

Bone Involvement in Rheumatoid Arthritis and Spondyloartritis: An Updated Review

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Bone Remodeling and Homeostasis in Health
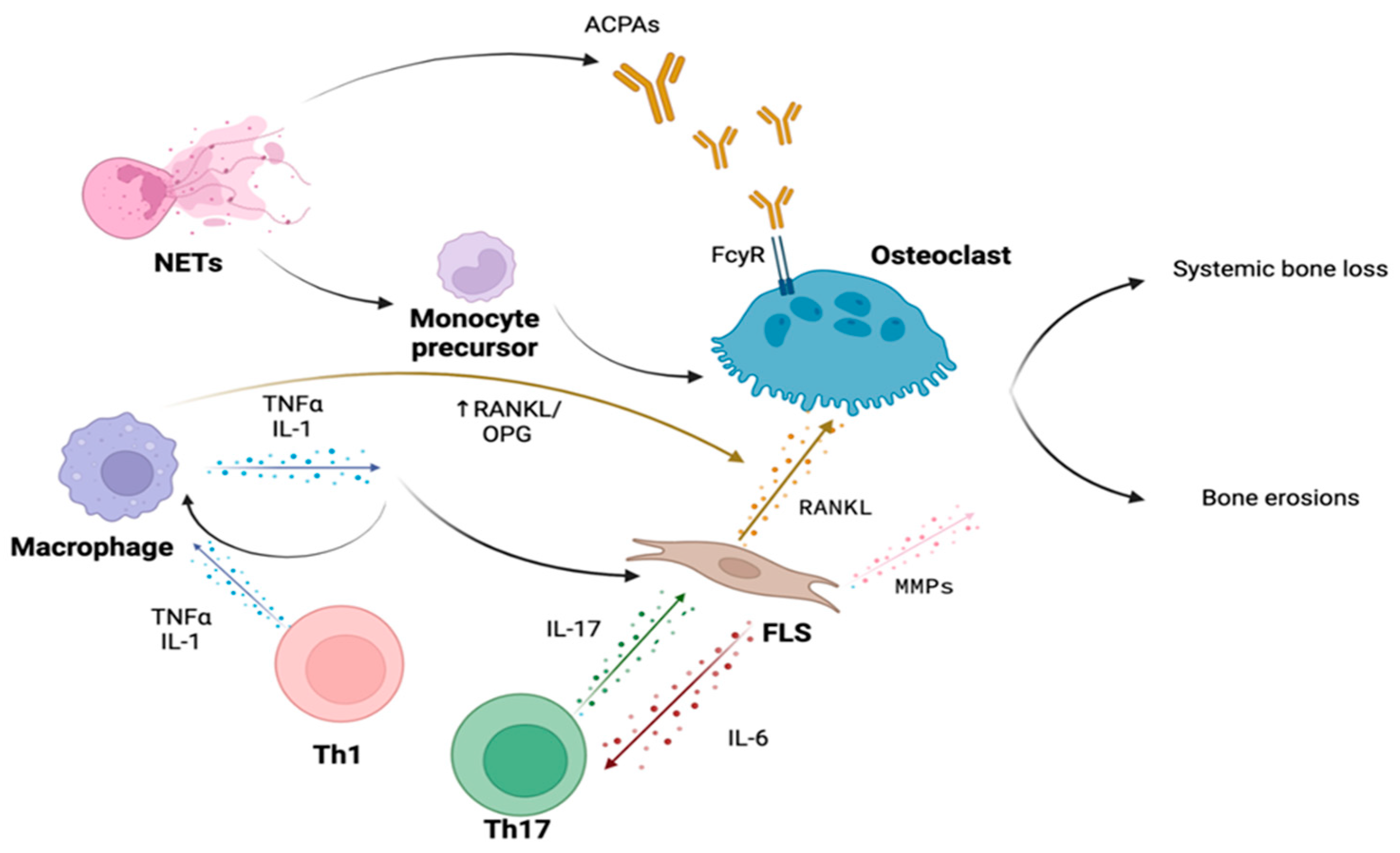
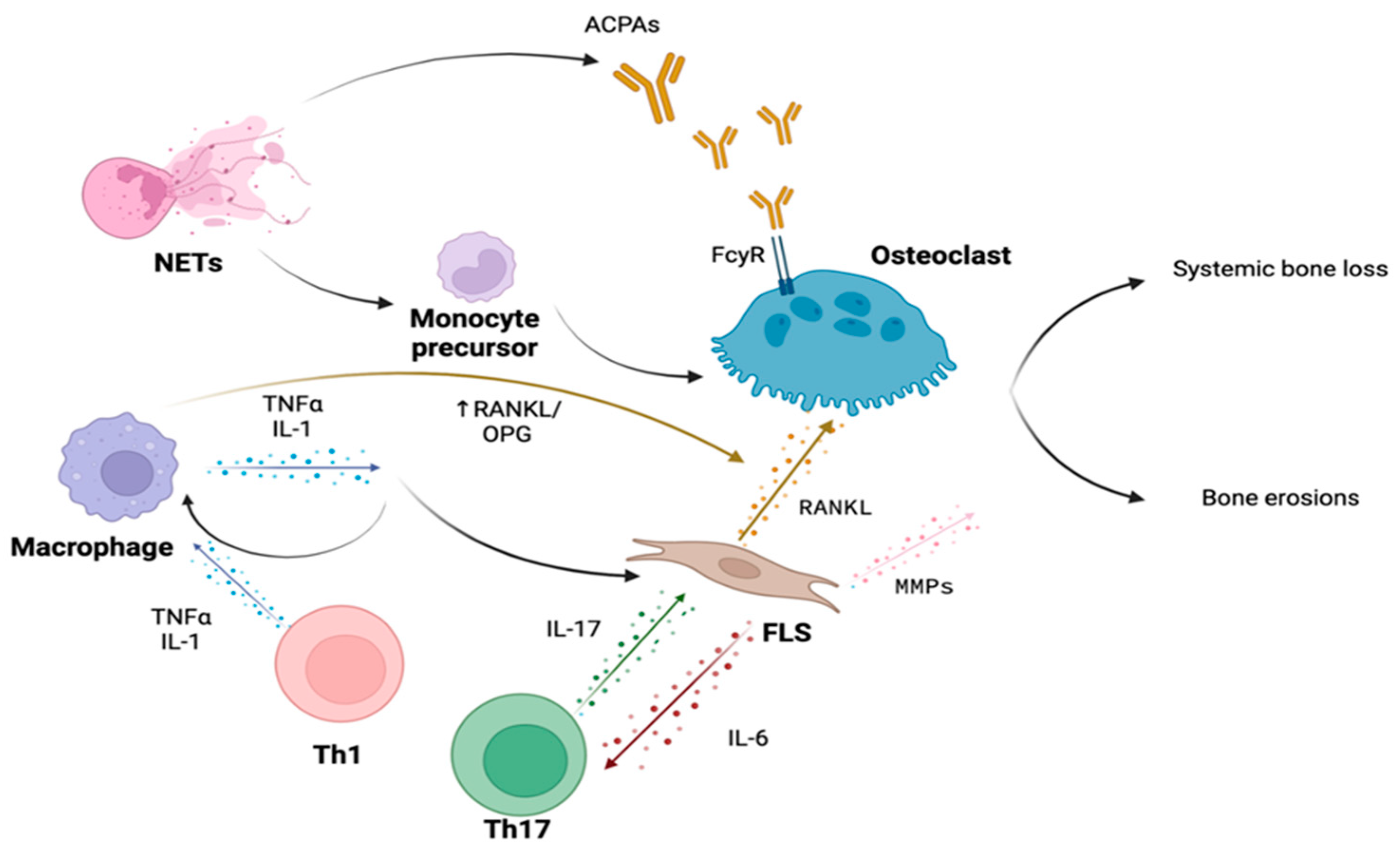
3. Bone Involvement in Rheumatoid Arthritis
3.1. Immune Cells
3.2. Cytokines
3.2.1. TNF-α
3.2.2. IL-6
3.2.3. OSCAR
3.3. Autoantibodies
3.4. Bone Erosions in Rheumatoid Arthritis
3.5. Phospholipase C Gamma and Rheumatoid Arthritis
3.6. Osteoporosis in Rheumatoid Arthritis
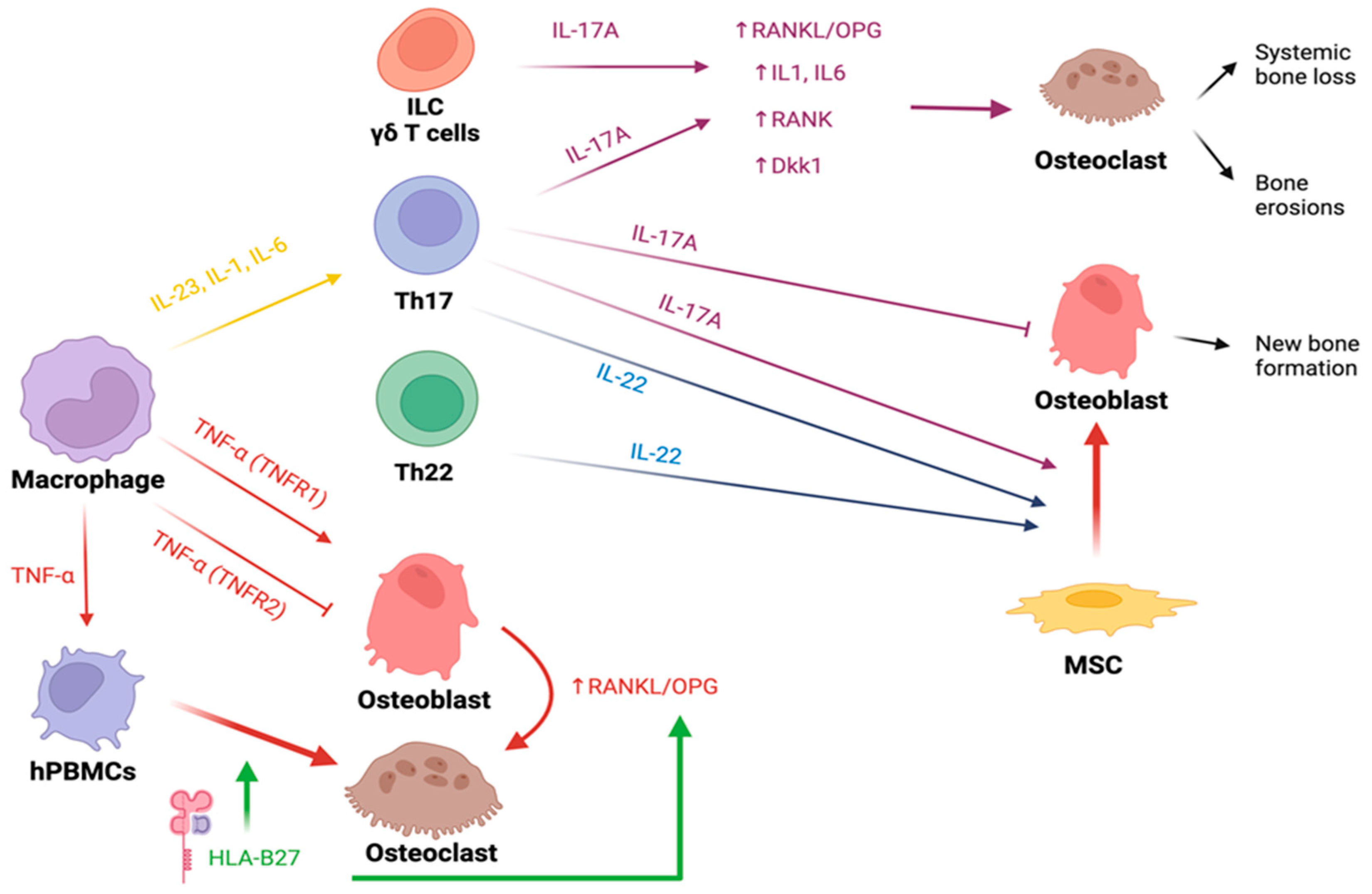
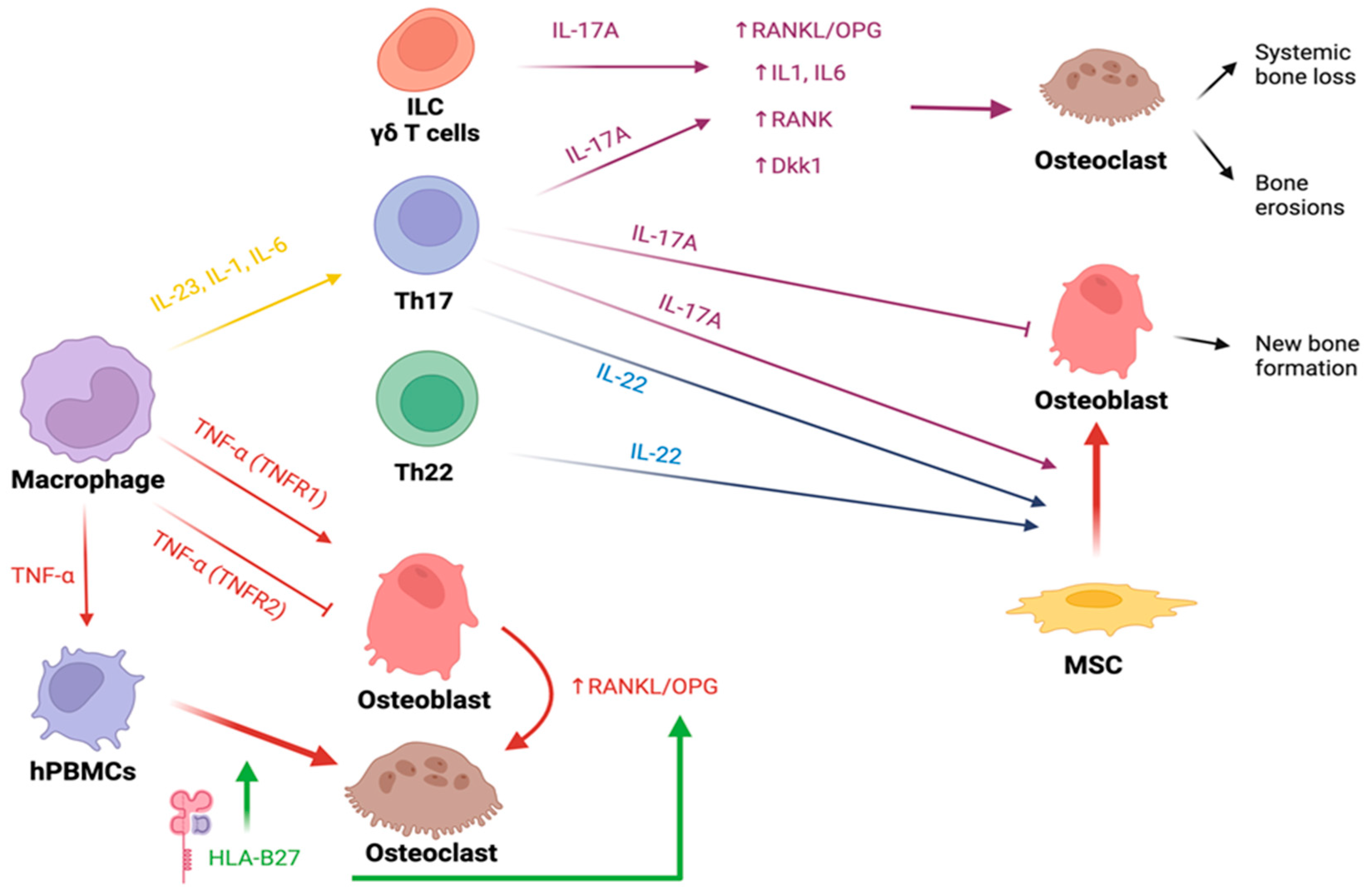
4. Bone Involvement in Spondyloarthritis
4.1. HLA-B27
4.2. Cytokines
4.2.1. IL-23 and IL-17
4.2.2. TNF-α
4.2.3. Other Cytokines
4.3. Erosions in Spondyloarthritis
4.4. New Bone Formation in Spondyloarthritis
4.5. The Enthesis Milieu
4.6. New Bone Formation Pathways
4.6.1. Wnt/ß-Catenin
4.6.2. Cytokines

{kind=link}
{kind=link}
| Molecule | Disease | Effect on Bone Metabolism |
|---|---|---|
| TNF-α | RA and SpA | Binding to TNFR1:
|
| IL-1 | RA | Promotion of osteoclastogenesis acting on bone marrow-derived macrophages [29] |
| IL-6 | RA | Promotion of osteoclastogenesis increasing RANKL expression in osteoblasts [85] |
| IL-17 |
|
|
| IL-23 | SpA | Stimulation of polarization into Th17. [182] Promotion of osteoclastogenesis increasing RANK expression in osteoclast precursors [27] |
| IL-22 | SpA | Promotion of osteogenetic differentiation and migration of osteoblasts precursors synergistically with IFN-γ and TNF [234,235] |
4.7. Osteoporosis in Spondyloarthritis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone Remodeling: An Operational Process Ensuring Survival and Bone Mechanical Competence. Bone Res. 2022, 10, 48. [Google Scholar] [CrossRef]
- Parfitt, A.M. Osteonal and Hemi-Osteonal Remodeling: The Spatial and Temporal Framework for Signal Traffic in Adult Human Bone. J. Cell. Biochem. 1994, 55, 273–286. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared Mechanisms and Crosstalk between the Immune and Bone Systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Yeo, L.; Toellner, K.-M.; Salmon, M.; Filer, A.; Buckley, C.D.; Raza, K.; Scheel-Toellner, D. Cytokine MRNA Profiling Identifies B Cells as a Major Source of RANKL in Rheumatoid Arthritis. Ann. Rheum. Dis. 2011, 70, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, C.; Pap, T.; Buckley, C.D.; Naylor, A.J. The Role of Stromal Cells in Inflammatory Bone Loss. Clin. Exp. Immunol. 2017, 189, 1–11. [Google Scholar] [CrossRef]
- Silva, B.C.; Bilezikian, J.P. Parathyroid Hormone: Anabolic and Catabolic Actions on the Skeleton. Curr. Opin. Pharmacol. 2015, 22, 41–50. [Google Scholar] [CrossRef]
- Nakashima, K.; de Crombrugghe, B. Transcriptional Mechanisms in Osteoblast Differentiation and Bone Formation. Trends Genet. 2003, 19, 458–466. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Titorencu, I.; Pruna, V.; Jinga, V.V.; Simionescu, M. Osteoblast Ontogeny and Implications for Bone Pathology: An Overview. Cell Tissue Res. 2014, 355, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.K.D.S.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 Is Required for the Proliferation of Osteoblast Progenitors and Induces Proliferation by Regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 13551. [Google Scholar] [CrossRef]
- Cawley, K.M.; Bustamante-Gomez, N.C.; Guha, A.G.; MacLeod, R.S.; Xiong, J.; Gubrij, I.; Liu, Y.; Mulkey, R.; Palmieri, M.; Thostenson, J.D.; et al. Local Production of Osteoprotegerin by Osteoblasts Suppresses Bone Resorption. Cell Rep. 2020, 32, 108052. [Google Scholar] [CrossRef]
- Westendorf, J.J.; Kahler, R.A.; Schroeder, T.M. Wnt Signaling in Osteoblasts and Bone Diseases. Gene 2004, 341, 19–39. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.; Di Matteo, B.; Chisari, E.; Cincinelli, G.; Angele, P.; Lattermann, C.; Filardo, G.; Vitale, N.D.; Selmi, C.; Kon, E. The Role of Wnt Pathway in the Pathogenesis of OA and Its Potential Therapeutic Implications in the Field of Regenerative Medicine. Biomed. Res. Int. 2018, 2018, 7402947. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The Amazing Osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Bellido, T. Osteocyte-Driven Bone Remodeling. Calcif. Tissue Int. 2014, 94, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.; Horowitz, M.; Choi, Y. Osteoimmunology: Interactions of the Bone and Immune System. Endocr. Rev. 2008, 29, 403–440. [Google Scholar] [CrossRef]
- Guder, C.; Gravius, S.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. Osteoimmunology: A Current Update of the Interplay Between Bone and the Immune System. Front. Immunol. 2020, 11, 58. [Google Scholar] [CrossRef]
- Rivollier, A.; Mazzorana, M.; Tebib, J.; Piperno, M.; Aitsiselmi, T.; Rabourdin-Combe, C.; Jurdic, P.; Servet-Delprat, C. Immature Dendritic Cell Transdifferentiation into Osteoclasts: A Novel Pathway Sustained by the Rheumatoid Arthritis Microenvironment. Blood 2004, 104, 4029–4037. [Google Scholar] [CrossRef]
- Speziani, C.; Rivollier, A.; Gallois, A.; Coury, F.; Mazzorana, M.; Azocar, O.; Flacher, M.; Bella, C.; Tebib, J.; Jurdic, P.; et al. Murine Dendritic Cell Transdifferentiation into Osteoclasts Is Differentially Regulated by Innate and Adaptive Cytokines. Eur. J. Immunol. 2007, 37, 747–757. [Google Scholar] [CrossRef]
- Maitra, R.; Follenzi, A.; Yaghoobian, A.; Montagna, C.; Merlin, S.; Cannizzo, E.S.; Hardin, J.A.; Cobelli, N.; Stanley, E.R.; Santambrogio, L. Dendritic Cell-Mediated in Vivo Bone Resorption. J. Immunol. 2010, 185, 1485–1491. [Google Scholar] [CrossRef]
- Wu, L.; Luo, Z.; Liu, Y.; Jia, L.; Jiang, Y.; Du, J.; Guo, L.; Bai, Y.; Liu, Y. Aspirin Inhibits RANKL-Induced Osteoclast Differentiation in Dendritic Cells by Suppressing NF-ΚB and NFATc1 Activation. Stem Cell Res. Ther. 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 Functions as an Osteoclastogenic Helper T Cell Subset That Links T Cell Activation and Bone Destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Frey, B.; Hess, A.; Zwerina, J.; Luther, J.; Nimmerjahn, F.; Engelke, K.; Kollias, G.; Hünig, T.; Schett, G.; et al. Regulatory T Cells Protect from Local and Systemic Bone Destruction in Arthritis. J. Immunol. 2010, 184, 7238–7246. [Google Scholar] [CrossRef] [PubMed]
- Flores-Borja, F.; Jury, E.C.; Mauri, C.; Ehrenstein, M.R. Defects in CTLA-4 Are Associated with Abnormal Regulatory T Cell Function in Rheumatoid Arthritis. Proc. Natl. Acad. Sci. USA 2008, 105, 19396–19401. [Google Scholar] [CrossRef]
- Hauser, B.; Zhao, S.; Visconti, M.R.; Riches, P.L.; Fraser, W.D.; Piec, I.; Goodson, N.J.; Ralston, S.H. Autoantibodies to Osteoprotegerin Are Associated with Low Hip Bone Mineral Density and History of Fractures in Axial Spondyloarthritis: A Cross-Sectional Observational Study. Calcif. Tissue Int. 2017, 101, 375–383. [Google Scholar] [CrossRef]
- Matsuda, K.; Shiba, N.; Hiraoka, K. New Insights into the Role of Synovial Fibroblasts Leading to Joint Destruction in Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 5173. [Google Scholar] [CrossRef]
- Moon, S.-J.; Ahn, I.E.; Jung, H.; Yi, H.; Kim, J.; Kim, Y.; Kwok, S.-K.; Park, K.-S.; Min, J.-K.; Park, S.-H.; et al. Temporal Differential Effects of Proinflammatory Cytokines on Osteoclastogenesis. Int. J. Mol. Med. 2013, 31, 769–777. [Google Scholar] [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 Mediates TNF-Induced Osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.-R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 Is a Master Regulator of Joint Remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ouyang, H.; Xie, Z.; Liang, Z.-H.; Wu, X.-W. Serum DKK-1 Level in the Development of Ankylosing Spondylitis and Rheumatic Arthritis: A Meta-Analysis. Exp. Mol. Med. 2016, 48, e228. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Wang, M.; Xia, Q.; Yang, J.; Wu, M.; Han, R.; Chen, M.; Hu, X.; Yuan, Y.; et al. The Serum Level of Dickkopf-1 in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Int. Immunopharmacol. 2018, 59, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Lamarque, R.; Gerbaix, M.; Caire, R.; Linossier, M.-T.; Laroche, N.; Thomas, M.; Thomas, T.; Vico, L.; Marotte, H. Early Sclerostin Expression Explains Bone Formation Inhibition before Arthritis Onset in the Rat Adjuvant-Induced Arthritis Model. Sci. Rep. 2018, 8, 3492. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.-M.; Liao, T.; Ye, Q.-L.; Wu, G.-C.; Zhang, Q.; Tao, S.-S.; Zhao, C.-N.; Wu, Q.; Dan, Y.-L.; Pan, H.-F.; et al. Increased Circulating Sclerostin Levels in Rheumatoid Arthritis Patients: An Updated Meta-Analysis. Z. Rheumatol. 2023, 82, 51–58. [Google Scholar] [CrossRef]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Böttcher, M.; Cromme, C.; König, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin Inhibition Promotes TNF-Dependent Inflammatory Joint Destruction. Sci. Transl. Med. 2016, 8, 330ra35. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- Singh, S.; Dutta, S.; Khasbage, S.; Kumar, T.; Sachin, J.; Sharma, J.; Varthya, S.B. A Systematic Review and Meta-Analysis of Efficacy and Safety of Romosozumab in Postmenopausal Osteoporosis. Osteoporos. Int. 2022, 33, 1–12. [Google Scholar] [CrossRef]
- Kobayakawa, T.; Miyazaki, A.; Kanayama, Y.; Hirano, Y.; Takahashi, J.; Suzuki, T.; Nakamura, Y. Comparable Efficacy of Denosumab and Romosozumab in Patients with Rheumatoid Arthritis Receiving Glucocorticoid Administration. Mod. Rheumatol. 2023, 33, 96–103. [Google Scholar] [CrossRef]
- Sims, N.A. Influences of the IL-6 Cytokine Family on Bone Structure and Function. Cytokine 2021, 146, 155655. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jiang, J.; Hong, R.; Xu, F.; Dai, S. Circulating IGFBP-3 and Interleukin 6 as Predictors of Osteoporosis in Postmenopausal Women: A Cross-Sectional Study. Mediat. Inflamm. 2023, 2023, 2613766. [Google Scholar] [CrossRef] [PubMed]
- Franchimont, N.; Wertz, S.; Malaise, M. Interleukin-6: An Osteotropic Factor Influencing Bone Formation? Bone 2005, 37, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bäckesjö, C.-M.; Haldosén, L.-A.; Lindgren, U. IL-6 Receptor Expression and IL-6 Effects Change during Osteoblast Differentiation. Cytokine 2008, 43, 165–173. [Google Scholar] [CrossRef]
- Herman, S.; Müller, R.B.; Krönke, G.; Zwerina, J.; Redlich, K.; Hueber, A.J.; Gelse, H.; Neumann, E.; Müller-Ladner, U.; Schett, G. Induction of Osteoclast-Associated Receptor, a Key Osteoclast Costimulation Molecule, in Rheumatoid Arthritis. Arthritis Rheum. 2008, 58, 3041–3050. [Google Scholar] [CrossRef]
- Crotti, T.N.; Dharmapatni, A.A.; Alias, E.; Zannettino, A.C.; Smith, M.D.; Haynes, D.R. The Immunoreceptor Tyrosine-Based Activation Motif (ITAM) -Related Factors Are Increased in Synovial Tissue and Vasculature of Rheumatoid Arthritic Joints. Arthritis Res. Ther. 2012, 14, R245. [Google Scholar] [CrossRef]
- Hecht, C.; Schett, G.; Finzel, S. The Impact of Rheumatoid Factor and ACPA on Bone Erosion in Rheumatoid Arthritis. Ann. Rheum. Dis. 2015, 74, e4. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of Osteoclastogenesis and Bone Loss by Human Autoantibodies against Citrullinated Vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Kleyer, A.; Finzel, S.; Rech, J.; Manger, B.; Krieter, M.; Faustini, F.; Araujo, E.; Hueber, A.J.; Harre, U.; Engelke, K.; et al. Bone Loss before the Clinical Onset of Rheumatoid Arthritis in Subjects with Anticitrullinated Protein Antibodies. Ann. Rheum. Dis. 2014, 73, 854–860. [Google Scholar] [CrossRef]
- Seeling, M.; Hillenhoff, U.; David, J.P.; Schett, G.; Tuckermann, J.; Lux, A.; Nimmerjahn, F. Inflammatory Monocytes and Fcγ Receptor IV on Osteoclasts Are Critical for Bone Destruction during Inflammatory Arthritis in Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 10729–10734. [Google Scholar] [CrossRef]
- Mathsson, L.; Lampa, J.; Mullazehi, M.; Rönnelid, J. Immune Complexes from Rheumatoid Arthritis Synovial Fluid Induce FcgammaRIIa Dependent and Rheumatoid Factor Correlated Production of Tumour Necrosis Factor-Alpha by Peripheral Blood Mononuclear Cells. Arthritis Res. Ther. 2006, 8, R64. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Bogliolo, L.; Vitolo, B.; Manzo, A.; Montecucco, C.; Caporali, R. Anti-Citrullinated Protein Antibodies and High Levels of Rheumatoid Factor Are Associated with Systemic Bone Loss in Patients with Early Untreated Rheumatoid Arthritis. Arthritis Res. Ther. 2016, 18, 226. [Google Scholar] [CrossRef] [PubMed]
- Amkreutz, J.A.M.P.; de Moel, E.C.; Theander, L.; Willim, M.; Heimans, L.; Nilsson, J.-Å.; Karlsson, M.K.; Huizinga, T.W.J.; Åkesson, K.E.; Jacobsson, L.T.H.; et al. Association Between Bone Mineral Density and Autoantibodies in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 921–930. [Google Scholar] [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.C.; van Veelen, P.A.; Levarht, N.E.W.; van der Helm-van Mil, A.H.M.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Oliveira, C.B.; Sandoval-Heglund, D.; Barrera-Vargas, A.; Merayo-Chalico, J.; Aguirre-Aguilar, E.; Kaplan, M.J.; Carmona-Rivera, C. Anti-Carbamylated LL37 Antibodies Promote Pathogenic Bone Resorption in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 715997. [Google Scholar] [CrossRef] [PubMed]
- Machold, K.P.; Stamm, T.A.; Nell, V.P.K.; Pflugbeil, S.; Aletaha, D.; Steiner, G.; Uffmann, M.; Smolen, J.S. Very Recent Onset Rheumatoid Arthritis: Clinical and Serological Patient Characteristics Associated with Radiographic Progression over the First Years of Disease. Rheumatology 2007, 46, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Svensson, B.; Andersson, M.L.E.; Gjertsson, I.; Hafström, I.; Ajeganova, S.; Forslind, K. Erosion-Free Rheumatoid Arthritis: Clinical and Conceptional Implications—A BARFOT Study. BMC Rheumatol. 2022, 6, 88. [Google Scholar] [CrossRef]
- Schett, G.; Gravallese, E. Bone Erosion in Rheumatoid Arthritis: Mechanisms, Diagnosis and Treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- Bromley, M.; Woolley, D.E. Histopathology of the Rheumatoid Lesion. Arthritis Rheum. 1984, 27, 857–863. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of Cell Types Responsible for Bone Resorption in Rheumatoid Arthritis and Juvenile Rheumatoid Arthritis. Am. J. Pathol. 1998, 152, 943–951. [Google Scholar]
- Kong, Y.-Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated T Cells Regulate Bone Loss and Joint Destruction in Adjuvant Arthritis through Osteoprotegerin Ligand. Nature 1999, 402, 43–47. [Google Scholar] [CrossRef]
- Berardi, S.; Corrado, A.; Maruotti, N.; Cici, D.; Cantatore, F.P. Osteoblast Role in the Pathogenesis of Rheumatoid Arthritis. Mol. Biol. Rep. 2021, 48, 2843–2852. [Google Scholar] [CrossRef]
- Harre, U.; Schett, G. Cellular and Molecular Pathways of Structural Damage in Rheumatoid Arthritis. Semin. Immunopathol. 2017, 39, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Caporali, R.; Manzo, A.; Vitolo, B.; Pitzalis, C.; Montecucco, C. Involvement of Subchondral Bone Marrow in Rheumatoid Arthritis: Lymphoid Neogenesis and in Situ Relationship to Subchondral Bone Marrow Osteoclast Recruitment. Arthritis Rheum. 2005, 52, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Allard-Chamard, H.; Carrier, N.; Dufort, P.; Durand, M.; de Brum-Fernandes, A.J.; Boire, G.; Komarova, S.V.; Dixon, S.J.; Harrison, R.E.; Manolson, M.F.; et al. Osteoclasts and Their Circulating Precursors in Rheumatoid Arthritis: Relationships with Disease Activity and Bone Erosions. Bone Rep. 2020, 12, 100282. [Google Scholar] [CrossRef] [PubMed]
- Crotti, T.N.; Flannery, M.; Walsh, N.C.; Fleming, J.D.; Goldring, S.R.; McHugh, K.P. NFATc1 Regulation of the Human Β3 Integrin Promoter in Osteoclast Differentiation. Gene 2006, 372, 92–102. [Google Scholar] [CrossRef]
- Nedeva, I.R.; Vitale, M.; Elson, A.; Hoyland, J.A.; Bella, J. Role of OSCAR Signaling in Osteoclastogenesis and Bone Disease. Front. Cell Dev. Biol. 2021, 9, 641162. [Google Scholar] [CrossRef]
- Chen, D.-Y.; Yao, L.; Chen, Y.-M.; Lin, C.-C.; Huang, K.-C.; Chen, S.-T.; Lan, J.-L.; Hsieh, S.-L.E. A Potential Role of Myeloid DAP12-Associating Lectin (MDL)-1 in the Regulation of Inflammation in Rheumatoid Arthritis Patients. PLoS ONE 2014, 9, e86105. [Google Scholar] [CrossRef]
- Jung, Y.-K.; Kang, Y.-M.; Han, S. Osteoclasts in the Inflammatory Arthritis: Implications for Pathologic Osteolysis. Immune Netw. 2019, 19, e2. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a Novel Chemokine-Dependent Molecular Mechanism Underlying Rheumatoid Arthritis-Associated Autoantibody-Mediated Bone Loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Gober, H.-J.; Sumiya, E.; Komatsu, N.; Okamoto, K.; Sawa, S.; Suematsu, A.; Suda, T.; Sato, K.; Takai, T.; et al. Immune Complexes Regulate Bone Metabolism through FcRγ Signalling. Nat. Commun. 2015, 6, 6637. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.; Cambridge, G. Rheumatoid Arthritis: The Predictable Effect of Small Immune Complexes in Which Antibody Is Also Antigen. Br. J. Rheumatol. 1998, 37, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.W.; Kang, E.H. Autoantibodies in Rheumatoid Arthritis: Rheumatoid Factors and Anticitrullinated Protein Antibodies. QJM Int. J. Med. 2010, 103, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive Effect of Anti-Citrullinated Protein Antibodies and Rheumatoid Factor on Bone Erosions in Patients with RA. Ann. Rheum. Dis. 2015, 74, 2151–2156. [Google Scholar] [CrossRef]
- Buckley, C.D.; Ospelt, C.; Gay, S.; Midwood, K.S. Location, Location, Location: How the Tissue Microenvironment Affects Inflammation in RA. Nat. Rev. Rheumatol. 2021, 17, 195–212. [Google Scholar] [CrossRef]
- Lu, M.-C.; Lai, N.-S.; Yu, H.-C.; Huang, H.-B.; Hsieh, S.-C.; Yu, C.-L. Anti-Citrullinated Protein Antibodies Bind Surface-Expressed Citrullinated Grp78 on Monocyte/Macrophages and Stimulate Tumor Necrosis Factor Alpha Production. Arthritis Rheum. 2010, 62, 1213–1223. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid Arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Weitzmann, M.N.; Ofotokun, I. Physiological and Pathophysiological Bone Turnover—Role of the Immune System. Nat. Rev. Endocrinol. 2016, 12, 518–532. [Google Scholar] [CrossRef]
- Luo, G.; Li, F.; Li, X.; Wang, Z.-G.; Zhang, B. TNF-α and RANKL Promote Osteoclastogenesis by Upregulating RANK via the NF-κB Pathway. Mol. Med. Rep. 2018, 17, 6605–6611. [Google Scholar] [CrossRef]
- Yao, Z.; Li, P.; Zhang, Q.; Schwarz, E.M.; Keng, P.; Arbini, A.; Boyce, B.F.; Xing, L. Tumor Necrosis Factor-α Increases Circulating Osteoclast Precursor Numbers by Promoting Their Proliferation and Differentiation in the Bone Marrow through Up-Regulation of c-Fms Expression. J. Biol. Chem. 2006, 281, 11846–11855. [Google Scholar] [CrossRef]
- Gilbert, L.; He, X.; Farmer, P.; Rubin, J.; Drissi, H.; van Wijnen, A.J.; Lian, J.B.; Stein, G.S.; Nanes, M.S. Expression of the Osteoblast Differentiation Factor RUNX2 (Cbfa1/AML3/Pebp2αA) Is Inhibited by Tumor Necrosis Factor-α. J. Biol. Chem. 2002, 277, 2695–2701. [Google Scholar] [CrossRef] [PubMed]
- Yeremenko, N.; Zwerina, K.; Rigter, G.; Pots, D.; Fonseca, J.E.; Zwerina, J.; Schett, G.; Baeten, D. Tumor Necrosis Factor and Interleukin-6 Differentially Regulate Dkk-1 in the Inflamed Arthritic Joint. Arthritis Rheumatol. 2015, 67, 2071–2075. [Google Scholar] [CrossRef] [PubMed]
- Srirangan, S.; Choy, E.H. The Role of Interleukin 6 in the Pathophysiology of Rheumatoid Arthritis. Ther. Adv. Musculoskelet. Dis. 2010, 2, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-Y.; Wu, H.-K.; Chen, Y.-H.; Hsu, Y.-P.; Cheng, M.-T.; Yu, C.-H.; Chen, S.-K. Interleukin-6 Transiently Promotes Proliferation of Osteoclast Precursors and Stimulates the Production of Inflammatory Mediators. Mol. Biol. Rep. 2022, 49, 3927–3937. [Google Scholar] [CrossRef]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 Directly Inhibits Osteoclast Differentiation by Suppressing Receptor Activator of NF-ΚB Signaling Pathways. J. Biol. Chem. 2008, 283, 11535–11540. [Google Scholar] [CrossRef]
- Takagawa, S.; Nakamura, F.; Kumagai, K.; Nagashima, Y.; Goshima, Y.; Saito, T. Decreased Semaphorin3A Expression Correlates with Disease Activity and Histological Features of Rheumatoid Arthritis. BMC Musculoskelet. Dis. 2013, 14, 40. [Google Scholar] [CrossRef]
- Akatsu, T.; Takahashi, N.; Udagawa, N.; Imamura, K.; Yamaguchi, A.; Sato, K.; Nagata, N.; Suda, T. Role of Prostaglandins in Interleukin-1-Induced Bone Resorption in Mice in Vitro. J. Bone Miner. Res. 1991, 6, 183–190. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The Mechanism of Osteoclast Differentiation Induced by IL-11. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef]
- Tanabe, N.; Maeno, M.; Suzuki, N.; Fujisaki, K.; Tanaka, H.; Ogiso, B.; Ito, K. IL-1α Stimulates the Formation of Osteoclast-like Cells by Increasing M-CSF and PGE2 Production and Decreasing OPG Production by Osteoblasts. Life Sci. 2005, 77, 615–626. [Google Scholar] [CrossRef]
- Jiang, Y.; Genant, H.K.; Watt, I.; Cobby, M.; Bresnihan, B.; Aitchison, R.; McCabe, D. A Multicenter, Double-Blind, Dose-Ranging, Randomized, Placebo-Controlled Study of Recombinant Human Interleukin-1 Receptor Antagonist in Patients with Rheumatoid Arthritis: Radiologic Progression and Correlation of Genant and Larsen Scores. Arthritis Rheum. 2000, 43, 1001–1009. [Google Scholar] [CrossRef]
- Gremese, E.; Tolusso, B.; Bruno, D.; Perniola, S.; Ferraccioli, G.; Alivernini, S. The Forgotten Key Players in Rheumatoid Arthritis: IL-8 and IL-17—Unmet Needs and Therapeutic Perspectives. Front. Med. 2023, 10, 956127. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E.; Suzuki, E.; Chao, C.-C.; Gorman, D.; Adda, S.; Maverakis, E.; Zarbalis, K.; Geissler, R.; Asio, A.; Blumenschein, W.M.; et al. IL-17A Gene Transfer Induces Bone Loss and Epidermal Hyperplasia Associated with Psoriatic Arthritis. Ann. Rheum. Dis. 2015, 74, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Nanke, Y.; Ichikawa, N.; Kobashigawa, T.; Mogi, M.; Kamatani, N.; Kotake, S. IL-17 Induces Osteoclastogenesis from Human Monocytes Alone in the Absence of Osteoblasts, Which Is Potently Inhibited by Anti-TNF-α Antibody: A Novel Mechanism of Osteoclastogenesis by IL-17. J. Cell Biochem. 2009, 108, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.G. IL-17 and IL-17-Producing Cells in Protection versus Pathology. Nat. Rev. Immunol. 2023, 23, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, B.; Bouvard, B.; Lequerre, T.; Lespessailles, E.; Marotte, H.; Pers, Y.-M.; Cortet, B. Implication of IL-17 in Bone Loss and Structural Damage in Inflammatory Rheumatic Diseases. Mediat. Inflamm. 2019, 2019, 8659302. [Google Scholar] [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation—Lessons from Rheumatoid Arthritis. Front. Immunol. 2019, 10, 353. [Google Scholar] [CrossRef]
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.; Ackermann, J.A.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of Autoantibody Activity by the IL-23–TH17 Axis Determines the Onset of Autoimmune Disease. Nat. Immunol. 2017, 18, 104–113. [Google Scholar] [CrossRef]
- Ciucci, T.; Ibáñez, L.; Boucoiran, A.; Birgy-Barelli, E.; Pène, J.; Abou-Ezzi, G.; Arab, N.; Rouleau, M.; Hébuterne, X.; Yssel, H.; et al. Bone Marrow Th17 TNFα Cells Induce Osteoclast Differentiation, and Link Bone Destruction to IBD. Gut 2015, 64, 1072–1081. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How Regulatory T Cells Work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Axmann, R.; Zwerina, J.; Polzer, K.; Gückel, E.; Skapenko, A.; Schulze-Koops, H.; Horwood, N.; Cope, A.; Schett, G. Treg Cells Suppress Osteoclast Formation: A New Link between the Immune System and Bone. Arthritis Rheum. 2007, 56, 4104–4112. [Google Scholar] [CrossRef]
- Fischer, L.; Herkner, C.; Kitte, R.; Dohnke, S.; Riewaldt, J.; Kretschmer, K.; Garbe, A.I. Foxp3+ Regulatory T Cells in Bone and Hematopoietic Homeostasis. Front. Endocrinol. 2019, 10, 578. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Hua, F.; Ding, W.; Ding, K.; Zhang, Y.; Xu, C. The Correlation between the Th17/Treg Cell Balance and Bone Health. Immun. Ageing 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Takayanagi, H. Mechanisms of Joint Destruction in Rheumatoid Arthritis—Immune Cell–Fibroblast–Bone Interactions. Nat. Rev. Rheumatol. 2022, 18, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving Concepts in Bone–Immune Interactions in Health and Disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Orr, C.; Vieira-Sousa, E.; Boyle, D.L.; Buch, M.H.; Buckley, C.D.; Cañete, J.D.; Catrina, A.I.; Choy, E.H.S.; Emery, P.; Fearon, U.; et al. Synovial Tissue Research: A State-of-the-Art Review. Nat. Rev. Rheumatol. 2017, 13, 463–475. [Google Scholar] [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring Synovial Homeostasis in Rheumatoid Arthritis by Targeting Fibroblast-like Synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef]
- Brentano, F.; Schorr, O.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Pre-B Cell Colony-Enhancing Factor/Visfatin, a New Marker of Inflammation in Rheumatoid Arthritis with Proinflammatory and Matrix-Degrading Activities. Arthritis Rheum. 2007, 56, 2829–2839. [Google Scholar] [CrossRef]
- Tu, J.; Huang, W.; Zhang, W.; Mei, J.; Zhu, C. Two Main Cellular Components in Rheumatoid Arthritis: Communication Between T Cells and Fibroblast-Like Synoviocytes in the Joint Synovium. Front. Immunol. 2022, 13, 922111. [Google Scholar] [CrossRef]
- Ouboussad, L.; Burska, A.N.; Melville, A.; Buch, M.H. Synovial Tissue Heterogeneity in Rheumatoid Arthritis and Changes with Biologic and Targeted Synthetic Therapies to Inform Stratified Therapy. Front. Med. 2019, 6, 45. [Google Scholar] [CrossRef]
- Buckley, C.; Filer, A. Fibroblasts and Fibroblastlike Synoviocytes. In Firestein and Kelley’s Textbook of Rheumatology; Elsevier, Inc.: Philadelphia, PA, USA, 2021; pp. 222–240. [Google Scholar]
- Wu, Z.; Ma, D.; Yang, H.; Gao, J.; Zhang, G.; Xu, K.; Zhang, L. Fibroblast-like Synoviocytes in Rheumatoid Arthritis: Surface Markers and Phenotypes. Int. Immunopharmacol. 2021, 93, 107392. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving Concepts of Rheumatoid Arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Grillet, B.; Pereira, R.V.S.; Van Damme, J.; Abu El-Asrar, A.; Proost, P.; Opdenakker, G. Matrix Metalloproteinases in Arthritis: Towards Precision Medicine. Nat. Rev. Rheumatol. 2023, 19, 363–377. [Google Scholar] [CrossRef]
- Noss, E.H.; Chang, S.K.; Watts, G.F.M.; Brenner, M.B. Cadherin-11 Engagement Modulates Matrix Metalloproteinase Production by Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheum. 2011, 63, 3768–3778. [Google Scholar] [CrossRef] [PubMed]
- Danks, L.; Komatsu, N.; Guerrini, M.M.; Sawa, S.; Armaka, M.; Kollias, G.; Nakashima, T.; Takayanagi, H. RANKL Expressed on Synovial Fibroblasts Is Primarily Responsible for Bone Erosions during Joint Inflammation. Ann. Rheum. Dis. 2016, 75, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Win, S.; Yan, M.; Huynh, N.C.-N.; Sawa, S.; Tsukasaki, M.; Terashima, A.; Pluemsakunthai, W.; Kollias, G.; Nakashima, T.; et al. Plasma Cells Promote Osteoclastogenesis and Periarticular Bone Loss in Autoimmune Arthritis. J. Clin. Investig. 2021, 131, e143060. [Google Scholar] [CrossRef]
- Juarez, M.; Toellner, D.S.; Karouzakis, E.; Hardy, R.; Yeo, L.; Bayley, R.; De Paz, B.; Raza, K.; Cooper, M.; Gay, S.; et al. Early Rheumatoid Arthritis and Resolving Fibroblasts Segregate According to Dickkopf Related Protein 1 Expression. Lancet 2013, 381, S57. [Google Scholar] [CrossRef]
- Zheng, L.; Hu, F.; Bian, W.; Li, Y.; Zhang, L.; Shi, L.; Ma, X.; Liu, Y.; Zhang, X.; Li, Z. Dickkopf-1 Perpetuated Synovial Fibroblast Activation and Synovial Angiogenesis in Rheumatoid Arthritis. Clin. Rheumatol. 2021, 40, 4279–4288. [Google Scholar] [CrossRef]
- Barranco, C. Sclerostin: A Novel Role in TNF Arthritis? Nat. Rev. Rheumatol. 2016, 12, 251. [Google Scholar] [CrossRef]
- Komatsu, N.; Takayanagi, H. Inflammation and Bone Destruction in Arthritis: Synergistic Activity of Immune and Mesenchymal Cells in Joints. Front. Imunol. 2012, 3, 7. [Google Scholar] [CrossRef]
- Chang, S.K.; Noss, E.H.; Chen, M.; Gu, Z.; Townsend, K.; Grenha, R.; Leon, L.; Lee, S.Y.; Lee, D.M.; Brenner, M.B. Cadherin-11 Regulates Fibroblast Inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 8402–8407. [Google Scholar] [CrossRef]
- Yellin, M.J.; Winikoff, S.; Fortune, S.M.; Baum, D.; Crow, M.K.; Lederman, S.; Chess, L. Ligation of CD40 on Fibroblasts Induces CD54 (ICAM-1) and CD106 (VCAM-1) up-Regulation and IL-6 Production and Proliferation. J. Leukoc. Biol. 1995, 58, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 Promotes Autoimmunity by Triggering a Positive-Feedback Loop via Interleukin-6 Induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Van Seventer, G.A.; Shimizu, Y.; Horgan, K.J.; Shaw, S. The LFA-1 Ligand ICAM-1 Provides an Important Costimulatory Signal for T Cell Receptor-Mediated Activation of Resting T Cells. J. Immunol. 1990, 144, 4579–4586. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Muraoka, S.; Kusunoki, N.; Masuoka, S.; Yamada, S.; Ogasawara, H.; Imai, T.; Akasaka, Y.; Tochigi, N.; Takahashi, H.; et al. Resistin Upregulates Chemokine Production by Fibroblast-like Synoviocytes from Patients with Rheumatoid Arthritis. Arthritis Res. Ther. 2017, 19, 263. [Google Scholar] [CrossRef]
- Cambier, S.; Gouwy, M.; Proost, P. The Chemokines CXCL8 and CXCL12: Molecular and Functional Properties, Role in Disease and Efforts towards Pharmacological Intervention. Cell. Mol. Immunol. 2023, 20, 217–251. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, B.; Jin, W.J.; Kim, H.-H.; Ha, H.; Lee, Z.H. Pathogenic Roles of CXCL10 Signaling through CXCR3 and TLR4 in Macrophages and T Cells: Relevance for Arthritis. Arthritis Res. Ther. 2017, 19, 163. [Google Scholar] [CrossRef]
- Hirota, K.; Hashimoto, M.; Yoshitomi, H.; Tanaka, S.; Nomura, T.; Yamaguchi, T.; Iwakura, Y.; Sakaguchi, N.; Sakaguchi, S. T Cell Self-Reactivity Forms a Cytokine Milieu for Spontaneous Development of IL-17+ Th Cells That Cause Autoimmune Arthritis. J. Exp. Med. 2007, 204, 41–47. [Google Scholar] [CrossRef]
- Burger, J.A.; Zvaifler, N.J.; Tsukada, N.; Firestein, G.S.; Kipps, T.J. Fibroblast-like Synoviocytes Support B-Cell Pseudoemperipolesis via a Stromal Cell–Derived Factor-1– and CD106 (VCAM-1)–Dependent Mechanism. J. Clin. Investig. 2001, 107, 305–315. [Google Scholar] [CrossRef]
- Bombardieri, M.; Kam, N.-W.; Brentano, F.; Choi, K.; Filer, A.; Kyburz, D.; McInnes, I.B.; Gay, S.; Buckley, C.; Pitzalis, C. A BAFF/APRIL-Dependent TLR3-Stimulated Pathway Enhances the Capacity of Rheumatoid Synovial Fibroblasts to Induce AID Expression and Ig Class-Switching in B Cells. Ann. Rheum. Dis. 2011, 70, 1857–1865. [Google Scholar] [CrossRef]
- Hasegawa, T.; Kikuta, J.; Sudo, T.; Matsuura, Y.; Matsui, T.; Simmons, S.; Ebina, K.; Hirao, M.; Okuzaki, D.; Yoshida, Y.; et al. Identification of a Novel Arthritis-Associated Osteoclast Precursor Macrophage Regulated by FoxM1. Nat. Immunol. 2019, 20, 1631–1643. [Google Scholar] [CrossRef]
- O’Neil, L.J.; Oliveira, C.B.; Wang, X.; Navarrete, M.; Barrera-Vargas, A.; Merayo-Chalico, J.; Aljahdali, R.; Aguirre-Aguilar, E.; Carlucci, P.; Kaplan, M.J.; et al. Neutrophil Extracellular Trap-Associated Carbamylation and Histones Trigger Osteoclast Formation in Rheumatoid Arthritis. Ann. Rheum. Dis. 2023, 82, 630–638. [Google Scholar] [CrossRef]
- Wigerblad, G.; Kaplan, M.J. Neutrophil Extracellular Traps in Systemic Autoimmune and Autoinflammatory Diseases. Nat. Rev. Immunol. 2023, 23, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Carlucci, P.M.; Goel, R.R.; James, E.; Brooks, S.R.; Rims, C.; Hoffmann, V.; Fox, D.A.; Buckner, J.H.; Kaplan, M.J. Neutrophil Extracellular Traps Mediate Articular Cartilage Damage and Enhance Cartilage Component Immunogenicity in Rheumatoid Arthritis. JCI Insight 2020, 5, e139388. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Silva, L.M.; Theofilou, V.I.; Greenwell-Wild, T.; Li, L.; Williams, D.W.; Ikeuchi, T.; Brenchley, L.; NIDCD/NIDCR Genomics and Computational Biology Core; Bugge, T.H.; et al. Neutrophil Extracellular Traps and Extracellular Histones Potentiate IL-17 Inflammation in Periodontitis. J. Exp. Med. 2023, 220, e20221751. [Google Scholar] [CrossRef] [PubMed]
- Harden, T.K.; Sondek, J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 355–379. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Bandyopadhyay, S.; Tyagi, K.; Roy, A. Recent advances in understanding the molecular role of phosphoinositide-specific phospholipase C gamma 1 as an emerging onco-driver and novel therapeutic target in human carcinogenesis. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188619. [Google Scholar] [CrossRef]
- Gresset, A.; Sondek, J.; Harden, T.K. The phospholipase C isozymes and their regulation. Subcell. Biochem. 2012, 58, 61–94. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Di, L.; Fu, G.; Chen, Y.; Bai, L.; Liu, J.; Feng, X.; McDonald, J.M.; Michalek, S.; et al. Phospholipase Cgamma2 mediates RANKL-stimulated lymph node organogenesis and osteoclastogenesis. J. Biol. Chem. 2008, 283, 29593–29601. [Google Scholar] [CrossRef]
- Kertész, Z.; Gyori, D.; Körmendi, S.; Fekete, T.; Kis-Tóth, K.; Jakus, Z.; Schett, G.; Rajnavölgyi, E.; Dobó-Nagy, C.; Mócsai, A. Phospholipase Cγ2 is required for basal but not oestrogen deficiency-induced bone resorption. Eur. J. Clin. Investig. 2012, 42, 49–60. [Google Scholar] [CrossRef]
- Glaser, D.L.; Kaplan, F.S. Osteoporosis. Definition and Clinical Presentation. Spine 1997, 22, 12S–16S. [Google Scholar] [CrossRef]
- Tong, J.-J.; Xu, S.-Q.; Zong, H.-X.; Pan, M.-J.; Teng, Y.-Z.; Xu, J.-H. Prevalence and Risk Factors Associated with Vertebral Osteoporotic Fractures in Patients with Rheumatoid Arthritis. Clin. Rheumatol. 2020, 39, 357–364. [Google Scholar] [CrossRef]
- Jin, S.; Hsieh, E.; Peng, L.; Yu, C.; Wang, Y.; Wu, C.; Wang, Q.; Li, M.; Zeng, X. Incidence of Fractures among Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Osteoporos. Int. 2018, 29, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Conforti, A.; Di Cola, I.; Pavlych, V.; Ruscitti, P.; Berardicurti, O.; Ursini, F.; Giacomelli, R.; Cipriani, P. Beyond the Joints, the Extra-Articular Manifestations in Rheumatoid Arthritis. Autoimmun. Rev. 2021, 20, 102735. [Google Scholar] [CrossRef] [PubMed]
- Hacquard-Bouder, C.; Ittah, M.; Breban, M. Animal Models of HLA-B27-Associated Diseases: New Outcomes. Jt. Bone Spine 2006, 73, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Bowness, P. HLA-B27. Annu. Rev. Immunol. 2015, 33, 29–48. [Google Scholar] [CrossRef]
- Gui, L.; Gu, J. The Study of the Effect of HLA-B27 on THP-1 Monocytic Cells Survival and Its Mechanism. Int. J. Rheum. Dis. 2023, 26, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Yong, S.-B.; Wei, J.C.-C. Ankylosing Spondylitis: History, Epidemiology, and HLA-B27. Int. J. Rheum. Dis. 2023, 26, 413–414. [Google Scholar] [CrossRef]
- Kavadichanda, C.G.; Geng, J.; Bulusu, S.N.; Negi, V.S.; Raghavan, M. Spondyloarthritis and the Human Leukocyte Antigen (HLA)-B*27 Connection. Front. Immunol. 2021, 12, 601518. [Google Scholar] [CrossRef]
- Layh-Schmitt, G.; Yang, E.Y.; Kwon, G.; Colbert, R.A. HLA-B27 Alters the Response to Tumor Necrosis Factor α and Promotes Osteoclastogenesis in Bone Marrow Monocytes From HLA-B27-Transgenic Rats: Cellular Effects of HLA-B27 Misfolding in Transgenic Rats. Arthritis Rheum. 2013, 65, 2123–2131. [Google Scholar] [CrossRef]
- Papet, I.; Yousfi, M.E.; Godin, J.-P.; Mermoud, A.-F.; Davicco, M.-J.; Coxam, V.; Breuillé, D.; Obled, C. HLA-B27 Rats Develop Osteopaenia through Increased Bone Resorption without Any Change in Bone Formation. J. Musculoskelet. Neuronal. Interact. 2008, 8, 251–256. [Google Scholar]
- Rauner, M.; Stupphann, D.; Haas, M.; Fert, I.; Glatigny, S.; Sipos, W.; Breban, M.; Pietschmann, P. The HLA-B27 Transgenic Rat, a Model of Spondyloarthritis, Has Decreased Bone Mineral Density and Increased RANKL to Osteoprotegerin MRNA Ratio. J. Rheumatol. 2009, 36, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Gamsjaeger, S.; Srivastava, A.K.; Wergedal, J.E.; Zwerina, J.; Klaushofer, K.; Paschalis, E.P.; Tatakis, D.N. Altered Bone Material Properties in HLA-B27 Rats Include Reduced Mineral to Matrix Ratio and Altered Collagen Cross-Links: BONE QUALITY IN HLA-B27 RATS. J. Bone Miner. Res. 2014, 29, 2382–2391. [Google Scholar] [CrossRef]
- Rauner, M.; Thiele, S.; Fert, I.; Araujo, L.M.; Layh-Schmitt, G.; Colbert, R.A.; Hofbauer, C.; Bernhardt, R.; Bürki, A.; Schwiedrzik, J.; et al. Loss of Bone Strength in HLA-B27 Transgenic Rats Is Characterized by a High Bone Turnover and Is Mainly Osteoclast-Driven. Bone 2015, 75, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Kim, H.-Y.; Lee, S.-H. Elevated Serum Levels of Soluble Receptor Activator of Nuclear Factors- B Ligand (SRANKL) and Reduced Bone Mineral Density in Patients with Ankylosing Spondylitis (AS). Rheumatology 2006, 45, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.Y.; Bealgey, K.W.; Fang, Y.; Gong, Y.M.; Bao, S. Interleukin-23: Immunological Roles and Clinical Implications. Int. J. Biochem. Cell Biol. 2009, 41, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Rizzo, A.; Triolo, G. Subclinical Gut Inflammation in Ankylosing Spondylitis. Curr. Opin. Rheumatol. 2016, 28, 89–96. [Google Scholar] [CrossRef]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Furuya, T.; Kobashigawa, T.; Kamatani, N.; Kotake, S. IL-23 Induces Human Osteoclastogenesis via IL-17 in Vitro, and Anti-IL-23 Antibody Attenuates Collagen-Induced Arthritis in Rats. Arthritis Res. Ther. 2007, 9, R96. [Google Scholar] [CrossRef]
- Li, X.; Kim, K.-W.; Cho, M.-L.; Ju, J.-H.; Kang, C.-M.; Oh, H.-J.; Min, J.-K.; Lee, S.-H.; Park, S.-H.; Kim, H.-Y. IL-23 Induces Receptor Activator of NF-ΚB Ligand Expression in Fibroblast-like Synoviocytes via STAT3 and NF-ΚB Signal Pathways. Immunol. Lett. 2010, 127, 100–107. [Google Scholar] [CrossRef]
- Chisălău, B.; Cringuș, L.-I.; Vreju, F.; Parvănescu, C.; Firulescu, S.; Dinescu, Ș.; Ciobanu, D.; Tica, A.; Sandu, R.; Siloși, I.; et al. New Insights into IL-17/IL-23 Signaling in Ankylosing Spondylitis (Review). Exp. Ther. Med. 2020, 20, 3493–3497. [Google Scholar] [CrossRef]
- Chalise, J.; Narendra, S.; Paudyal, B.; Magnusson, M. Interferon Alpha Inhibits Antigen-Specific Production of Proinflammatory Cytokines and Enhances Antigen-Specific Transforming Growth Factor Beta Production in Antigen-Induced Arthritis. Arthritis Res. Ther. 2013, 15, R143. [Google Scholar] [CrossRef]
- Tang, M.; Lu, L.; Yu, X. Interleukin-17A Interweaves the Skeletal and Immune Systems. Front. Immunol. 2021, 11, 625034. [Google Scholar] [CrossRef]
- Van Bezooijen, R.L.; Farih-Sips, H.C.M.; Papapoulos, S.E.; Löwik, C.W.G.M. Interleukin-17: A New Bone Acting Cytokine In Vitro. J. Bone Miner. Res. 1999, 14, 1513–1521. [Google Scholar] [CrossRef]
- Uluçkan, Ö.; Jimenez, M.; Karbach, S.; Jeschke, A.; Graña, O.; Keller, J.; Busse, B.; Croxford, A.L.; Finzel, S.; Koenders, M.; et al. Chronic Skin Inflammation Leads to Bone Loss by IL-17-Mediated Inhibition of Wnt Signaling in Osteoblasts. Sci. Transl. Med. 2016, 8, 330ra37. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, A.; Lembo, S.; Di Caprio, R.; Donnarumma, G.; Monfrecola, G.; Balato, N.; Ayala, F.; Balato, A. Psoriatic Cutaneous Inflammation Promotes Human Monocyte Differentiation into Active Osteoclasts, Facilitating Bone Damage. Eur. J. Immunol. 2017, 47, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Li, C.; Zhang, S.; Wong, P.; Cao, Y.; Griffith, J.F.; Zhang, X.; Gu, J.; Tam, L.-S. Effect of Biologics on Radiographic Progression of Peripheral Joint in Patients with Psoriatic Arthritis: Meta-Analysis. Rheumatology 2020, 59, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Osta, B.; Lavocat, F.; Eljaafari, A.; Miossec, P. Effects of Interleukin-17A on Osteogenic Differentiation of Isolated Human Mesenchymal Stem Cells. Front. Immunol. 2014, 5, 425. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor Necrosis Factor Alpha Stimulates Osteoclast Differentiation by a Mechanism Independent of the ODF/RANKL-RANK Interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Li, P.; Schwarz, E.M. The TNF-Alpha Transgenic Mouse Model of Inflammatory Arthritis. Springer Semin. Immunopathol. 2003, 25, 19–33. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Haas-Smith, S.A.; Li, P.; Hicks, D.G.; Schwarz, E.M. Mechanisms of TNF-α– and RANKL-Mediated Osteoclastogenesis and Bone Resorption in Psoriatic Arthritis. J. Clin. Investig. 2003, 111, 821–831. [Google Scholar] [CrossRef]
- Lambrecht, S.; Coudenys, J.; De Keyser, F.; Verbruggen, G.; Deforce, D.; Elewaut, D. Reduced Levels of the TGFb Family Member GDF15 in Spondyloarthritis versus Other Rheumatic Diseases. Ann. Rheum. Dis. 2011, 70, A88. [Google Scholar] [CrossRef]
- Hinoi, E.; Ochi, H.; Takarada, T.; Nakatani, E.; Iezaki, T.; Nakajima, H.; Fujita, H.; Takahata, Y.; Hidano, S.; Kobayashi, T.; et al. Positive Regulation of Osteoclastic Differentiation by Growth Differentiation Factor 15 Upregulated in Osteocytic Cells under Hypoxia. J. Bone Miner. Res. 2012, 27, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cui, Y.; Zhang, X.; Lin, H.; Zhang, G.; Zeng, H.; Zeng, Y. Increased Serum Levels of MIC1/GDF15 Correlated with Bone Erosion in Spondyloarthritis: A Pilot Study. Medicine 2018, 97, e13733. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Sirufo, M.M.; Suppa, M.; Ginaldi, L. IL-33/IL-31 Axis in Osteoporosis. Int. J. Mol. Sci. 2020, 21, 1239. [Google Scholar] [CrossRef] [PubMed]
- Perrigoue, J.G.; Li, J.; Zaph, C.; Goldschmidt, M.; Scott, P.; De Sauvage, F.J.; Pearce, E.J.; Ghilardi, N.; Artis, D. IL-31–IL-31R Interactions Negatively Regulate Type 2 Inflammation in the Lung. J. Exp. Med. 2007, 204, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Perrigoue, J.G.; Zaph, C.; Guild, K.; Du, Y.; Artis, D. IL-31-IL-31R Interactions Limit the Magnitude of Th2 Cytokine-Dependent Immunity and Inflammation Following Intestinal Helminth Infection. J. Immunol. 2009, 182, 6088–6094. [Google Scholar] [CrossRef]
- Rosine, N.; Etcheto, A.; Hendel-Chavez, H.; Seror, R.; Briot, K.; Molto, A.; Chanson, P.; Taoufik, Y.; Wendling, D.; Lories, R.; et al. Increase in Il-31 Serum Levels Is Associated with Reduced Structural Damage in Early Axial Spondyloarthritis. Sci. Rep. 2018, 8, 7731. [Google Scholar] [CrossRef]
- Lems, W.; Miceli-Richard, C.; Haschka, J.; Giusti, A.; Chistensen, G.L.; Kocijan, R.; Rosine, N.; Jørgensen, N.R.; Bianchi, G.; Roux, C. Bone Involvement in Patients with Spondyloarthropathies. Calcif. Tissue Int. 2022, 110, 393–420. [Google Scholar] [CrossRef]
- Rossini, M.; Viapiana, O.; Idolazzi, L.; Ghellere, F.; Fracassi, E.; Troplini, S.; Povino, M.R.; Kunnathully, V.; Adami, S.; Gatti, D. Higher Level of Dickkopf-1 Is Associated with Low Bone Mineral Density and Higher Prevalence of Vertebral Fractures in Patients with Ankylosing Spondylitis. Calcif. Tissue Int. 2016, 98, 438–445. [Google Scholar] [CrossRef]
- Szentpetery, A.; Heffernan, E.; Haroon, M.; Kilbane, M.; Gallagher, P.; McKenna, M.J.; FitzGerald, O. Striking Difference of Periarticular Bone Density Change in Early Psoriatic Arthritis and Rheumatoid Arthritis Following Anti-Rheumatic Treatment as Measured by Digital X-Ray Radiogrammetry. Rheumatology 2016, 55, 891–896. [Google Scholar] [CrossRef]
- Haynes, D.R. Osteoprotegerin Expression in Synovial Tissue from Patients with Rheumatoid Arthritis, Spondyloarthropathies and Osteoarthritis and Normal Controls. Rheumatology 2003, 42, 123–134. [Google Scholar] [CrossRef]
- Kavanaugh, A.; Puig, L.; Gottlieb, A.B.; Ritchlin, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; Song, M.; Zhu, Y.; Rahman, P.; et al. Maintenance of Clinical Efficacy and Radiographic Benefit Through Two Years of Ustekinumab Therapy in Patients with Active Psoriatic Arthritis: Results from a Randomized, Placebo-Controlled Phase III Trial. Arthritis Care Res. 2015, 67, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijde, D.; Landewé, R.B.; Mease, P.J.; McInnes, I.B.; Conaghan, P.G.; Pricop, L.; Ligozio, G.; Richards, H.B.; Mpofu, S. Brief Report: Secukinumab Provides Significant and Sustained Inhibition of Joint Structural Damage in a Phase III Study of Active Psoriatic Arthritis: Inhibition of joint structural damage with secukinumab. Arthritis Rheumatol. 2016, 68, 1914–1921. [Google Scholar] [CrossRef]
- Koenders, M.I.; Marijnissen, R.J.; Devesa, I.; Lubberts, E.; Joosten, L.A.B.; Roth, J.; Van Lent, P.L.E.M.; Van De Loo, F.A.; Van Den Berg, W.B. Tumor Necrosis Factor-Interleukin-17 Interplay Induces S100A8, Interleukin-1β, and Matrix Metalloproteinases, and Drives Irreversible Cartilage Destruction in Murine Arthritis: Rationale for Combination Treatment during Arthritis. Arthritis Rheum. 2011, 63, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Pool, B.; Smith, T.; Callon, K.E.; Lobo, M.; Taylor, W.J.; Jones, P.B.; Cornish, J.; McQueen, F.M. Circulating Mediators of Bone Remodeling in Psoriatic Arthritis: Implications for Disordered Osteoclastogenesis and Bone Erosion. Arthritis Res. Ther. 2010, 12, R164. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Li, Z.-C.; Sun, X.-L.; Liu, Y.-Y.; Shao, M.; Gan, Y.-Z.; Li, Y.-M.; Li, Y.-H.; Zhang, X.-W. Elevated Serum Dickkopf-1 Is a Biomarker for Bone Erosion in Patients with Psoriatic Arthritis. Chin. Med. J. 2021, 134, 2583–2588. [Google Scholar] [CrossRef] [PubMed]
- Fassio, A.; Idolazzi, L.; Viapiana, O.; Benini, C.; Vantaggiato, E.; Bertoldo, F.; Rossini, M.; Gatti, D. In Psoriatic Arthritis Dkk-1 and PTH Are Lower than in Rheumatoid Arthritis and Healthy Controls. Clin. Rheumatol. 2017, 36, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Kragstrup, T.W.; Andersen, T.; Heftdal, L.D.; Hvid, M.; Gerwien, J.; Sivakumar, P.; Taylor, P.C.; Senolt, L.; Deleuran, B. The IL-20 Cytokine Family in Rheumatoid Arthritis and Spondyloarthritis. Front. Immunol. 2018, 9, 2226. [Google Scholar] [CrossRef]
- Marijnissen, R.J.; Koenders, M.I.; Smeets, R.L.; Stappers, M.H.T.; Nickerson-Nutter, C.; Joosten, L.A.B.; Boots, A.M.H.; Van Den Berg, W.B. Increased Expression of Interleukin-22 by Synovial Th17 Cells during Late Stages of Murine Experimental Arthritis Is Controlled by Interleukin-1 and Enhances Bone Degradation: IL-1-Driven Regulation of IL-22 in Chronic Arthritis. Arthritis Rheum. 2011, 63, 2939–2948. [Google Scholar] [CrossRef]
- Sagiv, M.; Adawi, M.; Awisat, A.; Shouval, A.; Peri, R.; Sabbah, F.; Rosner, I.; Kessel, A.; Slobodin, G. The Association between Elevated Serum Interleukin-22 and the Clinical Diagnosis of Axial Spondyloarthritis: A Retrospective Study. Int. J. Rheum. Dis. 2022, 25, 56–60. [Google Scholar] [CrossRef]
- Poddubnyy, D.; Sieper, J. Mechanism of New Bone Formation in Axial Spondyloarthritis. Curr. Rheumatol. Rep. 2017, 19, 55. [Google Scholar] [CrossRef]
- Sieper, J.; van der Heijde, D. Review: Nonradiographic Axial Spondyloarthritis: New Definition of an Old Disease? Arthritis Rheum. 2013, 65, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Neerinckx, B.; Lories, R. Mechanisms, Impact and Prevention of Pathological Bone Regeneration in Spondyloarthritis. Curr. Opin. Rheumatol. 2017, 29, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Zheng, N.; Chen, S.-B.; Xiao, Z.-Y.; Wu, M.-Y.; Liu, Y.; Zeng, Q.-Y. Ten Years’ Experience with Needle Biopsy in the Early Diagnosis of Sacroiliitis. Arthritis Rheum. 2012, 64, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Appel, H.; Loddenkemper, C.; Grozdanovic, Z.; Ebhardt, H.; Dreimann, M.; Hempfing, A.; Stein, H.; Metz-Stavenhagen, P.; Rudwaleit, M.; Sieper, J. Correlation of Histopathological Findings and Magnetic Resonance Imaging in the Spine of Patients with Ankylosing Spondylitis. Arthritis Res. Ther. 2006, 8, R143. [Google Scholar] [CrossRef]
- Baraliakos, X.; Haibel, H.; Listing, J.; Sieper, J.; Braun, J. Continuous Long-Term Anti-TNF Therapy Does Not Lead to an Increase in the Rate of New Bone Formation over 8 Years in Patients with Ankylosing Spondylitis. Ann. Rheum. Dis. 2014, 73, 710–715. [Google Scholar] [CrossRef]
- Braun, J.; Baraliakos, X.; Deodhar, A.; Baeten, D.; Sieper, J.; Emery, P.; Readie, A.; Martin, R.; Mpofu, S.; Richards, H.B. Effect of Secukinumab on Clinical and Radiographic Outcomes in Ankylosing Spondylitis: 2-Year Results from the Randomised Phase III MEASURE 1 Study. Ann. Rheum. Dis. 2017, 76, 1070–1077. [Google Scholar] [CrossRef]
- Ramiro, S.; Heijde, D.V.D.; Tubergen, A.V.; Stolwijk, C.; Dougados, M.; Bosch, F.V.D.; Landewé, R. Higher Disease Activity Leads to More Structural Damage in the Spine in Ankylosing Spondylitis: 12-Year Longitudinal Data from the OASIS Cohort. Ann. Rheum. Dis. 2014, 73, 1455–1461. [Google Scholar] [CrossRef]
- van der Heijde, D.; Landewé, R.; Einstein, S.; Ory, P.; Vosse, D.; Ni, L.; Lin, S.L.; Tsuji, W.; Davis, J.C. Radiographic Progression of Ankylosing Spondylitis after up to Two Years of Treatment with Etanercept. Arthritis Rheum. 2008, 58, 1324–1331. [Google Scholar] [CrossRef]
- van Duivenvoorde, L.M.; Dorris, M.L.; Satumtira, N.; van Tok, M.N.; Redlich, K.; Tak, P.P.; Taurog, J.D.; Baeten, D.L. Relationship between Inflammation, Bone Destruction, and Osteoproliferation in the HLA-B27/Human Β2 -Microglobulin-Transgenic Rat Model of Spondylarthritis. Arthritis Rheum. 2012, 64, 3210–3219. [Google Scholar] [CrossRef]
- Llop, M.; Moreno, M.; Navarro-Compán, V.; Juanola, X.; de Miguel, E.; Almodóvar, R.; Quintana, E.C.; Sanz, J.S.; Beltrán, E.; Montesinos, M.D.R.; et al. Sustained Low Disease Activity Measured by ASDAS Slow Radiographic Spinal Progression in Axial Spondyloarthritis Patients Treated with TNF-Inhibitors: Data from REGISPONSERBIO. Arthritis Res. Ther. 2022, 24, 30. [Google Scholar] [CrossRef]
- van der Heijde, D.; Østergaard, M.; Reveille, J.D.; Baraliakos, X.; Kronbergs, A.; Sandoval, D.M.; Li, X.; Carlier, H.; Adams, D.H.; Maksymowych, W.P. Spinal Radiographic Progression and Predictors of Progression in Patients with Radiographic Axial Spondyloarthritis Receiving Ixekizumab Over 2 Years. J. Rheumatol. 2022, 49, 265–273. [Google Scholar] [CrossRef]
- Apostolakos, J.; Durant, T.J.; Dwyer, C.R.; Russell, R.P.; Weinreb, J.H.; Alaee, F.; Beitzel, K.; McCarthy, M.B.; Cote, M.P.; Mazzocca, A.D. The Enthesis: A Review of the Tendon-to-Bone Insertion. Muscles Ligaments Tendons J. 2014, 4, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.; Bridgewood, C.; Rowe, H.; Altaie, A.; Jones, E.; McGonagle, D. Cytokine “Fine Tuning” of Enthesis Tissue Homeostasis as a Pointer to Spondyloarthritis Pathogenesis with a Focus on Relevant TNF and IL-17 Targeted Therapies. Semin. Immunopathol. 2021, 43, 193–206. [Google Scholar] [CrossRef]
- Watad, A.; Rowe, H.; Russell, T.; Zhou, Q.; Anderson, L.K.; Khan, A.; Dunsmuir, R.; Loughenbury, P.; Borse, V.; Rao, A.; et al. Normal Human Enthesis Harbours Conventional CD4+ and CD8+ T Cells with Regulatory Features and Inducible IL-17A and TNF Expression. Ann. Rheum. Dis. 2020, 79, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Wakefield, R.J.; Tan, A.L.; D’Agostino, M.A.; Toumi, H.; Hayashi, K.; Emery, P.; Benjamin, M. Distinct Topography of Erosion and New Bone Formation in Achilles Tendon Enthesitis: Implications for Understanding the Link between Inflammation and Bone Formation in Spondylarthritis. Arthritis Rheum. 2008, 58, 2694–2699. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.; Lambrecht, S.; Verheugen, E.; Pauwels, E.; Kollias, G.; Armaka, M.; Verhoye, M.; Van der Linden, A.; Achten, R.; Lories, R.J.; et al. Proof of Concept: Enthesitis and New Bone Formation in Spondyloarthritis Are Driven by Mechanical Strain and Stromal Cells. Ann. Rheum. Dis. 2014, 73, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Schett, G. Effects of the IL-23–IL-17 Pathway on Bone in Spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Xie, Z.; Wang, P.; Li, J.; Li, M.; Cen, S.; Tang, S.; Liu, W.; Ye, G.; Li, Y.; et al. Enhanced Osteogenic Differentiation of Mesenchymal Stem Cells in Ankylosing Spondylitis: A Study Based on a Three-Dimensional Biomimetic Environment. Cell Death Dis. 2019, 10, 350. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, P.; Li, Y.; Deng, W.; Zhang, X.; Su, H.; Li, D.; Wu, Y.; Shen, H. Imbalance Between Bone Morphogenetic Protein 2 and Noggin Induces Abnormal Osteogenic Differentiation of Mesenchymal Stem Cells in Ankylosing Spondylitis. Arthritis Rheumatol. 2016, 68, 430–440. [Google Scholar] [CrossRef]
- Cui, H.; Li, Z.; Chen, S.; Li, X.; Chen, D.; Wang, J.; Li, Z.; Hao, W.; Zhong, F.; Zhang, K.; et al. CXCL12/CXCR4-Rac1–Mediated Migration of Osteogenic Precursor Cells Contributes to Pathological New Bone Formation in Ankylosing Spondylitis. Sci. Adv. 2022, 8, eabl8054. [Google Scholar] [CrossRef]
- Daoussis, D.; Liossis, S.N.C.; Solomou, E.E.; Tsanaktsi, A.; Bounia, K.; Karampetsou, M.; Yiannopoulos, G.; Andonopoulos, A.P. Evidence That Dkk-1 Is Dysfunctional in Ankylosing Spondylitis. Arthritis Rheum. 2010, 62, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Chen, C.; Wang, Z.-X.; Zhao, Y.; Jiang, L.-Q.; Fang, Y.; Zhang, R.-D.; Pan, H.-F.; Tao, S.-S. Serum DKK-1 Level in Ankylosing Spondylitis: Insights from Meta-Analysis and Mendelian Randomization. Front. Immunol. 2023, 14, 1193357. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Pavy, S.; Boudaoud, S.; Seror, R.; Goupille, P.; Chanson, P.; Heijde, D.V.D.; Gaalen, F.V.; Berenbaum, F.; Mariette, X.; et al. Increase in Dickkopf-1 Serum Level in Recent Spondyloarthritis. Data from the DESIR Cohort. PLoS ONE 2015, 10, e0134974. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Maksymowych, W.P.; Wordsworth, B.P.; Inman, R.D.; Danoy, P.; Rahman, P.; Stone, M.; Corr, M.; Gensler, L.S.; Gladman, D.; et al. Association Study of Genes Related to Bone Formation and Resorption and the Extent of Radiographic Change in Ankylosing Spondylitis. Ann. Rheum. Dis. 2015, 74, 1387–1393. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, G.; Wang, Y.; Yang, J.; Wang, Y.; Zhu, J.; Huang, F. Correlation between magnetic resonance imaging (MRI) findings and the new bone formation factor Dkk-1 in patients with spondyloarthritis. Clin. Rheumatol. 2019, 38, 465–475. [Google Scholar] [CrossRef]
- el Hamid, H.S.A.; Ibrahim, N.H.; Morsi, M.H.; Al-Tabbakh, A.-S.M.; EL-Melouk, M.S. Elevated Serum Dickkopf-1 Levels as a Biomarker for Disease Activity and Severity in Psoriatic Arthritis Patients. Egypt. J. Hosp. Med. 2022, 89, 6445. [Google Scholar] [CrossRef]
- Wahba, M.A.W.A.; El-Gazzar, N.M.; Elsharaby, R.M.; Tabra, S.A. DKK-1 in Psoriatic Arthritis: Correlation with Disease Activity and Enthesopathy. Reumatol. Clín. 2023, in press. [Google Scholar] [CrossRef]
- Aschermann, S.; Englbrecht, M.; Bergua, A.; Spriewald, B.M.; Said-Nahal, R.; Breban, M.; Schett, G.; Rech, J. Presence of HLA-B27 Is Associated with Changes of Serum Levels of Mediators of the Wnt and Hedgehog Pathway. Jt. Bone Spine 2016, 83, 43–46. [Google Scholar] [CrossRef]
- Saad, C.G.S.; Ribeiro, A.C.M.; Moraes, J.C.B.; Takayama, L.; Goncalves, C.R.; Rodrigues, M.B.; de Oliveira, R.M.; Silva, C.A.; Bonfa, E.; Pereira, R.M.R. Low Sclerostin Levels: A Predictive Marker of Persistent Inflammation in Ankylosing Spondylitis during Anti-Tumor Necrosis Factor Therapy? Arthritis Res. Ther. 2012, 14, R216. [Google Scholar] [CrossRef]
- Appel, H.; Ruiz-Heiland, G.; Listing, J.; Zwerina, J.; Herrmann, M.; Mueller, R.; Haibel, H.; Baraliakos, X.; Hempfing, A.; Rudwaleit, M.; et al. Altered Skeletal Expression of Sclerostin and Its Link to Radiographic Progression in Ankylosing Spondylitis. Arthritis Rheum. 2009, 60, 3257–3262. [Google Scholar] [CrossRef]
- Luchetti, M.M.; Ciccia, F.; Avellini, C.; Benfaremo, D.; Guggino, G.; Farinelli, A.; Ciferri, M.; Rossini, M.; Svegliati, S.; Spadoni, T.; et al. Sclerostin and Antisclerostin Antibody Serum Levels Predict the Presence of Axial Spondyloarthritis in Patients with Inflammatory Bowel Disease. J. Rheumatol. 2018, 45, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Sorour, N.E.; Abdel Hafeez, N.A.; Akl, E.M.; Mowafy, M.A.; Mohamed, A.A. Evaluation of Serum Level of Sclerostin in Patients with Psoriatic Arthritis. Benha J. Appl. Sci. 2021, 6, 11–15. [Google Scholar] [CrossRef]
- Fayed, A.; Elgohary, R.; Fawzy, M. Evaluating the Role of Serum Sclerostin as an Indicator of Activity and Damage in Egyptian Patients with Rheumatoid Arthritis: University Hospital Experience. Clin. Rheumatol. 2020, 39, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int. J. Mol. Sci. 2020, 21, 6665. [Google Scholar] [CrossRef]
- Daoussis, D.; Filippopoulou, A.; Liossis, S.-N.; Sirinian, C.; Klavdianou, K.; Bouris, P.; Karamanos, N.K.; Andonopoulos, A.P. Anti-TNFα Treatment Decreases the Previously Increased Serum Indian Hedgehog Levels in Patients with Ankylosing Spondylitis and Affects the Expression of Functional Hedgehog Pathway Target Genes. Semin. Arthritis Rheum. 2015, 44, 646–651. [Google Scholar] [CrossRef]
- González-Chávez, S.A.; Quiñonez-Flores, C.M.; Pacheco-Tena, C. Molecular Mechanisms of Bone Formation in Spondyloarthritis. Jt. Bone Spine 2016, 83, 394–400. [Google Scholar] [CrossRef]
- Lata, M.; Hettinghouse, A.S.; Liu, C. Targeting Tumor Necrosis Factor Receptors in Ankylosing Spondylitis. Ann. N. Y. Acad. Sci. 2019, 1442, 5–16. [Google Scholar] [CrossRef]
- Haroon, N.; Inman, R.D.; Learch, T.J.; Weisman, M.H.; Lee, M.; Rahbar, M.H.; Ward, M.M.; Reveille, J.D.; Gensler, L.S. The Impact of TNF-Inhibitors on Radiographic Progression in Ankylosing Spondylitis. Arthritis Rheum. 2013, 65, 2645–2654. [Google Scholar] [CrossRef]
- Koo, B.S.; Oh, J.S.; Park, S.Y.; Shin, J.H.; Ahn, G.Y.; Lee, S.; Joo, K.B.; Kim, T.-H. Tumour Necrosis Factor Inhibitors Slow Radiographic Progression in Patients with Ankylosing Spondylitis: 18-Year Real-World Evidence. Ann. Rheum. Dis. 2020, 79, 1327–1332. [Google Scholar] [CrossRef]
- Jo, S.; Wang, S.E.; Lee, Y.L.; Kang, S.; Lee, B.; Han, J.; Sung, I.-H.; Park, Y.-S.; Bae, S.-C.; Kim, T.-H. IL-17A Induces Osteoblast Differentiation by Activating JAK2/STAT3 in Ankylosing Spondylitis. Arthritis Res. Ther. 2018, 20, 115. [Google Scholar] [CrossRef]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-Producing Γδ T Cells Enhance Bone Regeneration. Nat. Commun. 2016, 7, 10928. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Park, J.-W.; Lee, J.-M.; Suh, J.-Y.; Lee, J.-K.; Chang, B.-S.; Um, H.-S.; Kim, J.-Y.; Lee, Y. IL-17 Inhibits Osteoblast Differentiation and Bone Regeneration in Rat. Arch. Oral Biol. 2014, 59, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Tok, M.N.; Duivenvoorde, L.M.; Kramer, I.; Ingold, P.; Pfister, S.; Roth, L.; Blijdorp, I.C.; Sande, M.G.H.; Taurog, J.D.; Kolbinger, F.; et al. Interleukin-17A Inhibition Diminishes Inflammation and New Bone Formation in Experimental Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Landewé, R.; Rahman, P.; Tahir, H.; Singhal, A.; Boettcher, E.; Navarra, S.; Readie, A.; Mpofu, S.; Delicha, E.M.; et al. Secukinumab Provides Sustained Improvement in Signs and Symptoms and Low Radiographic Progression in Patients with Psoriatic Arthritis: 2-Year (End-of-Study) Results from the FUTURE 5 Study. RMD Open 2021, 7, e001600. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Gensler, L.S.; Sieper, J.; Clark, M.; Calderon, C.; Wang, Y.; Zhou, Y.; Leu, J.H.; Campbell, K.; Sweet, K.; et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 258–270. [Google Scholar] [CrossRef]
- Baeten, D.; Østergaard, M.; Wei, J.C.-C.; Sieper, J.; Järvinen, P.; Tam, L.-S.; Salvarani, C.; Kim, T.-H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 Inhibitor, for Ankylosing Spondylitis: Results of a Randomised, Double-Blind, Placebo-Controlled, Proof-of-Concept, Dose-Finding Phase 2 Study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef]
- McGonagle, D.; Watad, A.; Sharif, K.; Bridgewood, C. Why Inhibition of IL-23 Lacked Efficacy in Ankylosing Spondylitis. Front. Immunol. 2021, 12, 614255. [Google Scholar] [CrossRef]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R.M. Interleukin-22: Immunobiology and Pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 Induces Spondyloarthropathy by Acting on ROR-Γt+ CD3+CD4−CD8− Entheseal Resident T Cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef]
- El-Zayadi, A.A.; Jones, E.A.; Churchman, S.M.; Baboolal, T.G.; Cuthbert, R.J.; El-Jawhari, J.J.; Badawy, A.M.; Alase, A.A.; El-Sherbiny, Y.M.; McGonagle, D. Interleukin-22 Drives the Proliferation, Migration and Osteogenic Differentiation of Mesenchymal Stem Cells: A Novel Cytokine That Could Contribute to New Bone Formation in Spondyloarthropathies. Rheumatology 2017, 56, 488–493. [Google Scholar] [CrossRef]
- Upadhyay, J.; Farr, O.M.; Mantzoros, C.S. The Role of Leptin in Regulating Bone Metabolism. Metabolism 2015, 64, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Hartl, A.; Sieper, J.; Syrbe, U.; Listing, J.; Hermann, K.-G.; Rudwaleit, M.; Poddubnyy, D. Serum Levels of Leptin and High Molecular Weight Adiponectin Are Inversely Associated with Radiographic Spinal Progression in Patients with Ankylosing Spondylitis: Results from the ENRADAS Trial. Arthritis Res. Ther. 2017, 19, 140. [Google Scholar] [CrossRef]
- Karsenty, G.; Yadav, V.K. Regulation of Bone Mass by Serotonin: Molecular Biology and Therapeutic Implications. Annu. Rev. Med. 2011, 62, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Klavdianou, K.; Liossis, S.-N.; Papachristou, D.J.; Theocharis, G.; Sirinian, C.; Kottorou, A.; Filippopoulou, A.; Andonopoulos, A.P.; Daoussis, D. Decreased Serotonin Levels and Serotonin-Mediated Osteoblastic Inhibitory Signaling in Patients With Ankylosing Spondylitis. J. Bone Min. Res. 2016, 31, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Clunie, G.; Horwood, N. Loss and Gain of Bone in Spondyloarthritis: What Drives These Opposing Clinical Features? Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20969260. [Google Scholar] [CrossRef]
- Ghozlani, I.; Ghazi, M.; Nouijai, A.; Mounach, A.; Rezqi, A.; Achemlal, L.; Bezza, A.; El Maghraoui, A. Prevalence and Risk Factors of Osteoporosis and Vertebral Fractures in Patients with Ankylosing Spondylitis. Bone 2009, 44, 772–776. [Google Scholar] [CrossRef]
- Puche-Larrubia, M.Á.; Ladehesa-Pineda, L.; Font-Ugalde, P.; Escudero-Contreras, A.; Moltó, A.; López-Medina, C.; Collantes-Estévez, E. Distribution of Comorbidities in Spondyloarthritis with Regard to the Phenotype and Psoriasis: Data from the ASAS-COMOSPA Study. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X2110452. [Google Scholar] [CrossRef]
- Orsolini, G.; Fassio, A.; Rossini, M.; Adami, G.; Giollo, A.; Caimmi, C.; Idolazzi, L.; Viapiana, O.; Gatti, D. Effects of biological and targeted synthetic DMARDs on bone loss in rheumatoid arthritis. Pharmacol. Res. 2019, 147, 104354. [Google Scholar] [CrossRef]
- Orsolini, G.; Bertoldi, I.; Rossini, M. Osteoimmunology in rheumatoid and psoriatic arthritis: Potential effects of tofacitinib on bone involvement. Clin. Rheumatol. 2020, 39, 727–736. [Google Scholar] [CrossRef]
| Cells | Effect on Bone Metabolism |
|---|---|
| DC | Differentiate into osteoclast-like cells stimulated by RANKL and M-CSF [23] |
| Th1 cells | Reduce osteoclastogenesis inhibiting RANK expression [23] |
| Th2 cells | Reduce osteoclastogenesis inhibiting RANK expression [23] |
| Th17 cells | Dual effect on bone metabolism (bone loss/bone formation) depending on the microenvironment, via IL-17 production |
| Th23 cells | Polarization into Th17 via IL23 production [27] |
| FLS | Promote bone loss via both RANKL-dependent and RANKL-independent pathways [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsini, F.; Crotti, C.; Cincinelli, G.; Di Taranto, R.; Amati, A.; Ferrito, M.; Varenna, M.; Caporali, R. Bone Involvement in Rheumatoid Arthritis and Spondyloartritis: An Updated Review. Biology 2023, 12, 1320. https://doi.org/10.3390/biology12101320
Orsini F, Crotti C, Cincinelli G, Di Taranto R, Amati A, Ferrito M, Varenna M, Caporali R. Bone Involvement in Rheumatoid Arthritis and Spondyloartritis: An Updated Review. Biology. 2023; 12(10):1320. https://doi.org/10.3390/biology12101320
Chicago/Turabian StyleOrsini, Francesco, Chiara Crotti, Gilberto Cincinelli, Raffaele Di Taranto, Andrea Amati, Matteo Ferrito, Massimo Varenna, and Roberto Caporali. 2023. "Bone Involvement in Rheumatoid Arthritis and Spondyloartritis: An Updated Review" Biology 12, no. 10: 1320. https://doi.org/10.3390/biology12101320
APA StyleOrsini, F., Crotti, C., Cincinelli, G., Di Taranto, R., Amati, A., Ferrito, M., Varenna, M., & Caporali, R. (2023). Bone Involvement in Rheumatoid Arthritis and Spondyloartritis: An Updated Review. Biology, 12(10), 1320. https://doi.org/10.3390/biology12101320