
The Role of the Intestinal Microbiome in Multiple Sclerosis—Lessons to Be Learned from Hippocrates

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Multiple Sclerosis
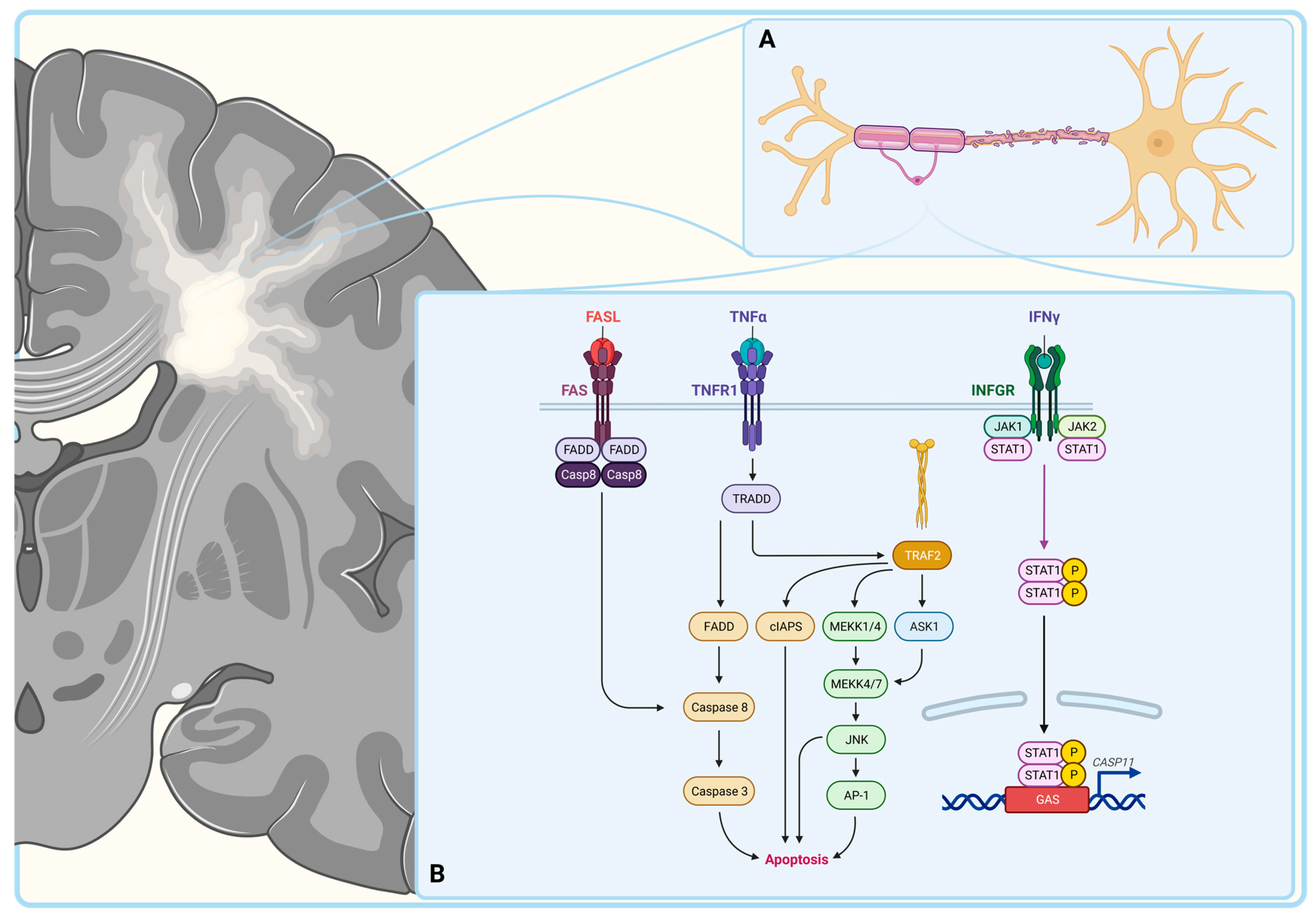
3. Autoimmunity in Multiple Sclerosis
3.1. Macrophages
3.2. T Lymphocytes
3.3. B Cells
3.4. Neutrophil Granulocytes
4. Microbiome as a Regulatory Factor of the Immune System
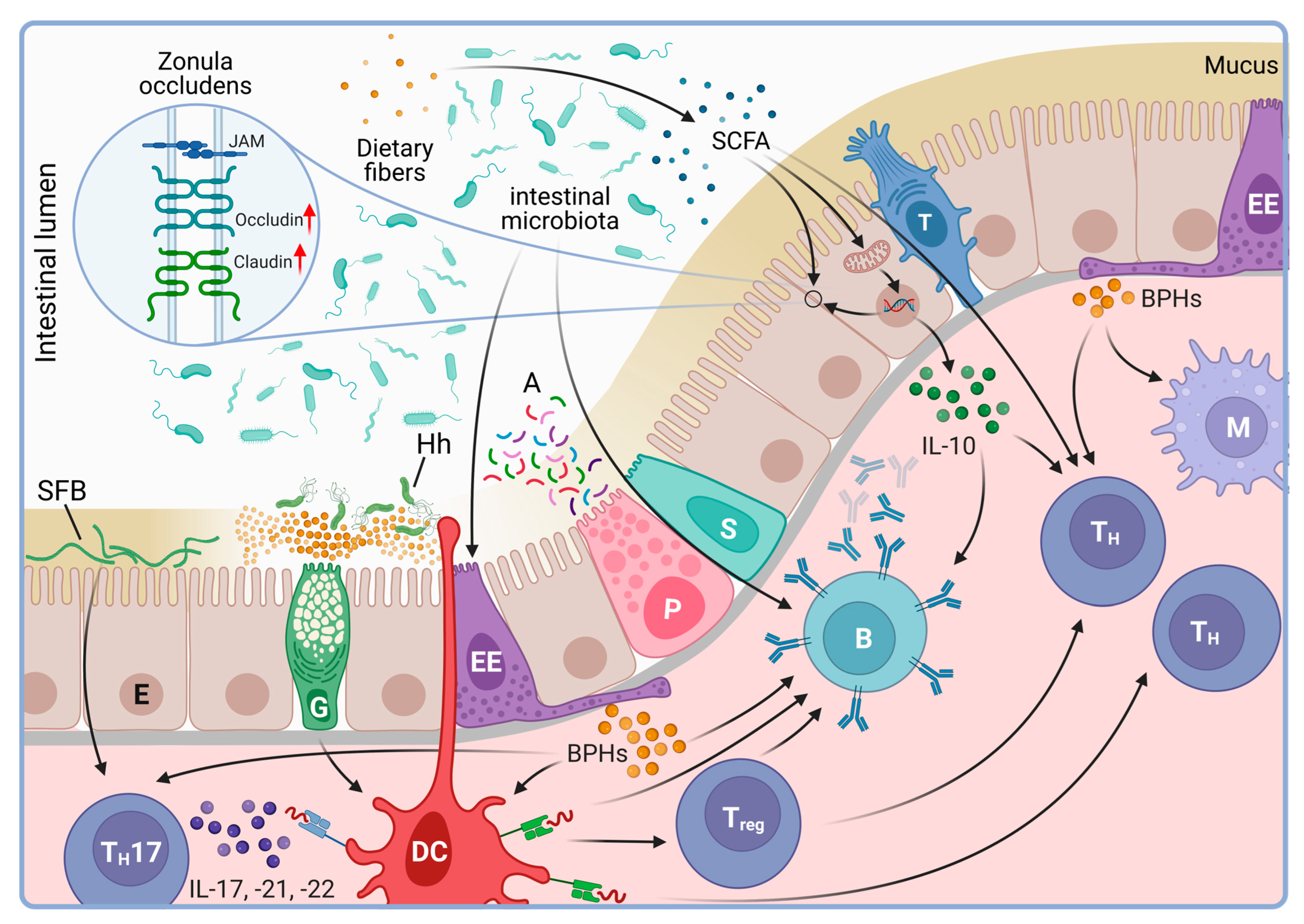
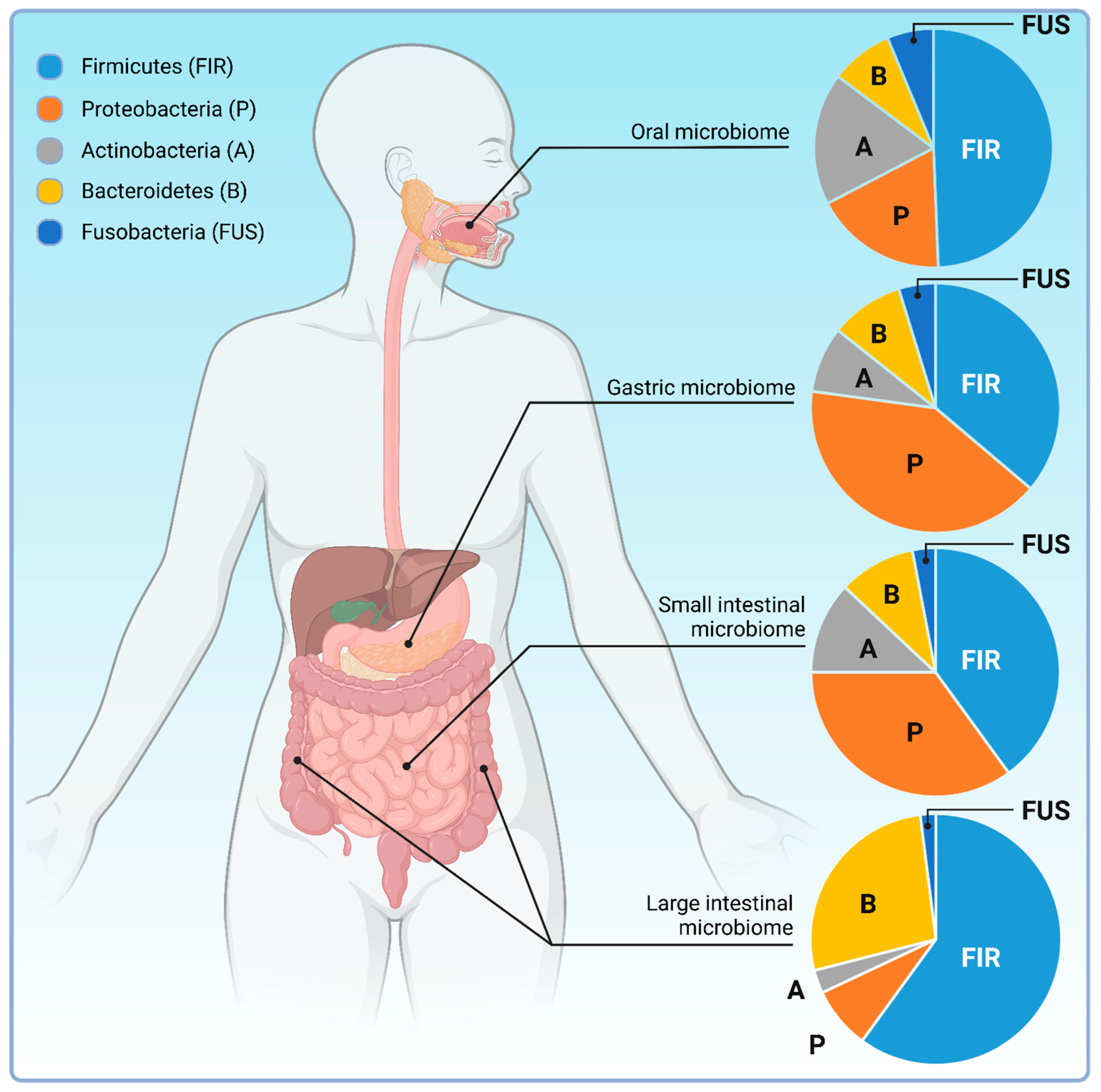
4.1. The Intestinal Microbiota
4.2. Origin of the GI Microbiome
4.3. Dietary Links
4.4. Microbiome and Disease
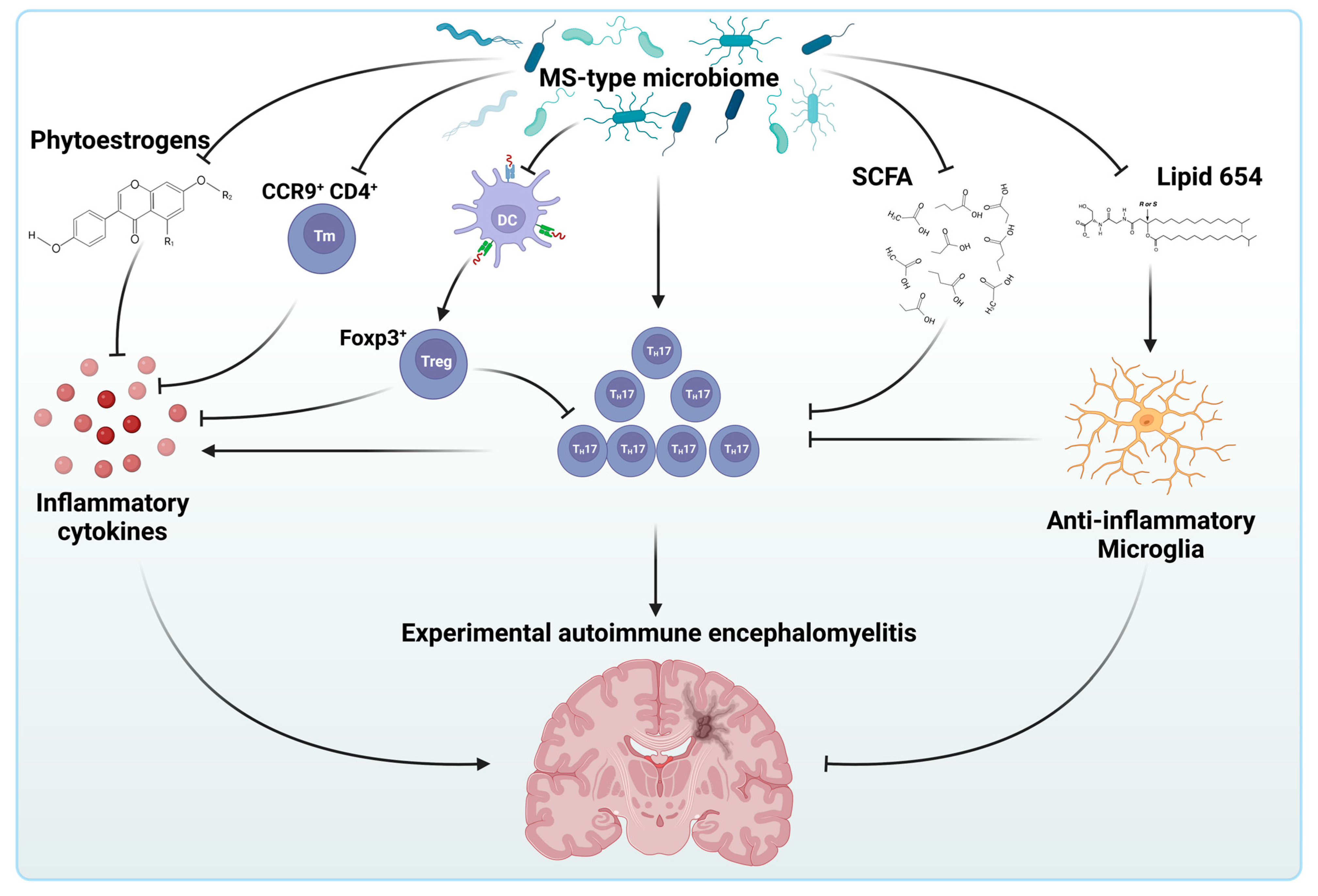
5. The Intestinal Microbiome and Multiple Sclerosis
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wylezinski, L.S.; Gray, J.D.; Polk, J.B.; Harmata, A.J.; Spurlock, C.F., 3rd. Illuminating an Invisible Epidemic: A Systemic Review of the Clinical and Economic Benefits of Early Diagnosis and Treatment in Inflammatory Disease and Related Syndromes. J. Clin. Med. 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Jones, H.B.; Feakes, R.; Gray, J.; Smaldon, N.; Chataway, J.; Robertson, N.; Clayton, D.; Goodfellow, P.N.; Compston, A. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat. Genet. 1996, 13, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zacharia, A.B. Therapeutics for multiple sclerosis symptoms. Mt. Sinai J. Med. 2011, 78, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.D.; D’Mellow, M.T.; Fowler, C.J. Urinary symptoms and the neurological features of bladder dysfunction in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 1993, 56, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.B.; Beck, C.A.; Williams, J.V.; Barbui, C.; Metz, L.M. Major depression in multiple sclerosis: A population-based perspective. Neurology 2003, 61, 1524–1527. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Gohil, K. Multiple Sclerosis: Progress, but No Cure. Pharm. Ther. 2015, 40, 604–605. [Google Scholar]
- de Waegh, S.; Brady, S.T. Altered slow axonal transport and regeneration in a myelin-deficient mutant mouse: The trembler as an in vivo model for Schwann cell-axon interactions. J. Neurosci. 1990, 10, 1855–1865. [Google Scholar] [CrossRef]
- McDonald, W.I.; Sears, T.A. Effect of demyelination on conduction in the central nervous system. Nature 1969, 221, 182–183. [Google Scholar] [CrossRef]
- Anlar, O.; Tombul, T.; Kisli, M. Peripheral sensory and motor abnormalities in patients with multiple sclerosis. Electromyogr. Clin. Neurophysiol. 2003, 43, 349–351. [Google Scholar]
- D’Souza, S.D.; Bonetti, B.; Balasingam, V.; Cashman, N.R.; Barker, P.A.; Troutt, A.B.; Raine, C.S.; Antel, J.P. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J. Exp. Med. 1996, 184, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.W.; Raine, C.S. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann. Neurol. 1988, 23, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Hisahara, S.; Yuan, J.; Momoi, T.; Okano, H.; Miura, M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J. Exp. Med. 2001, 193, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Isabwe, G.A.C.; Garcia Neuer, M.; de Las Vecillas Sanchez, L.; Lynch, D.M.; Marquis, K.; Castells, M. Hypersensitivity reactions to therapeutic monoclonal antibodies: Phenotypes and endotypes. J. Allergy Clin. Immunol. 2018, 142, 159–170 e152. [Google Scholar] [CrossRef] [PubMed]
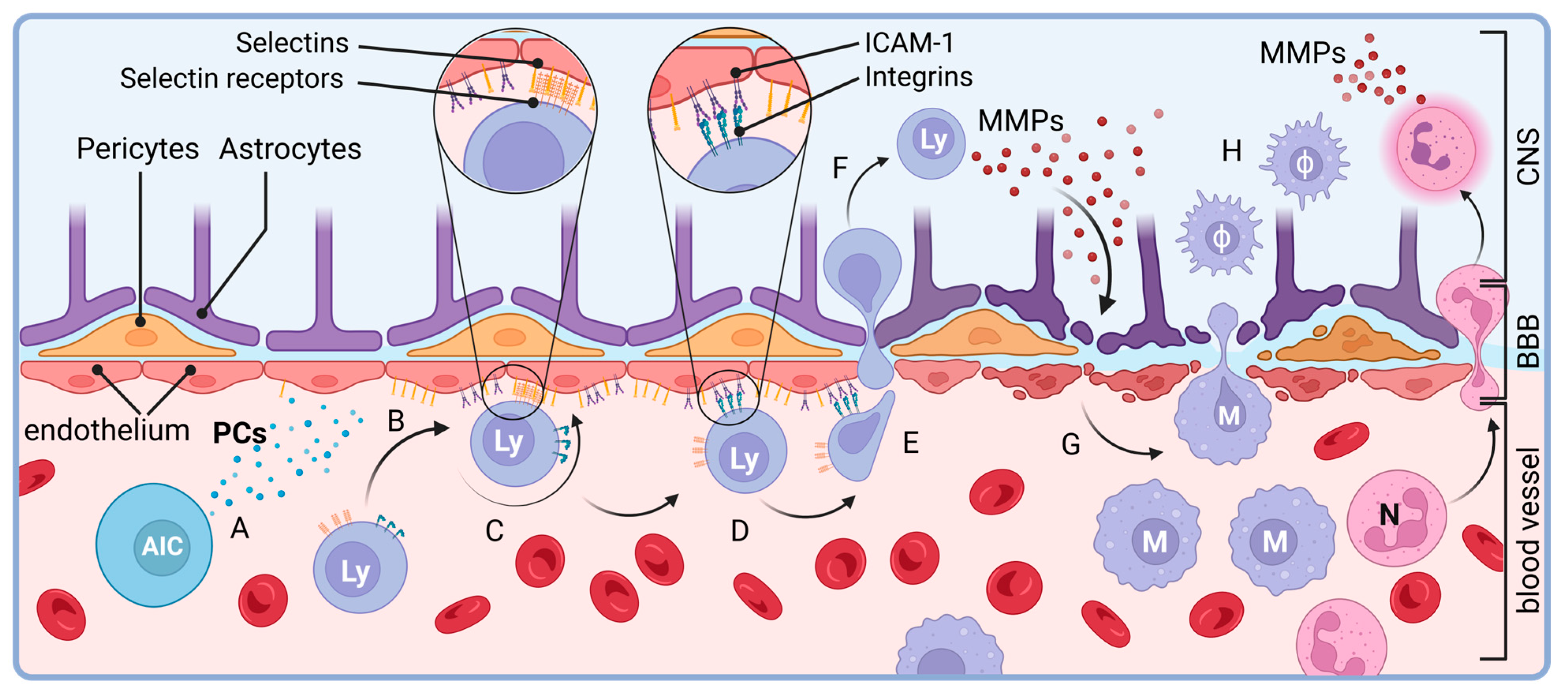
- Engelhardt, B. T cell migration into the central nervous system during health and disease: Different molecular keys allow access to different central nervous system compartments. Clin. Exp. Neuroimmunol. 2010, 1, 79–93. [Google Scholar] [CrossRef]
- Correale, J.; Gilmore, W.; McMillan, M.; Li, S.; McCarthy, K.; Le, T.; Weiner, L.P. Patterns of cytokine secretion by autoreactive proteolipid protein-specific T cell clones during the course of multiple sclerosis. J. Immunol. 1995, 154, 2959–2968. [Google Scholar] [CrossRef]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef]
- Kawachi, S.; Jennings, S.; Panes, J.; Cockrell, A.; Laroux, F.S.; Gray, L.; Perry, M.; van der Heyde, H.; Balish, E.; Granger, D.N.; et al. Cytokine and endothelial cell adhesion molecule expression in interleukin-10-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G734–G743. [Google Scholar] [CrossRef]
- Sellebjerg, F.; Sorensen, T.L. Chemokines and matrix metalloproteinase-9 in leukocyte recruitment to the central nervous system. Brain Res. Bull. 2003, 61, 347–355. [Google Scholar] [CrossRef]
- Bar-Or, A.; Nuttall, R.K.; Duddy, M.; Alter, A.; Kim, H.J.; Ifergan, I.; Pennington, C.J.; Bourgoin, P.; Edwards, D.R.; Yong, V.W. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 2003, 126 Pt 12, 2738–2749. [Google Scholar] [CrossRef]
- Chard, D.T.; Griffin, C.M.; Parker, G.J.; Kapoor, R.; Thompson, A.J.; Miller, D.H. Brain atrophy in clinically early relapsing-remitting multiple sclerosis. Brain 2002, 125, 327–337. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130 Pt 4, 1089–1104. [Google Scholar] [CrossRef]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134 Pt 9, 2755–2771. [Google Scholar] [CrossRef]
- Yandamuri, S.S.; Filipek, B.; Obaid, A.H.; Lele, N.; Thurman, J.M.; Makhani, N.; Nowak, R.J.; Guo, Y.; Lucchinetti, C.F.; Flanagan, E.P.; et al. MOGAD patient autoantibodies induce complement, phagocytosis, and cellular cytotoxicity. JCI Insight 2023, 8, e165373. [Google Scholar] [CrossRef]
- Steinman, L. Absence of “original antigenic sin” in autoimmunity provides an unforeseen platform for immune therapy. J. Exp. Med. 1999, 189, 1021–1024. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Zhang, J.; Witek, C.; Matsui, M.; Modabber, Y.; Ota, K.; Hafler, D.A. Clonal expansion and persistence of human T cells specific for an immunodominant myelin basic protein peptide. J. Immunol. 1994, 152, 5581–5592. [Google Scholar] [CrossRef]
- Pettinelli, C.B.; McFarlin, D.E. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: Requirement for Lyt 1+ 2− T lymphocytes. J. Immunol. 1981, 127, 1420–1423. [Google Scholar] [CrossRef]
- Gay, D.; Esiri, M. Blood-brain barrier damage in acute multiple sclerosis plaques. An immunocytological study. Brain 1991, 114 Pt 1B, 557–572. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Heppner, F.L.; Greter, M.; Marino, D.; Falsig, J.; Raivich, G.; Hovelmeyer, N.; Waisman, A.; Rulicke, T.; Prinz, M.; Priller, J.; et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005, 11, 146–152. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135 Pt 3, 886–899. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef]
- Izikson, L.; Klein, R.S.; Charo, I.F.; Weiner, H.L.; Luster, A.D. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J. Exp. Med. 2000, 192, 1075–1080. [Google Scholar] [CrossRef]
- Polman, C.H.; Dijkstra, C.D.; Sminia, T.; Koetsier, J.C. Immunohistological analysis of macrophages in the central nervous system of Lewis rats with acute experimental allergic encephalomyelitis. J. Neuroimmunol. 1986, 11, 215–222. [Google Scholar] [CrossRef]
- Bruck, W.; Sommermeier, N.; Bergmann, M.; Zettl, U.; Goebel, H.H.; Kretzschmar, H.A.; Lassmann, H. Macrophages in multiple sclerosis. Immunobiology 1996, 195, 588–600. [Google Scholar] [CrossRef]
- Fife, B.T.; Huffnagle, G.B.; Kuziel, W.A.; Karpus, W.J. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 899–905. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Mikita, J.; Dubourdieu-Cassagno, N.; Deloire, M.S.; Vekris, A.; Biran, M.; Raffard, G.; Brochet, B.; Canron, M.H.; Franconi, J.M.; Boiziau, C.; et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler. 2011, 17, 2–15. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Yu, J.; Feng, L.; Hou, S.; Liu, Y.; Guo, M.; Xie, Y.; Meng, J.; Zhang, H.; et al. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS ONE 2013, 8, e54841. [Google Scholar] [CrossRef]
- King, I.L.; Dickendesher, T.L.; Segal, B.M. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009, 113, 3190–3197. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Kondo, M.; Weissman, I.L. Role of interleukin-7 in T-cell development from hematopoietic stem cells. Immunol. Rev. 1998, 165, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Egerton, M.; Scollay, R.; Shortman, K. Kinetics of mature T-cell development in the thymus. Proc. Natl. Acad. Sci. USA 1990, 87, 2579–2582. [Google Scholar] [CrossRef] [PubMed]
- Stritesky, G.L.; Jameson, S.C.; Hogquist, K.A. Selection of self-reactive T cells in the thymus. Annu. Rev. Immunol. 2012, 30, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A.; Wolf, A.; Bezer, A.E. The Rapid Production of Acute Disseminated Encephalomyelitis in Rhesus Monkeys by Injection of Heterologous and Homologous Brain Tissue with Adjuvants. J. Exp. Med. 1947, 85, 117–130. [Google Scholar] [CrossRef]
- Paterson, P.Y. Transfer of allergic encephalomyelitis in rats by means of lymph node cells. J. Exp. Med. 1960, 111, 119–136. [Google Scholar] [CrossRef]
- Arnason, B.G.; Jankovic, B.D.; Waksman, B.H.; Wennersten, C. Role of the thymus in immune reactions in rats. II. Suppressive effect of thymectomy at birth on reactions of delayed (cellular) hypersensitivity and the circulating small lymphocyte. J. Exp. Med. 1962, 116, 177–186. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics, C.; Wellcome Trust Case Control, C.; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132 Pt 12, 3329–3341. [Google Scholar] [CrossRef]
- Voskuhl, R.R.; Martin, R.; Bergman, C.; Dalal, M.; Ruddle, N.H.; McFarland, H.F. T helper 1 (Th1) functional phenotype of human myelin basic protein-specific T lymphocytes. Autoimmunity 1993, 15, 137–143. [Google Scholar] [CrossRef]
- Milovanovic, J.; Arsenijevic, A.; Stojanovic, B.; Kanjevac, T.; Arsenijevic, D.; Radosavljevic, G.; Milovanovic, M.; Arsenijevic, N. Interleukin-17 in Chronic Inflammatory Neurological Diseases. Front. Immunol. 2020, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.W.; Hsieh, C.S. A two-step process for thymic regulatory T cell development. Immunity 2008, 28, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002, 169, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Stephens, L.A.; Anderton, S.M. Natural recovery and protection from autoimmune encephalomyelitis: Contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 2005, 175, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.T.; Korn, T.; Richard, J.; Ruzek, M.; Kohm, A.P.; Miller, S.; Nahill, S.; Oukka, M. Anti-thymocyte globulin (ATG) prevents autoimmune encephalomyelitis by expanding myelin antigen-specific FOXP3+ regulatory T cells. Int. Immunol. 2007, 19, 1003–1010. [Google Scholar] [CrossRef]
- Butti, E.; Bergami, A.; Recchia, A.; Brambilla, E.; Del Carro, U.; Amadio, S.; Cattalini, A.; Esposito, M.; Stornaiuolo, A.; Comi, G.; et al. IL4 gene delivery to the CNS recruits regulatory T cells and induces clinical recovery in mouse models of multiple sclerosis. Gene Ther. 2008, 15, 504–515. [Google Scholar] [CrossRef]
- Asseman, C.; Mauze, S.; Leach, M.W.; Coffman, R.L.; Powrie, F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999, 190, 995–1004. [Google Scholar] [CrossRef]
- Akdis, C.A.; Joss, A.; Akdis, M.; Faith, A.; Blaser, K. A molecular basis for T cell suppression by IL-10: CD28-associated IL-10 receptor inhibits CD28 tyrosine phosphorylation and phosphatidylinositol 3-kinase binding. FASEB J. 2000, 14, 1666–1668. [Google Scholar] [CrossRef]
- Joss, A.; Akdis, M.; Faith, A.; Blaser, K.; Akdis, C.A. IL-10 directly acts on T cells by specifically altering the CD28 co-stimulation pathway. Eur. J. Immunol. 2000, 30, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Das, M.P.; Howard, E.D.; Weiner, H.L.; Sobel, R.A.; Kuchroo, V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998, 161, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Astier, A.L.; Hafler, D.A. Abnormal Tr1 differentiation in multiple sclerosis. J. Neuroimmunol. 2007, 191, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, P.; Giorgio, A.; De Santi, L.; Zipoli, V.; Portaccio, E.; Amato, M.P.; Clerici, R.; Scarpini, E.; Moscato, G.; Iudice, A.; et al. Absence of cerebrospinal fluid oligoclonal bands is associated with delayed disability progression in relapsing-remitting MS patients treated with interferon-beta. J. Neurol. Sci. 2006, 244, 97–102. [Google Scholar] [CrossRef] [PubMed]
- de Seze, J.; Maillart, E.; Gueguen, A.; Laplaud, D.A.; Michel, L.; Thouvenot, E.; Zephir, H.; Zimmer, L.; Biotti, D.; Liblau, R. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front. Immunol. 2023, 14, 1004795. [Google Scholar] [CrossRef] [PubMed]
- Yuseff, M.I.; Lennon-Dumenil, A.M. B Cells use Conserved Polarity Cues to Regulate Their Antigen Processing and Presentation Functions. Front. Immunol. 2015, 6, 251. [Google Scholar] [CrossRef]
- Mitsdoerffer, M.; Lee, Y.; Jager, A.; Kim, H.J.; Korn, T.; Kolls, J.K.; Cantor, H.; Bettelli, E.; Kuchroo, V.K. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. USA 2010, 107, 14292–14297. [Google Scholar] [CrossRef]
- Dvorscek, A.R.; McKenzie, C.I.; Robinson, M.J.; Ding, Z.; Pitt, C.; O’Donnell, K.; Zotos, D.; Brink, R.; Tarlinton, D.M.; Quast, I. IL-21 has a critical role in establishing germinal centers by amplifying early B cell proliferation. EMBO Rep. 2022, 23, e54677. [Google Scholar] [CrossRef]
- Samuels, J.; Ng, Y.S.; Coupillaud, C.; Paget, D.; Meffre, E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med. 2005, 201, 1659–1667. [Google Scholar] [CrossRef]
- Sorensen, T.L.; Trebst, C.; Kivisakk, P.; Klaege, K.L.; Majmudar, A.; Ravid, R.; Lassmann, H.; Olsen, D.B.; Strieter, R.M.; Ransohoff, R.M.; et al. Multiple sclerosis: A study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J. Neuroimmunol. 2002, 127, 59–68. [Google Scholar] [CrossRef]
- Bogers, L.; Engelenburg, H.J.; Janssen, M.; Unger, P.A.; Melief, M.J.; Wierenga-Wolf, A.F.; Hsiao, C.C.; Mason, M.R.J.; Hamann, J.; van Langelaar, J.; et al. Selective emergence of antibody-secreting cells in the multiple sclerosis brain. EBioMedicine 2023, 89, 104465. [Google Scholar] [CrossRef] [PubMed]
- McGinley, A.M.; Sutton, C.E.; Edwards, S.C.; Leane, C.M.; DeCourcey, J.; Teijeiro, A.; Hamilton, J.A.; Boon, L.; Djouder, N.; Mills, K.H.G. Interleukin-17A Serves a Priming Role in Autoimmunity by Recruiting IL-1beta-Producing Myeloid Cells that Promote Pathogenic T Cells. Immunity 2020, 52, 342–356.e6. [Google Scholar] [CrossRef]
- Gijbels, K.; Proost, P.; Masure, S.; Carton, H.; Billiau, A.; Opdenakker, G. Gelatinase B is present in the cerebrospinal fluid during experimental autoimmune encephalomyelitis and cleaves myelin basic protein. J. Neurosci. Res. 1993, 36, 432–440. [Google Scholar] [CrossRef] [PubMed]
- McColl, S.R.; Staykova, M.A.; Wozniak, A.; Fordham, S.; Bruce, J.; Willenborg, D.O. Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 1998, 161, 6421–6426. [Google Scholar] [CrossRef] [PubMed]
- McQualter, J.L.; Darwiche, R.; Ewing, C.; Onuki, M.; Kay, T.W.; Hamilton, J.A.; Reid, H.H.; Bernard, C.C. Granulocyte macrophage colony-stimulating factor: A new putative therapeutic target in multiple sclerosis. J. Exp. Med. 2001, 194, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef]
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71. [Google Scholar] [CrossRef]
- Flachenecker, P. Epidemiology of neuroimmunological diseases. J. Neurol. 2006, 253, v2–v8. [Google Scholar] [CrossRef]
- Rosati, G. The prevalence of multiple sclerosis in the world: An update. Neurol. Sci. 2001, 22, 117–139. [Google Scholar] [CrossRef]
- Ebers, G.C. Environmental factors and multiple sclerosis. Lancet Neurol. 2008, 7, 268–277. [Google Scholar] [CrossRef]
- Kampman, M.; Wilsgaard, T.; Mellgren, S. Outdoor activities and diet in childhood and adolescence relate to MS risk above the Arctic Circle. J. Neurol. 2007, 254, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Lauer, K. Ecologic studies of multiple sclerosis. Neurology 1997, 49 (Suppl. S2), S18–S26. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D: The underappreciated D-lightful hormone that is important for skeletal and cellular health. Curr. Opin. Endocrinol. Diabetes Obes. 2002, 9, 87–98. [Google Scholar] [CrossRef]
- McGrath, J.J.; Féron, F.P.; Burne, T.H.; Mackay-Sim, A.; Eyles, D.W. Vitamin D3—Implications for brain development. J. Steroid Biochem. Mol. Biol. 2004, 89, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Waubant, E.; Chehoud, C.; Kuczynski, J.; DeSantis, T.Z.; Warrington, J.; Venkatesan, A.; Fraser, C.M.; Mowry, E.M. Gut microbiota in multiple sclerosis: Possible influence of immunomodulators. J. Investig. Med. 2015, 63, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.; Horowitz, R.E.; Levenson, S.M.; Popper, H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am. J. Pathol. 1963, 42, 471–483. [Google Scholar]
- Hapfelmeier, S.; Lawson, M.A.; Slack, E.; Kirundi, J.K.; Stoel, M.; Heikenwalder, M.; Cahenzli, J.; Velykoredko, Y.; Balmer, M.L.; Endt, K.; et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010, 328, 1705–1709. [Google Scholar] [CrossRef]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Shan, M.; Gentile, M.; Yeiser, J.R.; Walland, A.C.; Bornstein, V.U.; Chen, K.; He, B.; Cassis, L.; Bigas, A.; Cols, M.; et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 2013, 342, 447–453. [Google Scholar] [CrossRef]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, R.; Najar, T.A.; Mesa, K.R.; Grayson, A.; Kroehling, L.; Hao, Y.; Hao, S.; Pokrovskii, M.; Xu, M.; Talbot, J. A RORγt+ cell instructs gut microbiota-specific Treg cell differentiation. Nature 2022, 610, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pokrovskii, M.; Ding, Y.; Yi, R.; Au, C.; Harrison, O.J.; Galan, C.; Belkaid, Y.; Bonneau, R.; Littman, D.R. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018, 554, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Spindler, M.P.; Siu, S.; Mogno, I.; Li, Z.; Yang, C.; Mehandru, S.; Britton, G.J.; Faith, J.J. Human gut microbiota stimulate defined innate immune responses that vary from phylum to strain. Cell Host Microbe 2022, 30, 1481–1498.e5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.-Y.; Yeung, S.T.; Silva, H.M.; Li, D. Serum amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell 2020, 180, 79–91.e16. [Google Scholar] [CrossRef]
- Ladinsky, M.S.; Araujo, L.P.; Zhang, X.; Veltri, J.; Galan-Diez, M.; Soualhi, S.; Lee, C.; Irie, K.; Pinker, E.Y.; Narushima, S. Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science 2019, 363, eaat4042. [Google Scholar] [CrossRef]
- Nevo, S.; Kadouri, N.; Abramson, J. Tuft cells: From the mucosa to the thymus. Immunol. Lett. 2019, 210, 1–9. [Google Scholar] [CrossRef]
- Zwarycz, B.; Gracz, A.D.; Rivera, K.R.; Williamson, I.A.; Samsa, L.A.; Starmer, J.; Daniele, M.A.; Salter-Cid, L.; Zhao, Q.; Magness, S.T. IL22 Inhibits Epithelial Stem Cell Expansion in an Ileal Organoid Model. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 1–17. [Google Scholar] [CrossRef]
- Tang, W.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rong, X.; Zhao, G.; Zhou, Y.; Xiao, Y.; Ma, D.; Jin, X.; Wu, Y.; Yan, Y.; Yang, H. The microbial metabolite trimethylamine N-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab. 2022, 34, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, L.; Min, J.; Wang, J.; Wu, H.; Zeng, Y.; Chen, S.; Chu, Z. Butyrate interferes with the differentiation and function of human monocyte-derived dendritic cells. Cell Immunol. 2012, 277, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, B.G.; Kim, J.H.; Chun, J.; Im, J.P.; Kim, J.S. Sodium butyrate inhibits the NF-kappa B signaling pathway and histone deacetylation, and attenuates experimental colitis in an IL-10 independent manner. Int. Immunopharmacol. 2017, 51, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood J. Am. Soc. Hematol. 2020, 136, 501–515. [Google Scholar] [CrossRef]
- Chen, M.l.; Zhu, X.h.; Ran, L.; Lang, H.d.; Yi, L.; Mi, M.t. Trimethylamine-N-Oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef]
- Mirji, G.; Worth, A.; Bhat, S.A.; El Sayed, M.; Kannan, T.; Goldman, A.R.; Tang, H.-Y.; Liu, Q.; Auslander, N.; Dang, C.V. The microbiome-derived metabolite TMAO drives immune activation and boosts responses to immune checkpoint blockade in pancreatic cancer. Sci. Immunol. 2022, 7, eabn0704. [Google Scholar] [CrossRef]
- Leidy, J. A Flora and Fauna Within Living Animals; Smithsonian Institution: Washington, DC, USA, 1853. [Google Scholar]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70 (Suppl. S1), S38–S44. [Google Scholar] [CrossRef]
- Nissle, A. Über die Grundlagen einer neuen ursächlichen Bekämpfung der pathologischen Darmflora. Dtsch. Med. Wochenschr. 1916, 42, 1181–1184. [Google Scholar]
- Andersson, A.F.; Lindberg, M.; Jakobsson, H.; Backhed, F.; Nyren, P.; Engstrand, L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008, 3, e2836. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E.; Napolitano, M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, D.; Lehr, K.; Vilchez-Vargas, R.; Jonaitis, L.V.; Urba, M.; Kupcinskas, J.; Skieceviciene, J.; Link, A. Comparison of genomic and transcriptional microbiome analysis in gastric cancer patients and healthy individuals. World J. Gastroenterol. 2023, 29, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- King, C.H.; Desai, H.; Sylvetsky, A.C.; LoTempio, J.; Ayanyan, S.; Carrie, J.; Crandall, K.A.; Fochtman, B.C.; Gasparyan, L.; Gulzar, N.; et al. Baseline human gut microbiota profile in healthy people and standard reporting template. PLoS ONE 2019, 14, e0206484. [Google Scholar] [CrossRef] [PubMed]
- Vuik, F.; Dicksved, J.; Lam, S.Y.; Fuhler, G.M.; van der Laan, L.; van de Winkel, A.; Konstantinov, S.R.; Spaander, M.; Peppelenbosch, M.P.; Engstrand, L.; et al. Composition of the mucosa-associated microbiota along the entire gastrointestinal tract of human individuals. United Eur. Gastroenterol. J. 2019, 7, 897–907. [Google Scholar] [CrossRef]
- Korpela, K.; Blakstad, E.W.; Moltu, S.J.; Strommen, K.; Nakstad, B.; Ronnestad, A.E.; Braekke, K.; Iversen, P.O.; Drevon, C.A.; de Vos, W. Intestinal microbiota development and gestational age in preterm neonates. Sci. Rep. 2018, 8, 2453. [Google Scholar] [CrossRef]
- Rotimi, V.O.; Duerden, B.I. The development of the bacterial flora in normal neonates. J. Med. Microbiol. 1981, 14, 51–62. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Abrahamsson, T.R.; Jenmalm, M.C.; Harris, K.; Quince, C.; Jernberg, C.; Bjorksten, B.; Engstrand, L.; Andersson, A.F. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 2014, 63, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, A.M.; Bradley, A.K.; Oswald, S.; Drasar, B.S. Diet and the faecal microflora of infants, children and adults in rural Nigeria and urban U.K. J. Hyg. 1981, 86, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, X.; Li, D.; Li, L.; Zhang, D.; Peng, Y. Effects of different modes of delivery and feeding on intestinal flora of newborns and infants with different ages. Iran. J. Pediatr. 2019, 29, e88329. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef]
- Kaczmarek, J.L.; Liu, X.; Charron, C.S.; Novotny, J.A.; Jeffery, E.H.; Seifried, H.E.; Ross, S.A.; Miller, M.J.; Swanson, K.S.; Holscher, H.D. Broccoli consumption affects the human gastrointestinal microbiota. J. Nutr. Biochem. 2019, 63, 27–34. [Google Scholar] [CrossRef]
- Holscher, H.D.; Guetterman, H.M.; Swanson, K.S.; An, R.; Matthan, N.R.; Lichtenstein, A.H.; Novotny, J.A.; Baer, D.J. Walnut Consumption Alters the Gastrointestinal Microbiota, Microbially Derived Secondary Bile Acids, and Health Markers in Healthy Adults: A Randomized Controlled Trial. J. Nutr. 2018, 148, 861–867. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyoty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Hevia, A.; Milani, C.; Lopez, P.; Cuervo, A.; Arboleya, S.; Duranti, S.; Turroni, F.; Gonzalez, S.; Suarez, A.; Gueimonde, M.; et al. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 2014, 5, e01548-14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Chau, S.W.H.; Liu, Y.; Chan, J.W.Y.; Wang, J.; Ma, S.L.; Zhang, J.; Chan, P.K.S.; Yeoh, Y.K.; Chen, Z.; et al. Gut microbiome dysbiosis across early Parkinson’s disease, REM sleep behavior disorder and their first-degree relatives. Nat. Commun. 2023, 14, 2501. [Google Scholar] [CrossRef] [PubMed]
- Boertien, J.M.; Murtomaki, K.; Pereira, P.A.B.; van der Zee, S.; Mertsalmi, T.H.; Levo, R.; Nojonen, T.; Makinen, E.; Jaakkola, E.; Laine, P.; et al. Fecal microbiome alterations in treatment-naive de novo Parkinson’s disease. NPJ Park. Dis. 2022, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Jemimah, S.; Chabib, C.M.M.; Hadjileontiadis, L.; AlShehhi, A. Gut microbiome dysbiosis in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. PLoS ONE 2023, 18, e0285346. [Google Scholar] [CrossRef]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef]
- Laaker, C.; Hsu, M.; Fabry, Z.; Miller, S.D.; Karpus, W.J. Experimental Autoimmune Encephalomyelitis in the Mouse. Curr. Protoc. 2021, 1, e300. [Google Scholar] [CrossRef]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [PubMed]
- Cosorich, I.; Dalla-Costa, G.; Sorini, C.; Ferrarese, R.; Messina, M.J.; Dolpady, J.; Radice, E.; Mariani, A.; Testoni, P.A.; Canducci, F.; et al. High frequency of intestinal T(H)17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci. Adv. 2017, 3, e1700492. [Google Scholar] [CrossRef] [PubMed]
- Engen, S.A.; Valen Rukke, H.; Becattini, S.; Jarrossay, D.; Blix, I.J.; Petersen, F.C.; Sallusto, F.; Schenck, K. The oral commensal Streptococcus mitis shows a mixed memory Th cell signature that is similar to and cross-reactive with Streptococcus pneumoniae. PLoS ONE 2014, 9, e104306. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Siaw, Y.; Hart, A. Commentary: Is F aecalibacterium prausnitzii a potential treatment for maintaining remission in ulcerative colitis? Aliment. Pharmacol. Ther. 2013, 38, 551. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Composition and metabolic activities of bacterial biofilms colonizing food residues in the human gut. Appl. Environ. Microbiol. 2006, 72, 6204–6211. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef]
- Farrokhi, V.; Nemati, R.; Nichols, F.C.; Yao, X.; Anstadt, E.; Fujiwara, M.; Grady, J.; Wakefield, D.; Castro, W.; Donaldson, J.; et al. Bacterial lipodipeptide, Lipid 654, is a microbiome-associated biomarker for multiple sclerosis. Clin. Transl. Immunol. 2013, 2, e8. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.B.; Cervantes, J.L.; Maciejewski, M.W.; Farrokhi, V.; Nemati, R.; Yao, X.; Anstadt, E.; Fujiwara, M.; Wright, K.T.; Riddle, C.; et al. Serine lipids of Porphyromonas gingivalis are human and mouse Toll-like receptor 2 ligands. Infect. Immun. 2013, 81, 3479–3489. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Everett, C.; Barragan, J.A.; Vargas-Medrano, J.; Gadad, B.S.; Nichols, F.; Cervantes, J.L. Multiple Sclerosis-associated Bacterial Ligand 654. Arch. Med. Res. 2022, 53, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Jantaratnotai, N.; Utaisincharoen, P.; Sanvarinda, P.; Thampithak, A.; Sanvarinda, Y. Phytoestrogens mediated anti-inflammatory effect through suppression of IRF-1 and pSTAT1 expressions in lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2013, 17, 483–488. [Google Scholar] [CrossRef]
- Kadowaki, A.; Saga, R.; Lin, Y.; Sato, W.; Yamamura, T. Gut microbiota-dependent CCR9+CD4+ T cells are altered in secondary progressive multiple sclerosis. Brain 2019, 142, 916–931. [Google Scholar] [CrossRef]
- Guy-Grand, D.; Vassalli, P.; Eberl, G.; Pereira, P.; Burlen-Defranoux, O.; Lemaitre, F.; Di Santo, J.P.; Freitas, A.A.; Cumano, A.; Bandeira, A. Origin, trafficking, and intraepithelial fate of gut-tropic T cells. J. Exp. Med. 2013, 210, 1839–1854. [Google Scholar] [CrossRef]
- Kadowaki, A.; Miyake, S.; Saga, R.; Chiba, A.; Mochizuki, H.; Yamamura, T. Gut environment-induced intraepithelial autoreactive CD4+ T cells suppress central nervous system autoimmunity via LAG-3. Nat. Commun. 2016, 7, 11639. [Google Scholar] [CrossRef]
- Song, J.Y.; Larson, N.R.; Thati, S.; Torres-Vazquez, I.; Martinez-Rivera, N.; Subelzu, N.J.; Leon, M.A.; Rosa-Molinar, E.; Schoneich, C.; Forrest, M.L.; et al. Glatiramer acetate persists at the injection site and draining lymph nodes via electrostatically-induced aggregation. J. Control. Release 2019, 293, 36–47. [Google Scholar] [CrossRef]
- Messina, S.; Patti, F. The pharmacokinetics of glatiramer acetate for multiple sclerosis treatment. Expert. Opin. Drug Metab. Toxicol. 2013, 9, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K. Glatiramer Acetate 40 mg/mL in Relapsing-Remitting Multiple Sclerosis: A Review. CNS Drugs 2015, 29, 425–432. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, W.J.; Laman, J.D.; ‘t Hart, B.A. Modulation of multiple sclerosis and its animal model experimental autoimmune encephalomyelitis by food and gut microbiota. Front. Immunol. 2017, 8, 1081. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Ditrio, L.E.; Burroughs, A.R.; Foureau, D.M.; Haque-Begum, S.; Kasper, L.H. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2009, 183, 6041–6050. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Haque-Begum, S.; Kasper, L.H. Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes 2010, 1, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Planas, R.; Santos, R.; Tomas-Ojer, P.; Cruciani, C.; Lutterotti, A.; Faigle, W.; Schaeren-Wiemers, N.; Espejo, C.; Eixarch, H.; Pinilla, C.; et al. GDP-l-fucose synthase is a CD4+ T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Simala-Grant, J.L.; Taylor, D.E. Fucosylation in prokaryotes and eukaryotes. Glycobiology 2006, 16, 158R–184R. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Kanitani, H.; Nemoto, K.; Ono, T.; Miyake, Y. Cross-reactivity between human sialyl Lewis(x) oligosaccharide and common causative oral bacteria of infective endocarditis. FEMS Immunol. Med. Microbiol. 1995, 12, 159–164. [Google Scholar] [CrossRef]
- Lee, H.S.; Choe, G.; Kim, W.H.; Kim, H.H.; Song, J.; Park, K.U. Expression of Lewis antigens and their precursors in gastric mucosa: Relationship with Helicobacter pylori infection and gastric carcinogenesis. J. Pathol. 2006, 209, 88–94. [Google Scholar] [CrossRef]
- Yokota, S.; Amano, K.; Hayashi, S.; Kubota, T.; Fujii, N.; Yokochi, T. Human antibody response to Helicobacter pylori lipopolysaccharide: Presence of an immunodominant epitope in the polysaccharide chain of lipopolysaccharide. Infect. Immun. 1998, 66, 3006–3011. [Google Scholar] [CrossRef]
- Garcia-Vallejo, J.J.; Ilarregui, J.M.; Kalay, H.; Chamorro, S.; Koning, N.; Unger, W.W.; Ambrosini, M.; Montserrat, V.; Fernandes, R.J.; Bruijns, S.C.; et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J. Exp. Med. 2014, 211, 1465–1483. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ha, S.; Kim, M.; Kim, S.W.; Yun, J.; Ozcan, S.; Hwang, H.; Ji, I.J.; Yin, D.; Webster, M.J.; et al. Spatial and temporal diversity of glycome expression in mammalian brain. Proc. Natl. Acad. Sci. USA 2020, 117, 28743–28753. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Multiple Sclerosis Collaborators. Global, regional, and national burden of multiple sclerosis 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, V.; Bakshi, R. Quality of life in patients with multiple sclerosis: The impact of fatigue and depression. J. Neurol. Sci. 2002, 205, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Sumelahti, M.L.; Tienari, P.J.; Wikstrom, J.; Palo, J.; Hakama, M. Increasing prevalence of multiple sclerosis in Finland. Acta Neurol. Scand. 2001, 103, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.H.; Williams, D.B.; Day, S.; Macaskill, P.; McLeod, J.G. Progressive increase in incidence and prevalence of multiple sclerosis in Newcastle, Australia: A 35-year study. J. Neurol. Sci. 2003, 213, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Garvey, P.B.; Zhang, P.; Nelson, S.; Bagby, G.; Summer, W.R.; Schwarzenberger, P.; Shellito, J.E.; Kolls, J.K. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 2001, 25, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Schnyder-Candrian, S.; Togbe, D.; Couillin, I.; Mercier, I.; Brombacher, F.; Quesniaux, V.; Fossiez, F.; Ryffel, B.; Schnyder, B. Interleukin-17 is a negative regulator of established allergic asthma. J. Exp. Med. 2006, 203, 2715–2725. [Google Scholar] [CrossRef]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef]
- Drekonja, D.; Reich, J.; Gezahegn, S.; Greer, N.; Shaukat, A.; MacDonald, R.; Rutks, I.; Wilt, T.J. Fecal Microbiota Transplantation for Clostridium difficile Infection: A Systematic Review. Ann. Intern. Med. 2015, 162, 630–638. [Google Scholar] [CrossRef]
- Varga, A.; Kocsis, B.; Sipos, D.; Kasa, P.; Vigvari, S.; Pal, S.; Dembrovszky, F.; Farkas, K.; Peterfi, Z. How to Apply FMT More Effectively, Conveniently and Flexible—A Comparison of FMT Methods. Front. Cell Infect. Microbiol. 2021, 11, 657320. [Google Scholar] [CrossRef] [PubMed]
- Vigvari, S.; Nemes, Z.; Vincze, A.; Solt, J.; Sipos, D.; Feiszt, Z.; Kovacs, B.; Bartos, B.; Peterfi, Z. Faecal microbiota transplantation in Clostridium difficile infections. Infect. Dis. 2015, 47, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Al, K.F.; Craven, L.J.; Gibbons, S.; Parvathy, S.N.; Wing, A.C.; Graf, C.; Parham, K.A.; Kerfoot, S.M.; Wilcox, H.; Burton, J.P.; et al. Fecal microbiota transplantation is safe and tolerable in patients with multiple sclerosis: A pilot randomized controlled trial. Mult. Scler. J. Exp. Transl. Clin. 2022, 8, 20552173221086662. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Genus |
|---|---|
| Bacteroidetes | Pedobacteria Flavobacterium |
| Firmicutes | Dorea Balutia Streptococcus |
| Proteobacteria | Mycoplana Acinetobacter Pseudomonas |
| Actinobacteria | Eggerthella |
| Verrucomicrobia | Akkermansia |
| Phylum | Genus |
|---|---|
| Bacteroidetes | Bacteroides Prevotella Parabacteroides |
| Firmicutes | Coprobacillus Lactobacillus Clostridium Anaerostipes Faecalibacterium |
| Proteobacteria | Haemophylus Sutterella |
| Actinobacteria | Adlercreutzia Collinsella |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sayed, M.M.; Mohak, S.; Gala, D.; Fabian, R.; Peterfi, Z.; Fabian, Z. The Role of the Intestinal Microbiome in Multiple Sclerosis—Lessons to Be Learned from Hippocrates. Biology 2023, 12, 1463. https://doi.org/10.3390/biology12121463
El-Sayed MM, Mohak S, Gala D, Fabian R, Peterfi Z, Fabian Z. The Role of the Intestinal Microbiome in Multiple Sclerosis—Lessons to Be Learned from Hippocrates. Biology. 2023; 12(12):1463. https://doi.org/10.3390/biology12121463
Chicago/Turabian StyleEl-Sayed, Mohamed Mahmoud, Sidhesh Mohak, Dhir Gala, Reka Fabian, Zoltan Peterfi, and Zsolt Fabian. 2023. "The Role of the Intestinal Microbiome in Multiple Sclerosis—Lessons to Be Learned from Hippocrates" Biology 12, no. 12: 1463. https://doi.org/10.3390/biology12121463
APA StyleEl-Sayed, M. M., Mohak, S., Gala, D., Fabian, R., Peterfi, Z., & Fabian, Z. (2023). The Role of the Intestinal Microbiome in Multiple Sclerosis—Lessons to Be Learned from Hippocrates. Biology, 12(12), 1463. https://doi.org/10.3390/biology12121463