, G.; Bartido, S.; Lewis, J.S.; Schoder, H.; Zanzonico, P.; Scher, H.I.; Sadelain, M.; et al. Chimeric antigen receptor (CAR+) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients with castrate metastatic prostate cancer. J. Clin. Oncol. 2013, 31, 72. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation and Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Lucas, P.J.; Kim, S.J.; Melby, S.J.; Gress, R.E. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J. Exp. Med. 2000, 191, 1187–1196. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- McKean, M.; Carabasi, M.H.; Stein, M.N.; Schweizer, M.T.; Luke, J.J.; Narayan, V.; Parikh, R.A.; Pachynski, R.K.; Zhang, J.; Peddareddigari, V.G.R.; et al. Safety and early efficacy results from a phase 1, multicenter trial of PSMA-targeted armored CAR T cells in patients with advanced mCRPC. J. Clin. Oncol. 2022, 40, 94. [Google Scholar] [CrossRef]
- Barrett, D.; Chagin, K.; Fountaine, T.J.; Moore, A.; Ka, M.; Gladney, W.; Verma, B.; Luo, Y.; Hui, D.; Peddareddigari, V.G.R. TmPSMA-02: A CD2 endodomain containing double armored PSMA CAR T with enhanced efficacy and lower immune toxicity. J. Clin. Oncol. 2022, 40, 158. [Google Scholar] [CrossRef]
- Slovin, S.; Dorff, T.B.; Falchook, G.S.; Wei, X.X.; Gao, X.; McKay, R.R.; Oh, D.Y.; Wibmer, A.G.; Spear, M.A.; McCaigue, J.; et al. Phase 1 study of P-PSMA-101 CAR-T cells in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 98. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J. Cancer Res. Clin. Oncol. 2022, 148, 2643–2652. [Google Scholar] [CrossRef]
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J. Hematol. Oncol. 2021, 14, 191. [Google Scholar] [CrossRef]
- Heczey, A.; Courtney, A.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Xu, X.; Ngai, H.; Di Piero, E.; Sher, A.; et al. Natural Killer T Cells Expressing a GD2-CAR and IL-15 Are Safe and Can Induce Complete Remission in Children with Relapsed Neuroblastoma—A First-in-Human, Phase 1 Trial. Mol. Ther. 2021, 29, 198. [Google Scholar]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Zhou, W.T.; Jin, W.L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef]
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct. Target Ther. 2021, 6, 125. [Google Scholar] [CrossRef]
- Hu, G.; Li, G.; Wen, W.; Ding, W.; Zhou, Z.; Zheng, Y.; Huang, T.; Ren, J.; Chen, R.; Zhu, D.; et al. Case report: B7-H3 CAR-T therapy partially controls tumor growth in a basal cell carcinoma patient. Front. Oncol. 2022, 12, 956593. [Google Scholar] [CrossRef]
- Vitanza, N.; Wilson, A.; Gust, J.; Huang, W.; Perez, F.; Albert, C.; Pinto, N.; Gardner, R.; Orentas, R.; Berens, M.; et al. IMMU-11. Clinical updates and correlative findings from the first patient with DIPG treated with intracranial CAR T cells. Neuro-Oncol. 2021, 23, i29. [Google Scholar] [CrossRef]
- Pinto, N.R.; Albert, C.M.; Taylor, M.; Wilson, A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; Narayanaswany, P.; Wu, V.; Brown, C.; et al. STRIVE-02: A first-in-human phase 1 trial of systemic B7H3 CAR T cells for children and young adults with relapsed/refractory solid tumors. J. Clin. Oncol. 2022, 40, 10011. [Google Scholar] [CrossRef]
- Tang, X.; Liu, F.; Liu, Z.; Cao, Y.; Zhang, Z.; Wang, Y.; Huang, J.; Fan, S.; Zhao, S.; Chen, Y.; et al. Bioactivity and safety of B7-H3-targeted chimeric antigen receptor T cells against anaplastic meningioma. Clin. Transl. Immunol. 2020, 9, e1137. [Google Scholar] [CrossRef]
- Li, D.; English, H.; Hong, J.; Liang, T.; Merlino, G.; Day, C.P.; Ho, M. A novel PD-L1-targeted shark V(NAR) single-domain-based CAR-T cell strategy for treating breast cancer and liver cancer. Mol. Ther. Oncolytics 2022, 24, 849–863. [Google Scholar] [CrossRef]
- Yang, C.Y.; Fan, M.H.; Miao, C.H.; Liao, Y.J.; Yuan, R.H.; Liu, C.L. Engineering Chimeric Antigen Receptor T Cells against Immune Checkpoint Inhibitors PD-1/PD-L1 for Treating Pancreatic Cancer. Mol. Ther. Oncolytics 2020, 17, 571–585. [Google Scholar] [CrossRef]
- Bajor, M.; Graczyk-Jarzynka, A.; Marhelava, K.; Burdzinska, A.; Muchowicz, A.; Goral, A.; Zhylko, A.; Soroczynska, K.; Retecki, K.; Krawczyk, M.; et al. PD-L1 CAR effector cells induce self-amplifying cytotoxic effects against target cells. J. Immunother. Cancer 2022, 10, e002500. [Google Scholar] [CrossRef]
- Liu, W.N.; So, W.Y.; Harden, S.L.; Fong, S.Y.; Wong, M.X.Y.; Tan, W.W.S.; Tan, S.Y.; Ong, J.K.L.; Rajarethinam, R.; Liu, M.; et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci. Adv. 2022, 8, eadd1187. [Google Scholar] [CrossRef]
- Robbins, Y.; Greene, S.; Friedman, J.; Clavijo, P.E.; Van Waes, C.; Fabian, K.P.; Padget, M.R.; Abdul Sater, H.; Lee, J.H.; Soon-Shiong, P.; et al. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. Elife 2020, 9, e54854. [Google Scholar] [CrossRef]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. CD19-specific CAR T Cells that Express a PD-1/CD28 Chimeric Switch-Receptor are Effective in Patients with PD-L1-positive B-Cell Lymphoma. Clin. Cancer Res. 2021, 27, 473–484. [Google Scholar] [CrossRef]
- Hickman, T.L.; Choi, E.; Whiteman, K.R.; Muralidharan, S.; Pai, T.; Johnson, T.; Parikh, A.; Friedman, T.; Gilbert, M.; Shen, B.; et al. BOXR1030, an anti-GPC3 CAR with exogenous GOT2 expression, shows enhanced T cell metabolism and improved anti-cell line derived tumor xenograft activity. PLoS ONE 2022, 17, e0266980. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xing, C.; Jiang, S.; Yu, K.; Dai, S.; Kong, H.; Jin, Y.; Shan, Y.; Yang, W.; Wang, Z.; et al. Long term complete response of advanced hepatocellular carcinoma to glypican-3 specific chimeric antigen receptor T-Cells plus sorafenib, a case report. Front. Immunol. 2022, 13, 963031. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Fu, Q.; Zhao, Q.; Zheng, Y.; Liu, L.; Li, Z.; Dai, X.; Wang, H.; Zhu, X.; Zhao, P.; et al. Phase I trial of fourth-generation chimeric antigen receptor T-cells targeting glypican-3 for advanced hepatocellular carcinoma. J. Clin. Oncol. 2021, 39, 4088. [Google Scholar] [CrossRef]
- Obajdin, J.; Davies, D.M.; Maher, J. Engineering of chimeric natural killer cell receptors to develop precision adoptive immunotherapies for cancer. Clin. Exp. Immunol. 2020, 202, 11–27. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Sallman, D.A.; Brayer, J.B.; Poire, X.; Havelange, V.; Awada, A.; Lewalle, P.; Odunsi, K.; Wang, E.S.; Lonez, C.; Lequertier, T.; et al. Results from the Completed Dose-Escalation of the Hematological Arm of the Phase I Think Study Evaluating Multiple Infusions of NKG2D-Based CAR T-Cells as Standalone Therapy in Relapse/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome Patients. Blood 2019, 134, 3826. [Google Scholar] [CrossRef]
- Sallman, D.A.; Al-Homsi, A.S.; Davila, M.; Kerre, T.; Moors, I.; Poire, X.; Lehmann, F.F. Results from the Phase I Clinical Studies Evaluating CYAD-01, a first-generation NKG2D CAR T-cell Product in Relapsed or Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome Patients. Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Sallman, D.A.; Brayer, J.; Sagatys, E.M.; Lonez, C.; Breman, E.; Agaugue, S.; Verma, B.; Gilham, D.E.; Lehmann, F.F.; Davila, M.L. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica 2018, 103, e424–e426. [Google Scholar] [CrossRef]
- Brayer, J.B.; Sallman, D.A.; Kerre, T.; Poire, X.; Havelange, V.; Lewalle, P.; Wang, E.S.; Selleslag, D.; Anguille, S.; Beguin, Y.; et al. Results and perspectives from Phase 1 studies assessing the safety and clinical activity of multiple doses of a NKG2D-based CAR-T therapy, CYAD-01, in metastatic solid tumors. J. Immunother. Cancer 2018, 6, 114. [Google Scholar]
- Shaza, L.; Hendlisz, A.; Awada, A.; Canon, J.L.; Carrasco, J.; Cutsem, E.V.; Dekervel, J.; Alcantar-Orozco, E.; Renard, F.; Cerf, E.; et al. Results from the completed dose-escalation phase I SHRINK study evaluating the autologous NKG2D-based CAR T-cell therapy CYAD-01 in metastatic colorectal cancer patients. Lung 2019, 3, 33. [Google Scholar]
- Al-Homsi, A.S.; Purev, E.; Lewalle, P.; Abdul-Hay, M.; Pollyea, D.A.; Salaroli, A.; Lequertier, T.; Alcantar-Orozco, E.; Borghese, F.; Lonez, C.; et al. Interim Results from the Phase I Deplethink Trial Evaluating the Infusion of a NKG2D CAR T-Cell Therapy Post a Non-Myeloablative Conditioning in Relapse or Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome Patients. Blood 2019, 134, 3844. [Google Scholar] [CrossRef]
- Prenen, H.; Dekervel, J.; Hendlisz, A.; Anguille, S.; Awada, A.; Cerf, E.; Lonez, C.; Breman, E.; Dheur, M.-S.; Alcantar-Orozco, E.; et al. Updated data from alloSHRINK phase I first-in-human study evaluating CYAD-101, an innovative non-gene edited allogeneic CAR-T in mCRC. J. Clin. Oncol. 2021, 39, 74. [Google Scholar] [CrossRef]
- Michaux, A.; Mauen, S.; Breman, E.; Dheur, M.S.; Twyffels, L.; Saerens, L.; Jacques-Hespel, C.; Gauthy, E.; Agaugue, S.; Gilham, D.E.; et al. Clinical Grade Manufacture of CYAD-101, a NKG2D-based, First in Class, Non-Gene-edited Allogeneic CAR T-Cell Therapy. J. Immunother. 2022, 45, 150–161. [Google Scholar] [CrossRef]
- Ijaz, A.; Khan, A.Y.; Malik, S.U.; Faridi, W.; Fraz, M.A.; Usman, M.; Tariq, M.J.; Durer, S.; Durer, C.; Russ, A.; et al. Significant Risk of Graft-versus-Host Disease with Exposure to Checkpoint Inhibitors before and after Allogeneic Transplantation. Biol. Blood Marrow Transpl. 2019, 25, 94–99. [Google Scholar] [CrossRef]
- Ono, H.; Sakamoto, H.; Yoshida, T.; Saeki, N. Prostate stem cell antigen is expressed in normal and malignant human brain tissues. Oncol. Lett. 2018, 15, 3081–3084. [Google Scholar] [CrossRef]
- Dorff, T.B.; Blanchard, S.; Martirosyan, H.; Adkins, L.; Dhapola, G.; Moriarty, A.; Wagner, J.R.; Chaudhry, A.; D’Apuzzo, M.; Kuhn, P.; et al. Phase 1 study of PSCA-targeted chimeric antigen receptor (CAR) T cell therapy for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 91. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef]
- Katz, S.C.; Hardaway, J.; Prince, E.; Guha, P.; Cunetta, M.; Moody, A.; Wang, L.J.; Armenio, V.; Espat, N.J.; Junghans, R.P. HITM-SIR: Phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene Ther. 2020, 27, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tran, B.; Haanen, J.; Hurwitz, M.; Sacher, A.; Agarwal, N.; Tannir, N.; Budde, E.; Harrison, S.; Klobuch, S.; et al. 558 CTX130 allogeneic CRISPR-Cas9–engineered chimeric antigen receptor (CAR) T cells in patients with advanced clear cell renal cell carcinoma: Results from the phase 1 COBALT-RCC study. J. ImmunoTherapy Cancer 2022, 10, A584. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Antonucci, L.; Canciani, G.; Mastronuzzi, A.; Carai, A.; Del Baldo, G.; Del Bufalo, F. CAR-T Therapy for Pediatric High-Grade Gliomas: Peculiarities, Current Investigations and Future Strategies. Front. Immunol. 2022, 13, 867154. [Google Scholar] [CrossRef]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients with Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Kohn, E.C.; LoRusso, P.; Houston, N.D.; Coleman, R.L.; Buzoianu, M.; Robbie, G.; Lechleider, R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Invest New Drugs 2013, 31, 77–84. [Google Scholar] [CrossRef]
- Gan, H.K.; Parakh, S.; Lee, F.T.; Tebbutt, N.C.; Ameratunga, M.; Lee, S.T.; O’Keefe, G.J.; Gong, S.J.; Vanrenen, C.; Caine, J.; et al. A phase 1 safety and bioimaging trial of antibody DS-8895a against EphA2 in patients with advanced or metastatic EphA2 positive cancers. Investig. New Drugs 2022, 40, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Britschgi, C.; Schuberth, P.; Bankel, L.; Nguyen-Kim, T.D.L.; Gulati, P.; Weder, W.; Opitz, I.; Lauk, O.; Caviezel, C.; et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: First report of FAPME, a phase I clinical trial. Ann. Oncol. 2021, 32, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Luo, T.; Lu, Z.; Zhang, H.; Tong, C.; Wang, S.; Tang, F.; Ai, G. 737MO—EpCAM-targeted CAR-T cell therapy in patients with advanced colorectal and gastric cancers. Ann. Oncol. 2022, 33, S331–S355. [Google Scholar] [CrossRef]
- Wang, S.; Fang, J.; Wang, H.; Xu, Q. 177 A severe cytokine release syndrome with respiratory failure in recurrent mesothelioma induced by EpCAM CAR-T cells infusion: A case report. J. ImmunoTherapy Cancer 2020, 8, A105. [Google Scholar] [CrossRef]
- Qin, D.; Li, D.; Zhang, B.; Chen, Y.; Liao, X.; Li, X.; Alexander, P.B.; Wang, Y.; Li, Q.J. Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology 2020, 9, 1806009. [Google Scholar] [CrossRef]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef]
- Li, C.; Yang, N.; Li, H.; Wang, Z. Robo1-specific chimeric antigen receptor natural killer cell therapy for pancreatic ductal adenocarcinoma with liver metastasis. J. Cancer Res. Ther. 2020, 16, 393–396. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
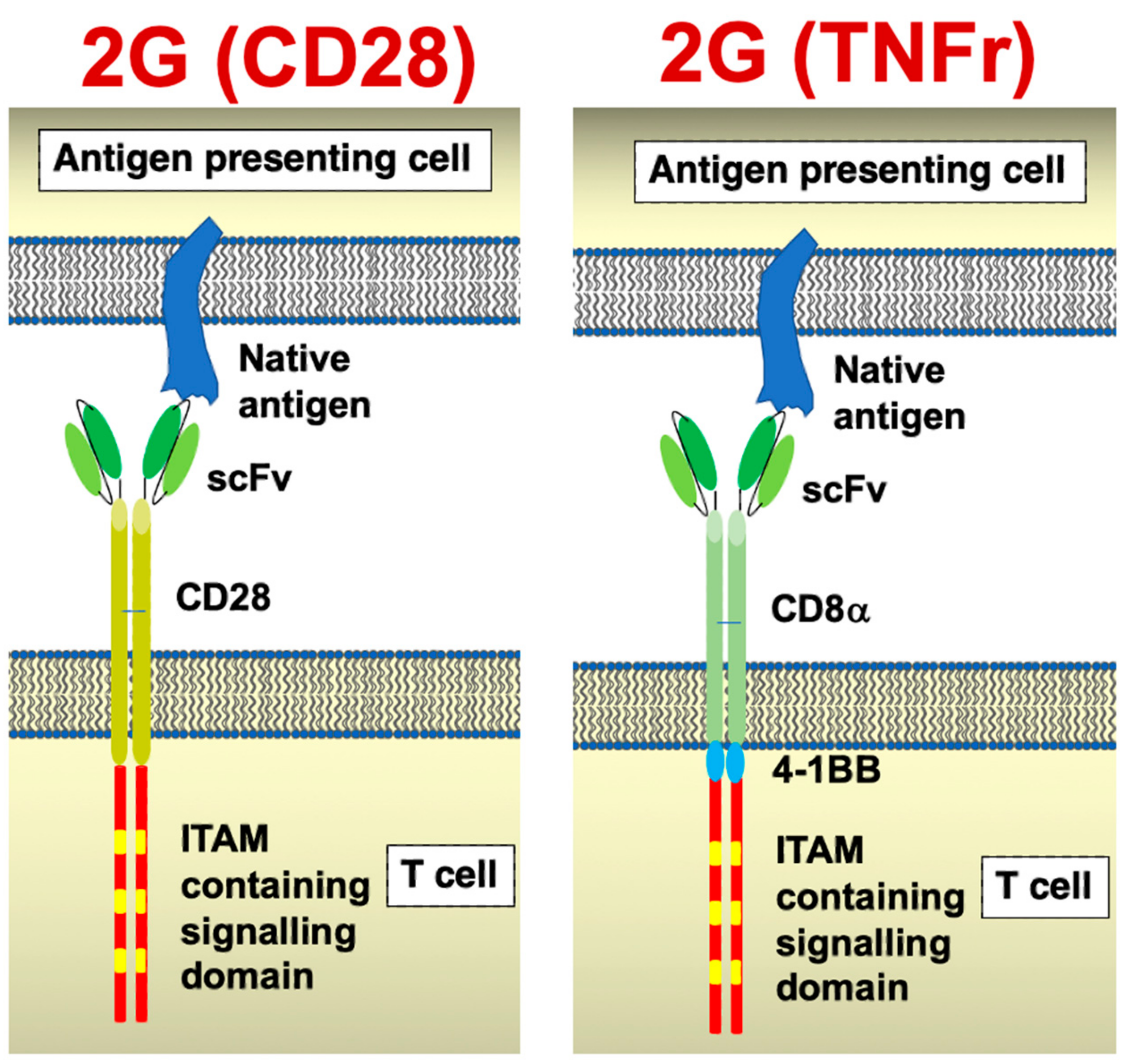
- Muliaditan, T.; Halim, L.; Whilding, L.M.; Draper, B.; Achkova, D.Y.; Kausar, F.; Glover, M.; Bechman, N.; Arulappu, A.; Sanchez, J.; et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep. Med. 2021, 2, 100457. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Mesothelin+ tumours | Ruijin Hospital | mRNA electroporated CAR T-cells | NCT04981691 |
| Mesothelin+ tumours | 2nd Affiliated Hospital Guangzhou Medical University | One of many targets under study | NCT03198052 |
| Mesothelin+ tumours | Shanghai Pudong Hospital | NCT05531708 | |
| Ovarian cancer | Weijia Fang | NCT05372692 | |
| Pancreatic cancer | Shenzhen BinDeBio Ltd. | NCT03638193 | |
| Mesothelin+ tumours | University of Pennsylvania | Investigating different routes of administration | NCT03054298 |
| Gastric cancer | Shenzhen BinDeBio Ltd. | One of multiple targets and indications | NCT03941626 |
| Mesothelin+ tumours | Shanghai Mengchao Cancer Hospital | CAR T-cells secrete PD1 nanobodies | NCT05373147 |
| Mesothelin+ tumours | TCR2 Therapeutics | See text for description of CAR; Also incorporates a PD1/CD28 switch receptor | NCT05451849 |
| Ovarian cancer | Shanghai 6th People’s Hospital | NCT03814447 | |
| Mesothelin+ tumours | TCR2 Therapeutics | See text for description of CAR | NCT03907852 |
| Mesothelin+ tumours | 1st Affil. Hospital with Nanjing Medical University | Cells co-express CD19 CAR and truncated EGF receptor safety switch | NCT05166070 |
| Pancreatic cancer | 1st Affil. Hospital of Harbin Medical University | Explores influence of gut microbiome on CAR T-cell metabolism and function | NCT04203459 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| CNS tumours | Baylor College of Medicine | Intracranial | NCT02442297 |
| Lung cancer 1 | Guanghou Med. University | Systemic or regional | NCT3198052 |
| Solid tumours | Bellicum Pharmaceuticals | Dual switch CAR T-cells with rimiducid | NCT04650451 |
| Paediatric CNS tumours | Seattle Children’s Hospital | Intracranial [31] | NCT03500991 |
| HER2 3+ solid tumours | Triumvira | TAC 2 comprising HER2-specific DARPin fused to aCD3ε scFv and CD4 spacer, transmembrane and endodomain | NCT04727151 |
| Brain/leptomeningeal malignancy | City of Hope Medical Center | Intracranial | NCT03696030 |
| Breast cancer | Shenzen Geno-Immune Medical Institute | CAR also targets GD2 and CD44v6 | NCT04430595 |
| Multiple solid tumours | Baylor College of Medicine | Combination of CAR T-cells with oncolytic adenovirus | NCT03740256 |
| Serosal cavity metastases | Sichuan University | CAR also targets PD-L1 | NCT04684459 |
| Multiple solid tumours | Shanghai PerHum Therapeutics | Intravenous delivery | NCT04511871 |
| Brain tumours 1 | Xuanwu Hospital | Patients may also receive anti-PD-L1 | NCT03423992 |
| Sarcoma | Baylor College of Medicine | Patients also receive immune checkpoint blockade | NCT04995003 |
| Ependymoma | Pediatric brain tumor consortium | Intravenous. May be followed by surgery | NCT04903080 |
| Brain tumours | City of Hope Medical Center | Intracranial | NCT03389230 |
| Solid tumours | Carisma Therapeutics | CAR-engineered macrophages administered intravenously | NCT04660929 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| SCCHN 1 | King’s College London | Intratumoural delivery of panErbB CAR T-cells | NCT01818323 |
| NSCLC 2 | Sun Yat-Sen University | CXCR5 armoured CAR T-cells | NCT04153799 |
| NSCLC | 2nd Affiliated Hospital Guangzhou Medical University | CXCR5 armoured CAR T-cells | NCT05060796 |
| Solid tumours | Seattle Children’s Hospital | EGFR806 antibody | NCT03618381 |
| Solid tumours | Chinese PLA General Hospital | TGF-β receptor knockout | NCT04976218 |
| Paediatric CNS tumours | Seattle Children’s Hospital | EGFR806 antibody—intracranial delivery | NCT03638167 |
| Solid tumours | 2nd Affiliated Hospital Guangzhou Medical University | One of many targets under study | NCT03198052 |
| Lung and TNBC | 2nd Affiliated Hospital Guangzhou Medical University | CAR also targets B7-H3 | NCT05341492 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Glioma | Xuanwu Hospital, Beijing | One of many targets under study | NCT03423992 |
| Glioblastoma | 2nd Affiliated Massachusetts General Hospital | CAR T-cells co-express an EGFR-specific T-cell engager | NCT05024175 |
| Multiple solid tumours | Shenzhen BinDeBio | One of many targets under study | NCT03941626 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Solid tumours | 2nd Affiliated Hospital Guangzhou Medical University | One of multiple targets | NCT03198052 |
| Breast cancer | Minerva Biotechnologies Corp. | Targets MUC1* | NCT04020575 |
| Solid tumours | Poseida Therapeutics | Allogeneic CAR T-cells targeted against MUC1-C | NCT05239143 |
| Sarcomas | Shenzhen Geno-Immune Medical Institute | One of multiple targets | NCT03356782 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Gastric and pancreatic cancer | Changhai Hospital | Collaboration with CARsgen—current trial status unknown | NCT03159819 |
| Solid tumours | CARSgen Pharmaceuticals | Reference [68] | NCT03874897 |
| Lung cancer | 2nd Affiliated Hospital Guangzhou Medical University | One of many targets under study | NCT03198052 |
| Multiple solid tumours | Suzhou Immunofoco Biotechnology Co. | One of many targets under study | NCT05472857 |
| Gastric, GOJ 1 and PDAC 2 | Shenzhen Fifth People’s Hospital | NCT05277987 | |
| Solid tumours | CARSgen Pharmaceuticals | NCT04404595 | |
| Pancreatic and gastric cancers | Shenzhen University General Hospital | NCT05620732 | |
| Solid tumours | Shanghai East Hospital | Developed by Nanjing Legend Biotech Co. | NCT04467853 |
| Gastric, GOJ and PDAC | CARsgen Therapeutics Co. | NCT04581473 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Ovarian, fallopian tube and peritoneal cancer | University of Pennsylvania | Enrolment requires ≥2 + FRα staining in ≥70% of tumour cells. Cells are administered via an intraperitoneal catheter | NCT03585764 |
| Osteosarcoma | Seattle Children’s Hospital/Umoja BioPharma | Fluorescein-specific (universal) CAR T-cells plus folate–fluorescein conjugate | NCT05312411 |
| Ovarian, NSCLC 1 and RCC 2 | Instil Bio | Tumour-infiltrating lymphocytes engineered to express a FRα-specific CD28 + CD40 co-stimulatory receptor | NCT05397093 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Glioma | Xuanwu Hospital | Patients may also receive anti-PDL1 antibody | NCT03423992 |
| Paediatric malignant brain tumours | City of Hope Medical Center | Systemic lymphodepletion followed by intraventricular CAR T-cells | NCT04510051 |
| Various adult brain tumours | City of Hope Medical Center | Intraventricular CAR T-cells | NCT04661384 |
| Malignant glioma | CellabMED | Intravenous delivery | NCT05540873 |
| Melanoma | Jonsson Comprehensive Cancer Center | Intravenous delivery post lymphodepletion | NCT04119024 |
| Glioblastoma | City of Hope Medical Center | Intracranial CAR T-cells administered with systemic nivolumab (anti-PD1) and Ipilimumab (anti-CTLA-4) | NCT04003649 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| PSMA+ tumours | Shenzen Geno-Immune Medical Institute | Includes cases in which PSMA is demonstrated in tumour stroma | NCT04429451 |
| Tumours that co-express PSMA and GD2 | Shenzen Geno-Immune Medical Institute | Bispecific CAR T-cells | NCT05437315 |
| Tumours that co-express PSMA and CD70 | Shenzen Geno-Immune Medical Institute | Bispecific CAR T-cells | NCT05437341 |
| mCRPC 1 | Affil. Hospital of Xuzhou Medical University | Administered in combination with IL-2 | NCT05354375 |
| mCRPC and salivary gland cancer | Poseida | Transgenes delivered using PiggyBac. Vector also contains iCaspase-9 suicide gene | NCT04727151 |
| mCRPC | Zhejiang University | Non-viral gene transfer. Vector includes PD1 inhibitory system. | NCT04768608 |
| mCRPC | AvenCell Europe | Universal CAR in combination with PSMA-specific antibody derivative | NCT04633148 |
| mCRPC | Tmunity | CAR containing CD2 co-stimulatory domain. Dual armouring with dnTGF-βR and PD1/CD28 switch receptor | NCT05489991 |
| Sarcomas | Shenzen Geno-Immune Medical Institute | Multiple CAR T-cells combined targeting antigens that include PSMA | NCT04433221 |
| Neuroblastoma | Shenzen Geno-Immune Medical Institute | CAR also targets GD2 and CD276 | NCT04637503 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| GD2+ Brain Tumours | Baylor College of Medicine | Armoured with constitutively active IL-7 receptor | NCT04099797 |
| GD2+ non-brain Tumours | Baylor College of Medicine | Armoured with constitutively active IL-7 receptor | NCT03635632 |
| Tumours that co-express GD2 and CD70 | Shenzen Geno-Immune Medical Institute | Bispecific CAR T-cells | NCT05438368 |
| Tumours that co-express GD2 and CD56 | Shenzen Geno-Immune Medical Institute | Bispecific CAR T-cells | NCT05437328 |
| Tumours that co-express GD2 and PSMA | Shenzen Geno-Immune Medical Institute | Bispecific CAR T-cells | NCT05437315 |
| Neuroblastoma Osteosarcoma | UNC Lineberger Comprehensive Cancer Center | Armoured with IL-15 and inducible caspase-9 | NCT03721068 |
| Neuroblastoma | Shenzen Geno-Immune Medical Institute | Trispecific CAR T-cells directed against GD2, PSMA and CD276 (B7-H3) | NCT04637503 |
| B-NHL 1 | 7th Affil. Hospital of Sun Yat-sen University | One of multiple targets | NCT04429438 |
| Lung Cancer | UNC Lineberger Comprehensive Cancer Center | Armoured with IL-15 and inducible caspase-9 | NCT05620342 |
| DIPG/DMG 2 | Crystal Mackall MD | Dasatinib-containing culture system [109] | NCT04196413 |
| GD2+ tumours | Bambino Gesu Hospital and Research Institute | Armoured with inducible caspase-9 | NCT03373097 |
| Neuroblastoma Osteosarcoma | National Cancer Institute | Dasatinib-containing culture system | NCT04539366 |
| Breast Cancer | 7th Affil. Hospital of Sun Yat-sen University | One of multiple targets. GD2 considered a breast cancer stem cell marker | NCT04430595 |
| Neuroblastoma | Baylor College of Medicine | IL-15 armoured NKT-cells | NCT03294954 |
| Glioma | Xuanwu Hospital | One of multiple targets | NCT03423992 |
| Sarcomas | Shenzen Geno-Immune Medical Institute | One of multiple targets | NCT03356782 |
| Sarcomas | Shenzen Geno-Immune Medical Institute | One of multiple targets. Combination treatment with chemotherapy and/ or tumour vaccines | NCT04433221 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Glioblastoma | UNC Lineberger Comprehensive Cancer Center of Medicine | Intraventricular infusion | NCT05366179 |
| Ovarian cancer | UNC Lineberger Comprehensive Cancer Center of Medicine | Intraperitoneal infusion | NCT04670068 |
| Paediatric CNS tumours | Seattle Children’s Hospital | Intracranial delivery. Methotrexate selection system for CAR T-cells. Reference [113] | NCT04185038 |
| Solid tumours | 2nd Affil. Hospital of Guangzhou Medical University | One of multiple targets | NCT03198052 |
| Glioblastoma | Beijing Tiantan Hospital | NCT05241392 | |
| Paediatric solid tumours | St. Jude’s Children’s Research Hospital | NCT04897321 | |
| Solid tumours in children and young adults | Seattle Children’s Hospital | In one arm, patients receive T-cells that co-express the B7-H3 CAR with a CD19 CAR in an effort to increase expansion and persistence. Reference [114] | NCT04483778 |
| Hepatocellular carcinoma | Affil. Hospital of Xuzhou Medical University | Transhepatic arterial infusion | NCT05323201 |
| Ovarian carcinoma | Affil. Hospital of Xuzhou Medical University | Intraperitoneal infusion | NCT05211557 |
| Glioblastoma Multiforme | Crystal Mackall MD | Locoregional delivery | NCT05474378 |
| Solid tumours | Shenzen Geno-Immune Medical Institute | NCT04432649 | |
| Glioblastoma | 2nd Affil. Hospital School of Medicine, Zhejiang University | Intracerebral CAR T-cells are administered between temozolomide cycles | NCT04077866 |
| Solid tumours | 1st Peoples Hospital of Lianyungang | NCT05515185 | |
| Lung cancer TNBC 1 | 2nd Affil. Hospital of Guangzhou Medical University | Also targeted against EGFR | NCT05341492 |
| Melanoma Lung cancer Colorectal cancer | 4th Hospital of Hebei Medical University | NCT05190185 | |
| Pancreatic cancer | Shenzhen University General Hospital | NCT05143151 | |
| Neuroblastoma Osteosarcoma Gastric and Lung cancer | PersonGen BioTherapeutics | Intravenous and intra-tumoural delivery | NCT04864821 |
| Neuroblastoma | Shenzen Geno-Immune Medical Institute | One of multiple targets | NCT04637503 |
| Baylor College of Medicine | IL-15 armoured NKT-cells | NCT03294954 | |
| Glioma | Xuanwu Hospital | One of multiple targets | NCT03423992 |
| Sarcomas | Shenzen Geno-Immune Medical Institute | One of multiple targets | NCT03356782 |
| Sarcomas | Shenzen Geno-Immune Medical Institute | One of multiple targets. Combination treatment with chemotherapy and/or tumour vaccines | NCT04433221 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Childhood tumours that express Glypican-3 | Baylor College of Medicine | IL-15 armoured CAR T-cells iCaspase-9 suicide gene | NCT04377932 |
| Hepatocellular carcinoma | Baylor College of Medicine | IL-15 armoured CAR T-cells iCaspase-9 suicide gene | NCT05103631 |
| Hepatocellular carcinoma | Affil. Nanjing Drum Tower Hospital | NCT04121273 | |
| Glypican-3+ tumours | SOTIO | Glutamic oxaloacetic transaminase 2 armouring [122] | NCT05354375 |
| Hepatocellular carcinoma | National Cancer Institute | NCT05003895 | |
| Hepatocellular carcinoma | Tongji University | NCT05070156 | |
| Hepatocellular carcinoma | 2nd Affiliated Hospital Guangzhou Medical University | In CD4+ T-cells, CAR also targets TGF-β and secretes IL-7/CCL19 and scFvs against PD1, CTLA4 and Tigit. In CD8+ T-cells, a Glypican-3/Dap10 CAR is expressed and both PD1 and HPK are knocked down | NCT03198546 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| NKG2D ligand+ tumours | Fudan University | NCT05131763 | |
| Liver metastatic colorectal carcinoma | 3rd Affiliated Hospital Guangzhou Medical University | Hepatic artery infusion | NCT05248048 |
| NKG2D ligand+ tumours | Jianming Xu | NCT05382377 | |
| NKG2D ligand+/ Claudin 18.2+ tumours | Jianming Xu | Also targets Claudin-18.2 | NCT05583201 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Prostate cancer | City of Hope Medical Center | NCT03873805 | |
| Prostate cancer | Bellicum Pharmaceuticals | Incorporates rimiducid inducible co-stimulatory domain | NCT02744287 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Colorectal cancer | Changhai Hospital | NCT05240950 | |
| CEA+ cancer | Chongqing Precision Biotech Co. | NCT05538195 | |
| CEA+ cancer | Chongqing Precision Biotech Co. | NCT04348643 | |
| CEA+ cancer | Chongqing Precision Biotech Co. | NCT05538195 | |
| CEA+ cancer | Weijia Fang | Intravenous or intraperitoneal administration | NCT05396300 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| CD70+ cancer | Chongqing Precision Biotech Co. | Intravenous or intraperitoneal delivery | NCT05468190 |
| CD70+ renal cell carcinoma | Chongqing Precision Biotech Co. | NCT05420519 | |
| CD70+ cancer | Chongqing Precision Biotech Co. | Intravenous or intraperitoneal delivery | NCT05420545 |
| CD70+ cancer | Weijia Fang | Intravenous or intraperitoneal delivery | NCT05518253 |
| CD70+ clear cell renal carcinoma | CRISPR Therapeutics | Allogeneic CRISPR-Cas9 engineered T-cells | NCT04438083 |
| CD70+ cancer | National Cancer Institute | Targeted using CD27 | NCT02830724 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Glioma | Xuanwu Hospital, Beijing | One of multiple targets; administered with or without anti-PD1 | NCT03423992 |
| Sarcomas | Shenzhen Geno-Immune Medical Institute | One of multiple targets | NCT03356782 |
| Disease | Sponsor | Notes | Identifier |
|---|---|---|---|
| Solid tumours | Sichuan University | Nasopharyngeal, breast and gastric cancers main focus | NCT02915445 |
| Gastrointestinal cancers | Zhejiang University | Hepatocellular, colorectal, gastric and pancreatic cancers | NCT05028933 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maher, J.; Davies, D.M. CAR Based Immunotherapy of Solid Tumours—A Clinically Based Review of Target Antigens. Biology 2023, 12, 287. https://doi.org/10.3390/biology12020287
Maher J, Davies DM. CAR Based Immunotherapy of Solid Tumours—A Clinically Based Review of Target Antigens. Biology. 2023; 12(2):287. https://doi.org/10.3390/biology12020287
Chicago/Turabian StyleMaher, John, and David M. Davies. 2023. "CAR Based Immunotherapy of Solid Tumours—A Clinically Based Review of Target Antigens" Biology 12, no. 2: 287. https://doi.org/10.3390/biology12020287
APA StyleMaher, J., & Davies, D. M. (2023). CAR Based Immunotherapy of Solid Tumours—A Clinically Based Review of Target Antigens. Biology, 12(2), 287. https://doi.org/10.3390/biology12020287