
Microglia and Astrocytes in Amyotrophic Lateral Sclerosis: Disease-Associated States, Pathological Roles, and Therapeutic Potential

 , , , and
, , , and 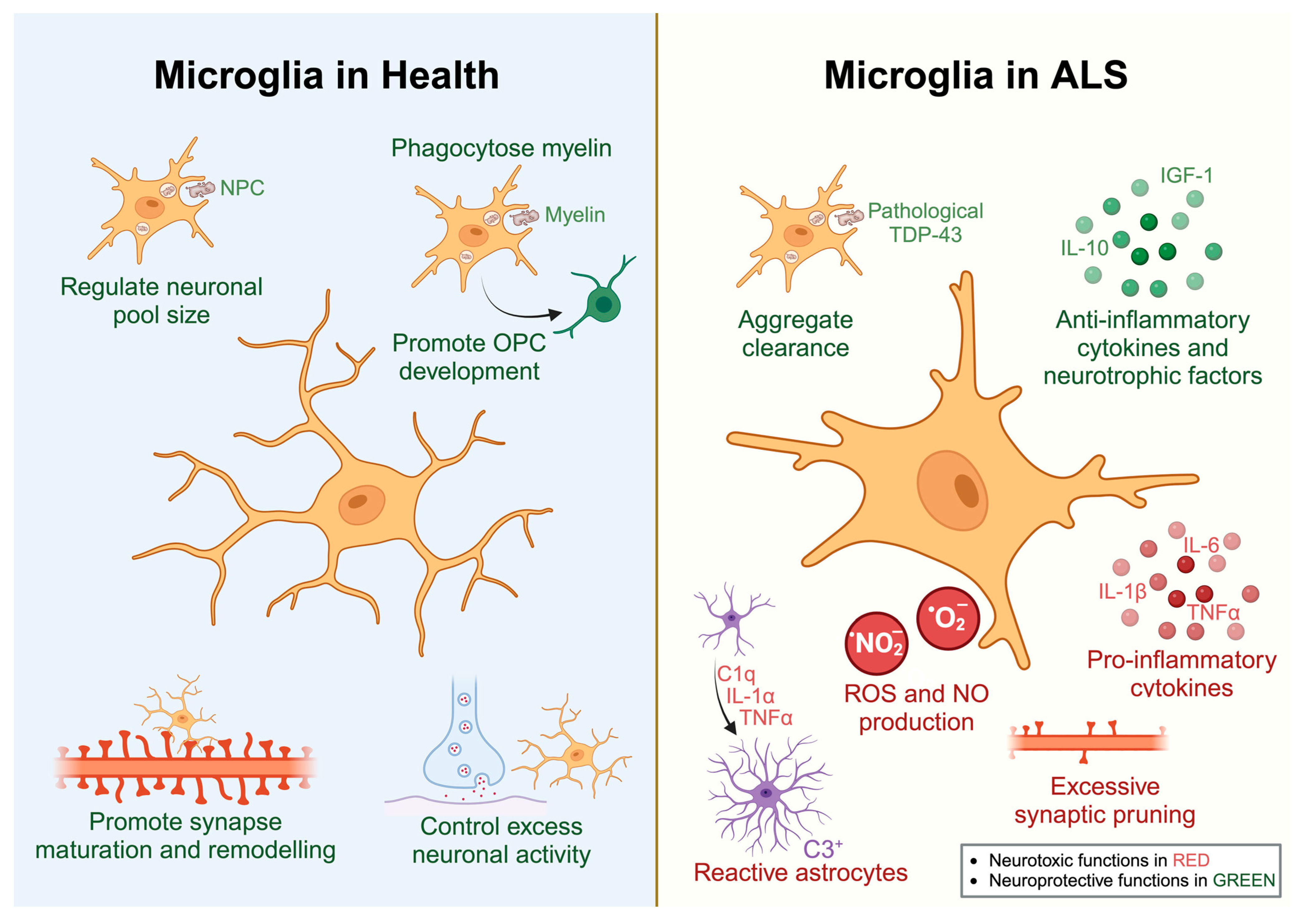{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Microglia and Astrocytes: Their Physiological Roles and Disease-Associated States
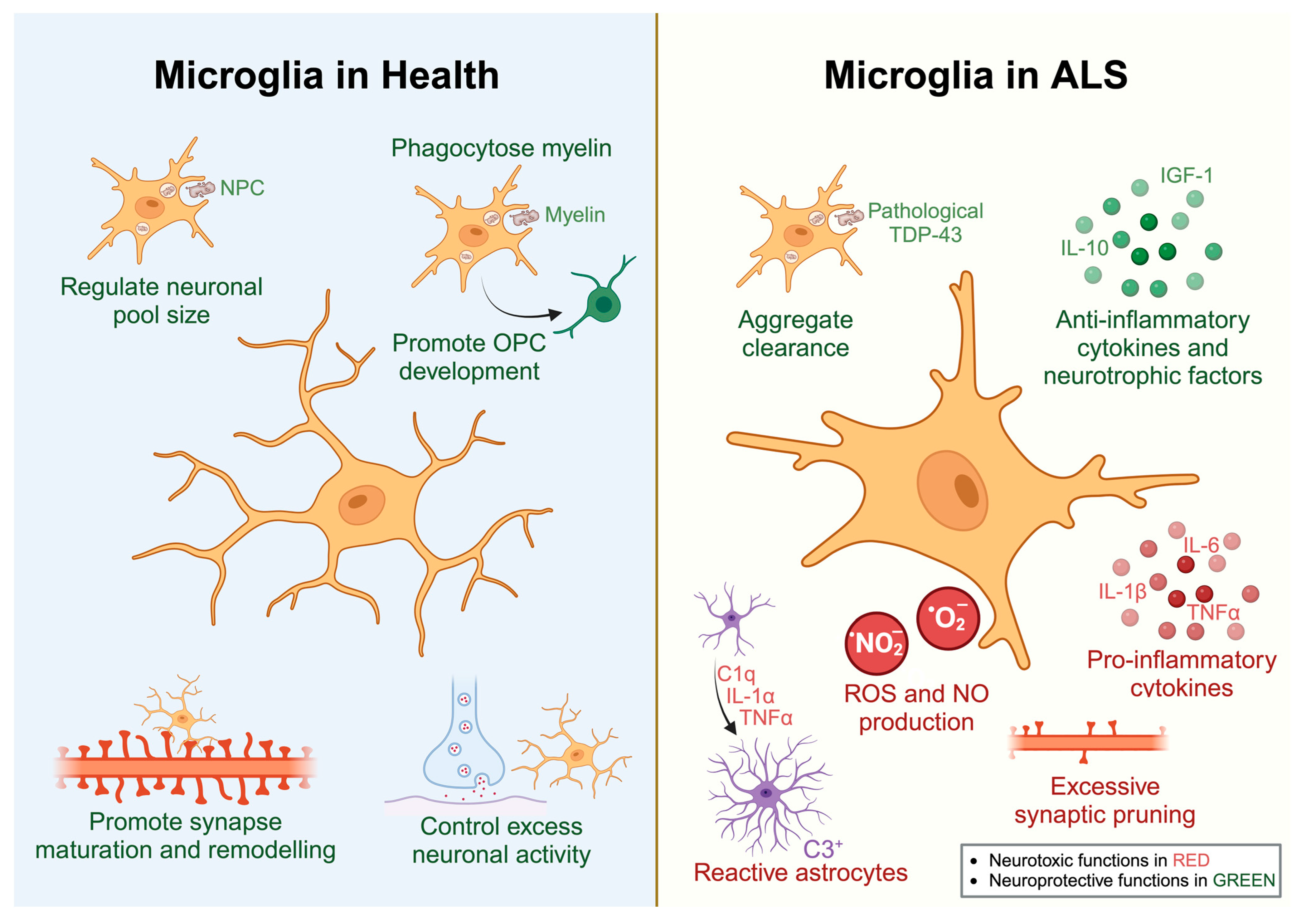
2.1. Microglia
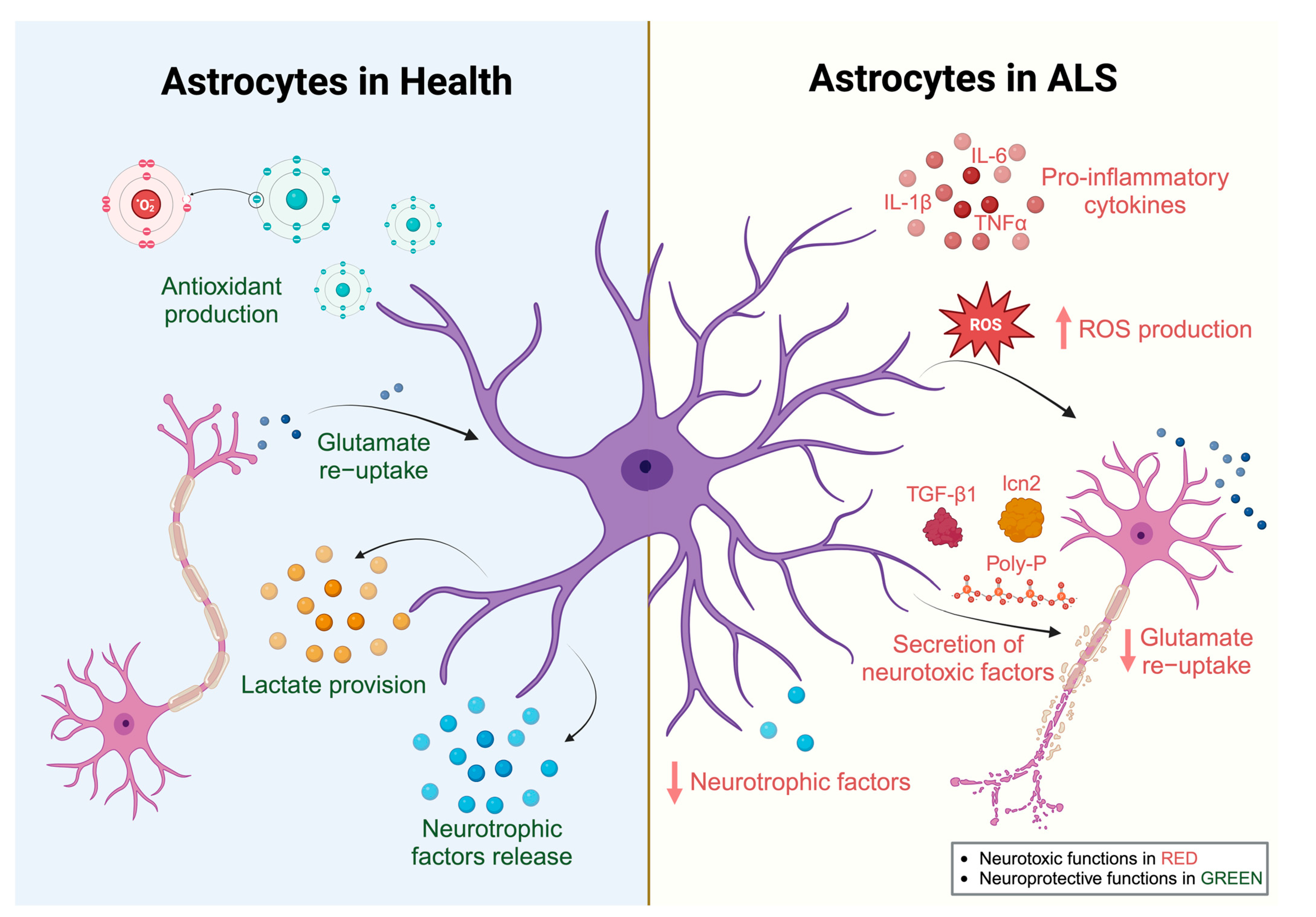
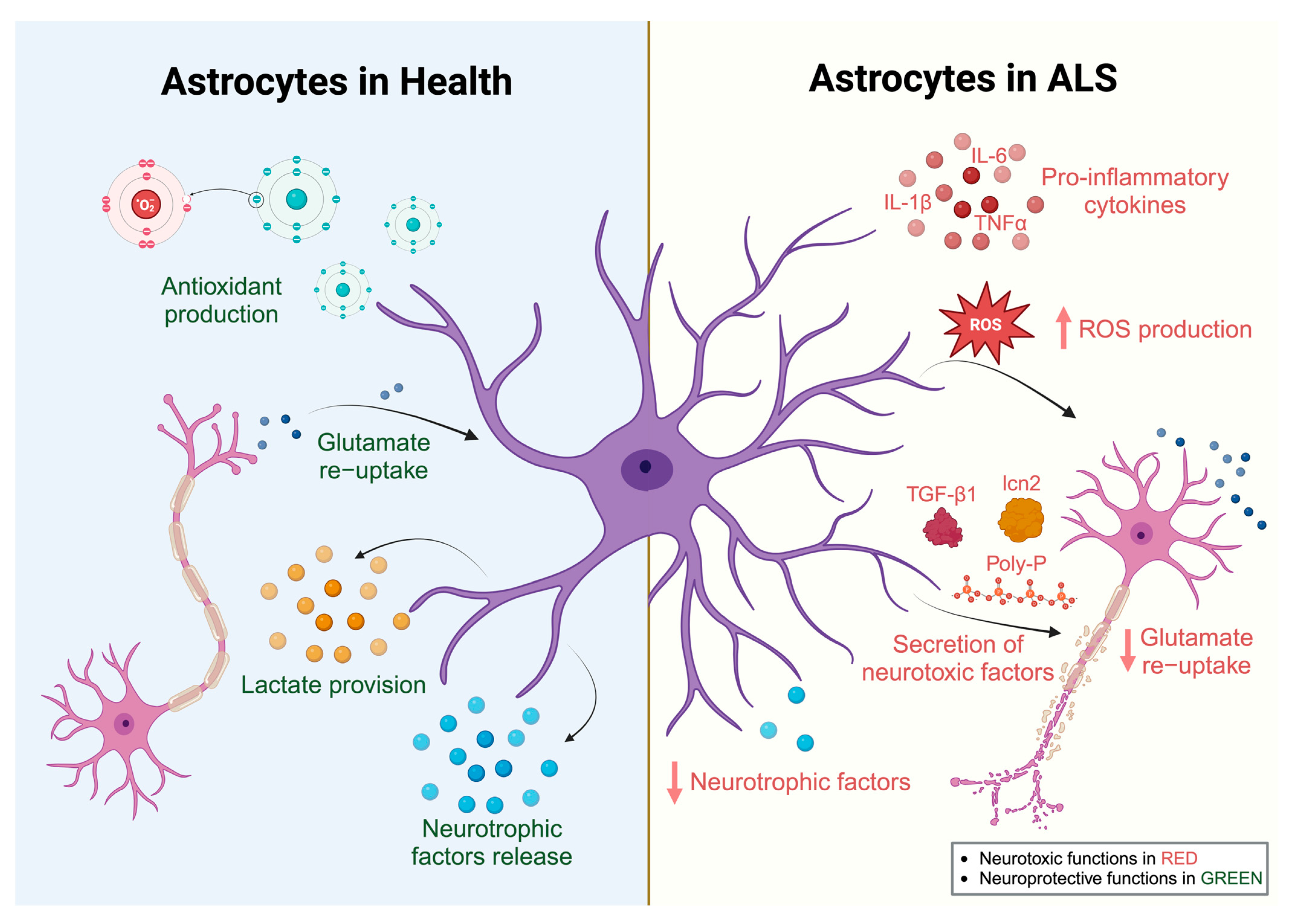
2.2. Astrocytes
3. Microglial and Astrocytic Reactivity in Human ALS
4. Role of Microglia and Astrocytes in ALS Rodent Models
4.1. Glial Studies from SOD1 Rodent Models
4.2. Glial Studies from TARDBP Rodent Models
4.3. Glial Studies from C9ORF72 Rodent Models
4.4. Insights from Human iPSC-Derived Microglia and Astrocytes That Model ALS
5. Candidate Therapeutics for ALS Targeting Microglia or Astrocytes
5.1. Masitinib
5.2. RIPK1 Inhibitors
5.3. CSF1R Inhibitors
5.4. MN-166 (Ibudilast)
5.5. Complement Cascade Inhibitors
5.6. Astrocyte Transplantation
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene Discovery in Amyotrophic Lateral Sclerosis: Implications for Clinical Management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, J.R.; van Bruggen, R.; Park, J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Mol. Cells 2018, 41, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Brás, J.; Hardy, J. SnapShot: Genetics of ALS and FTD. Cell 2015, 160, 798–798.e1. [Google Scholar] [CrossRef]
- Izrael, M.; Slutsky, S.G.; Revel, M. Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front. Neurosci. 2020, 14, 824. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.S.; van Bruggen, R.; Kim, J.R.; Chen, X.X.L.; Chan, C.; Lee, J.; Cho, W.I.; Zhao, M.; Arndt, C.; Maksimovic, K.; et al. Selective Neuronal Degeneration in MATR3 S85C Knock-in Mouse Model of Early-Stage ALS. Nat. Commun. 2020, 11, 5304. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Maksimovic, K.; Lee, J.; Khan, M.; Masuda, R.; Park, J. Selective Loss of MATR3 in Spinal Interneurons, Upper Motor Neurons and Hippocampal CA1 Neurons in a MATR3 S85C Knock-In Mouse Model of Amyotrophic Lateral Sclerosis. Biology 2022, 11, 298. [Google Scholar] [CrossRef]
- Ebstein, S.Y.; Yagudayeva, I.; Shneider, N.A. Mutant TDP-43 Causes Early-Stage Dose-Dependent Motor Neuron Degeneration in a TARDBP Knockin Mouse Model of ALS. Cell Rep. 2019, 26, 364–373.e4. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.; Chen, G.; Cui, S. Dissecting the Dual Role of the Glial Scar and Scar-Forming Astrocytes in Spinal Cord Injury. Front. Cell. Neurosci. 2020, 14, 78. [Google Scholar] [CrossRef]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual Roles of Astrocytes in Plasticity and Reconstruction after Traumatic Brain Injury. Cell Commun. Signal. CCS 2020, 18, 62. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The Dual Role of Astrocyte Activation and Reactive Gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Gamdzyk, M.; Lenahan, C.; Tang, J.; Tan, S.; Zhang, J.H. The Dual Role of Microglia in Blood-Brain Barrier Dysfunction after Stroke. Curr. Neuropharmacol. 2020, 18, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Onuska, K.M. The Dual Role of Microglia in the Progression of Alzheimer’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 1608–1610. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia States and Nomenclature: A Field at Its Crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-Resident Macrophages Originate from Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of Microglia in Central Nervous System Infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New Insights on the Role of Microglia in Synaptic Pruning in Health and Disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.-M.; Akriditou, M.-A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance during Adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-Related Myelin Degradation Burdens the Clearance Function of Microglia during Aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White Matter Aging Drives Microglial Diversity. Neuron 2021, 109, 1100–1117.e10. [Google Scholar] [CrossRef] [PubMed]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative Feedback Control of Neuronal Activity by Microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernández-Klett, F.; Lin, G.; Sagar, N.; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A New Fate Mapping System Reveals Context-Dependent Random or Clonal Expansion of Microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local Self-Renewal Can Sustain CNS Microglia Maintenance and Function throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Tremblay, M.-È.; Wake, H. Never-Resting Microglia: Physiological Roles in the Healthy Brain and Pathological Implications. Front. Cell. Neurosci. 2014, 8, 240. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.-P.; et al. A Lymphocyte-Microglia-Astrocyte Axis in Chronic Active Multiple Sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, L.; Hu, Z.; Dufort, C.; Hession, C.C.; Walker, A.J.; Niu, K.; Zhu, H.; Liu, N.; Liu, J.S.; Levin, J.Z.; et al. A RIPK1-Regulated Inflammatory Microglial State in Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2025102118. [Google Scholar] [CrossRef] [PubMed]
- De Andrade Costa, A.; Chatterjee, J.; Cobb, O.; Sanapala, S.; Scheaffer, S.; Guo, X.; Dahiya, S.; Gutmann, D.H. RNA Sequence Analysis Reveals ITGAL/CD11A as a Stromal Regulator of Murine Low-Grade Glioma Growth. Neuro Oncol. 2022, 24, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single Cell RNA Sequencing of Human Microglia Uncovers a Subset Associated with Alzheimer’s Disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Liu, Y.U.; Zhao, S.; Zhang, L.; Bosco, D.B.; Pang, Y.-P.; Zhong, J.; Sheth, U.; Martens, Y.A.; Zhao, N.; et al. TREM2 Interacts with TDP-43 and Mediates Microglial Neuroprotection against TDP-43-Related Neurodegeneration. Nat. Neurosci. 2022, 25, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854.e9. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Meilandt, W.J.; Xie, L.; Gandham, V.D.; Ngu, H.; Barck, K.H.; Rezzonico, M.G.; Imperio, J.; Lalehzadeh, G.; Huntley, M.A.; et al. Trem2 Restrains the Enhancement of Tau Accumulation and Neurodegeneration by β-Amyloid Pathology. Neuron 2021, 109, 1283–1301.e6. [Google Scholar] [CrossRef] [PubMed]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 Deficiency Exacerbates Tau Pathology through Dysregulated Kinase Signaling in a Mouse Model of Tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 Deficiency Attenuates Neuroinflammation and Protects against Neurodegeneration in a Mouse Model of Tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef]
- Sayed, F.A.; Telpoukhovskaia, M.; Kodama, L.; Li, Y.; Zhou, Y.; Le, D.; Hauduc, A.; Ludwig, C.; Gao, F.; Clelland, C.; et al. Differential Effects of Partial and Complete Loss of TREM2 on Microglial Injury Response and Tauopathy. Proc. Natl. Acad. Sci. USA 2018, 115, 10172–10177. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 Function Impedes Tau Seeding in Neuritic Plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M.; et al. TREM2 Drives Microglia Response to Amyloid-β via SYK-Dependent and -Independent Pathways. Cell 2022, 185, 4153–4169.e19. [Google Scholar] [CrossRef] [PubMed]
- Ennerfelt, H.; Frost, E.L.; Shapiro, D.A.; Holliday, C.; Zengeler, K.E.; Voithofer, G.; Bolte, A.C.; Lammert, C.R.; Kulas, J.A.; Ulland, T.K.; et al. SYK Coordinates Neuroprotective Microglial Responses in Neurodegenerative Disease. Cell 2022, 185, 4135–4152.e22. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J. Astrocyte Heterogeneity in the Adult Central Nervous System. Front. Cell. Neurosci. 2018, 12, 401. [Google Scholar] [CrossRef] [PubMed]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte-Neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter Release from Astrocytes: Functional, Developmental and Pathological Implications in the Brain. Front. Neurosci. 2016, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Covelo, A.; Araque, A. Neuronal Activity Determines Distinct Gliotransmitter Release from a Single Astrocyte. eLife 2018, 7, e32237. [Google Scholar] [CrossRef] [PubMed]
- Kawanabe, R.; Yoshihara, K.; Hatada, I.; Tsuda, M. Activation of Spinal Dorsal Horn Astrocytes by Noxious Stimuli Involves Descending Noradrenergic Signaling. Mol. Brain 2021, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, L.; Echeverría, A.; Georgiou, A.; Kuhn, B. Ca+ Activity Maps of Astrocytes Tagged by Axoastrocytic AAV Transfer. Sci. Adv. 2022, 8, eabe5371. [Google Scholar] [CrossRef]
- Lines, J.; Martin, E.D.; Kofuji, P.; Aguilar, J.; Araque, A. Astrocytes Modulate Sensory-Evoked Neuronal Network Activity. Nat. Commun. 2020, 11, 3689. [Google Scholar] [CrossRef]
- Zhou, Z.; Okamoto, K.; Onodera, J.; Hiragi, T.; Andoh, M.; Ikawa, M.; Tanaka, K.F.; Ikegaya, Y.; Koyama, R. Astrocytic CAMP Modulates Memory via Synaptic Plasticity. Proc. Natl. Acad. Sci. USA 2021, 118, e2016584118. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The Role of Astrocytic Glutamate Transporters GLT-1 and GLAST in Neurological Disorders: Potential Targets for Neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- Mederos, S.; Perea, G. GABAergic-Astrocyte Signaling: A Refinement of Inhibitory Brain Networks. Glia 2019, 67, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial Influence on the Blood Brain Barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Pham, L.-D.D.; Arai, K.; Lo, E.H. Reactive Astrocytes Promote Adhesive Interactions between Brain Endothelium and Endothelial Progenitor Cells via HMGB1 and Beta-2 Integrin Signaling. Stem Cell Res. 2014, 12, 531–538. [Google Scholar] [CrossRef]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.V. Glial Scar Borders Are Formed by Newly Proliferated, Elongated Astrocytes That Interact to Corral Inflammatory and Fibrotic Cells via STAT3-Dependent Mechanisms after Spinal Cord Injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-Associated Astrocytes in Alzheimer’s Disease and Aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-Specific and Region-Specific Transcriptomics in the Multiple Sclerosis Model: Focus on Astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 Ion Channel Deficits Contribute to Neuronal Dysfunction in Huntington’s Disease Model Mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 Astrocyte Conversion by Microglia Is Neuroprotective in Models of Parkinson’s Disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of Neuropathology-Associated Reactive Astrocytes: A Systematic Review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte Contribution to Dysfunction, Risk and Progression in Neurodegenerative Disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef]
- Zou, S.; Lan, Y.-L.; Wang, H.; Zhang, B.; Sun, Y.-G. The Potential Roles of Aquaporin 4 in Amyotrophic Lateral Sclerosis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2019, 40, 1541–1549. [Google Scholar] [CrossRef]
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Ugawa, Y.; Nishizawa, M.; Takahashi, H.; Kakita, A. Characteristics of Aquaporin Expression Surrounding Senile Plaques and Cerebral Amyloid Angiopathy in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 750–759. [Google Scholar] [CrossRef]
- Takahashi, K.; Kong, Q.; Lin, Y.; Stouffer, N.; Schulte, D.A.; Lai, L.; Liu, Q.; Chang, L.-C.; Dominguez, S.; Xing, X.; et al. Restored Glial Glutamate Transporter EAAT2 Function as a Potential Therapeutic Approach for Alzheimer’s Disease. J. Exp. Med. 2015, 212, 319–332. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, X.; Jiao, Z.; Liu, Y.; Zhang, X.; Qu, S. Generation of a Novel Mouse Model of Parkinson’s Disease via Targeted Knockdown of Glutamate Transporter GLT-1 in the Substantia Nigra. ACS Chem. Neurosci. 2020, 11, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Venugopal, S.; Majid, S.; Ahn, I.S.; Diamante, G.; Hong, J.; Yang, X.; Chandler, S.H. Single-Cell RNA-Seq Analysis of the Brainstem of Mutant SOD1 Mice Reveals Perturbed Cell Types and Pathways of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2020, 141, 104877. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of Glia from Alzheimer’s Mice Reveals Inflammation and Dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sun, Y.; Ling, S.-C.; Ferraiuolo, L.; McAlonis-Downes, M.; Zou, Y.; Drenner, K.; Wang, Y.; Ditsworth, D.; Tokunaga, S.; et al. Translational Profiling Identifies a Cascade of Damage Initiated in Motor Neurons and Spreading to Glia in Mutant SOD1-Mediated ALS. Proc. Natl. Acad. Sci. USA 2015, 112, E6993–E7002. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Toledo, J.B.; Van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.-Y.; Trojanowski, J.Q. Microglial Activation Correlates with Disease Progression and Upper Motor Neuron Clinical Symptoms in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef] [PubMed]
- Dols-Icardo, O.; Montal, V.; Sirisi, S.; López-Pernas, G.; Cervera-Carles, L.; Querol-Vilaseca, M.; Muñoz, L.; Belbin, O.; Alcolea, D.; Molina-Porcel, L.; et al. Motor Cortex Transcriptome Reveals Microglial Key Events in Amyotrophic Lateral Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e829. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Tondo, G.; Iaccarino, L.; Cerami, C.; Vanoli, G.E.; Presotto, L.; Masiello, V.; Coliva, A.; Salvi, F.; Bartolomei, I.; Mosca, L.; et al. 11 C-PK11195 PET-Based Molecular Study of Microglia Activation in SOD1 Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1513–1523. [Google Scholar] [CrossRef]
- Fujita, K.; Kato, T.; Yamauchi, M.; Ando, M.; Honda, M.; Nagata, Y. Increases in Fragmented Glial Fibrillary Acidic Protein Levels in the Spinal Cords of Patients with Amyotrophic Lateral Sclerosis. Neurochem. Res. 1998, 23, 169–174. [Google Scholar] [CrossRef]
- Schiffer, D.; Cordera, S.; Cavalla, P.; Migheli, A. Reactive Astrogliosis of the Spinal Cord in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1996, 139, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.D.; Stephenson, D.T.; Wright, S. Reactive Astrogliosis Is Widespread in the Subcortical White Matter of Amyotrophic Lateral Sclerosis Brain. J. Neuropathol. Exp. Neurol. 1991, 50, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.; Venkatesh, S.; Hasan, R.; Herb, J.T.; de Paiva Lopes, K.; Küçükali, F.; Byrska-Bishop, M.; Evani, U.S.; Narzisi, G.; Fagegaltier, D.; et al. Integrative Transcriptomic Analysis of the Amyotrophic Lateral Sclerosis Spinal Cord Implicates Glial Activation and Suggests New Risk Genes. Nat. Neurosci. 2023, 26, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, J.; Jossan, S.S.; Bergström, M.; Oreland, L.; Walum, E.; Aquilonius, S.M. Monoamine Oxidase-B in Astrocytes. Glia 1993, 8, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Furumoto, S.; Kudo, Y.; Yanai, K.; Villemagne, V.L.; Okamura, N. Imaging of Reactive Astrogliosis by Positron Emission Tomography. Front. Neurosci. 2022, 16, 807435. [Google Scholar] [CrossRef]
- Johansson, A.; Engler, H.; Blomquist, G.; Scott, B.; Wall, A.; Aquilonius, S.-M.; Långström, B.; Askmark, H. Evidence for Astrocytosis in ALS Demonstrated by [11C](L)-Deprenyl-D2 PET. J. Neurol. Sci. 2007, 255, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An Adverse Property of a Familial ALS-Linked SOD1 Mutation Causes Motor Neuron Disease Characterized by Vacuolar Degeneration of Mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic Mice Expressing an Altered Murine Superoxide Dismutase Gene Provide an Animal Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 689–693. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor Neuron Degeneration in Mice That Express a Human Cu, Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Pramatarova, A.; Laganière, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-Specific Expression of Mutant Superoxide Dismutase 1 in Transgenic Mice Does Not Lead to Motor Impairment. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 3369–3374. [Google Scholar] [CrossRef]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 Mutants in Postnatal Motoneurons Does Not Cause Motoneuron Pathology or Motoneuron Disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [CrossRef]
- Wang, L.; Sharma, K.; Deng, H.-X.; Siddique, T.; Grisotti, G.; Liu, E.; Roos, R.P. Restricted Expression of Mutant SOD1 in Spinal Motor Neurons and Interneurons Induces Motor Neuron Pathology. Neurobiol. Dis. 2008, 29, 400–408. [Google Scholar] [CrossRef]
- Jaarsma, D.; Teuling, E.; Haasdijk, E.D.; De Zeeuw, C.I.; Hoogenraad, C.C. Neuron-Specific Expression of Mutant Superoxide Dismutase Is Sufficient to Induce Amyotrophic Lateral Sclerosis in Transgenic Mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2075–2088. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-Type Microglia Extend Survival in PU.1 Knockout Mice with Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhao, W.; Beers, D.R.; Yen, A.A.; Xie, W.; Henkel, J.S.; Appel, S.H. Mutant SOD1(G93A) Microglia Are More Neurotoxic Relative to Wild-Type Microglia. J. Neurochem. 2007, 102, 2008–2019. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-ΚB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef]
- Martínez-Muriana, A.; Mancuso, R.; Francos-Quijorna, I.; Olmos-Alonso, A.; Osta, R.; Perry, V.H.; Navarro, X.; Gomez-Nicola, D.; López-Vales, R. CSF1R Blockade Slows the Progression of Amyotrophic Lateral Sclerosis by Reducing Microgliosis and Invasion of Macrophages into Peripheral Nerves. Sci. Rep. 2016, 6, 25663. [Google Scholar] [CrossRef] [PubMed]
- Gowing, G.; Philips, T.; Van Wijmeersch, B.; Audet, J.-N.; Dewil, M.; Van Den Bosch, L.; Billiau, A.D.; Robberecht, W.; Julien, J.-P. Ablation of Proliferating Microglia Does Not Affect Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis Caused by Mutant Superoxide Dismutase. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 10234–10244. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a Neuroprotective to a Neurotoxic Microglial Phenotype in a Mouse Model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Gravel, M.; Béland, L.-C.; Soucy, G.; Abdelhamid, E.; Rahimian, R.; Gravel, C.; Kriz, J. IL-10 Controls Early Microglial Phenotypes and Disease Onset in ALS Caused by Misfolded Superoxide Dismutase 1. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 1031–1048. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as Determinants of Disease Progression in Inherited Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Wang, L.; Gutmann, D.H.; Roos, R.P. Astrocyte Loss of Mutant SOD1 Delays ALS Disease Onset and Progression in G85R Transgenic Mice. Hum. Mol. Genet. 2011, 20, 286–293. [Google Scholar] [CrossRef]
- Lepore, A.C.; Rauck, B.; Dejea, C.; Pardo, A.C.; Rao, M.S.; Rothstein, J.D.; Maragakis, N.J. Focal Transplantation-Based Astrocyte Replacement Is Neuroprotective in a Model of Motor Neuron Disease. Nat. Neurosci. 2008, 11, 1294–1301. [Google Scholar] [CrossRef]
- Rochat, C.; Bernard-Marissal, N.; Källstig, E.; Pradervand, S.; Perrin, F.E.; Aebischer, P.; Raoul, C.; Schneider, B.L. Astrocyte-Targeting RNA Interference against Mutated Superoxide Dismutase 1 Induces Motoneuron Plasticity and Protects Fast-Fatigable Motor Units in a Mouse Model of Amyotrophic Lateral Sclerosis. Glia 2022, 70, 842–857. [Google Scholar] [CrossRef]
- Papadeas, S.T.; Kraig, S.E.; O’Banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes Carrying the Superoxide Dismutase 1 (SOD1G93A) Mutation Induce Wild-Type Motor Neuron Degeneration in Vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808. [Google Scholar] [CrossRef]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted Expression of G86R Cu/Zn Superoxide Dismutase in Astrocytes Results in Astrocytosis but Does Not Cause Motoneuron Degeneration. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 660–665. [Google Scholar] [CrossRef]
- Rojas, F.; Cortes, N.; Abarzua, S.; Dyrda, A.; van Zundert, B. Astrocytes Expressing Mutant SOD1 and TDP43 Trigger Motoneuron Death That Is Mediated via Sodium Channels and Nitroxidative Stress. Front. Cell. Neurosci. 2014, 8, 24. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-Cell Autonomous Effect of Glia on Motor Neurons in an Embryonic Stem Cell-Based ALS Model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes Expressing ALS-Linked Mutated SOD1 Release Factors Selectively Toxic to Motor Neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Boulting, G.L.; Bobrowicz, S.; Eggan, K.C. Human Embryonic Stem Cell-Derived Motor Neurons Are Sensitive to the Toxic Effect of Glial Cells Carrying an ALS-Causing Mutation. Cell Stem Cell 2008, 3, 637–648. [Google Scholar] [CrossRef]
- Stotz, S.C.; Scott, L.O.; Drummond-Main, C.; Avchalumov, Y.; Girotto, F.; Davidsen, J.; Gómez-Gárcia, M.R.; Rho, J.M.; Pavlov, E.V.; Colicos, M.A. Inorganic Polyphosphate Regulates Neuronal Excitability through Modulation of Voltage-Gated Channels. Mol. Brain 2014, 7, 42. [Google Scholar] [CrossRef]
- Arredondo, C.; Cefaliello, C.; Dyrda, A.; Jury, N.; Martinez, P.; Díaz, I.; Amaro, A.; Tran, H.; Morales, D.; Pertusa, M.; et al. Excessive Release of Inorganic Polyphosphate by ALS/FTD Astrocytes Causes Non-Cell-Autonomous Toxicity to Motoneurons. Neuron 2022, 110, 1656–1670.e12. [Google Scholar] [CrossRef]
- Hassanian, S.M.; Avan, A.; Ardeshirylajimi, A. Inorganic Polyphosphate: A Key Modulator of Inflammation. J. Thromb. Haemost. JTH 2017, 15, 213–218. [Google Scholar] [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef]
- Rojas, F.; Gonzalez, D.; Cortes, N.; Ampuero, E.; Hernández, D.E.; Fritz, E.; Abarzua, S.; Martinez, A.; Elorza, A.A.; Alvarez, A.; et al. Reactive Oxygen Species Trigger Motoneuron Death in Non-Cell-Autonomous Models of ALS through Activation of c-Abl Signaling. Front. Cell. Neurosci. 2015, 9, 203. [Google Scholar] [CrossRef]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-Derived TGF-Β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L.; Martorana, F.; Guidotti, G.; Rossi, D. Dysregulation of Astrocytic HMGB1 Signaling in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 622. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.-S.; Dhull, D.K.; Nalini, A.; Vijayalakshmi, K.; Sathyaprabha, T.N.; Alladi, P.A.; Raju, T.R. Astroglia Acquires a Toxic Neuroinflammatory Role in Response to the Cerebrospinal Fluid from Amyotrophic Lateral Sclerosis Patients. J. Neuroinflamm. 2016, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Hoye, M.L.; Regan, M.R.; Jensen, L.A.; Lake, A.M.; Reddy, L.V.; Vidensky, S.; Richard, J.-P.; Maragakis, N.J.; Rothstein, J.D.; Dougherty, J.D.; et al. Motor Neuron-Derived MicroRNAs Cause Astrocyte Dysfunction in Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2018, 141, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Hoye, M.L.; Koval, E.D.; Wegener, A.J.; Hyman, T.S.; Yang, C.; O’Brien, D.R.; Miller, R.L.; Cole, T.; Schoch, K.M.; Shen, T.; et al. MicroRNA Profiling Reveals Marker of Motor Neuron Disease in ALS Models. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 5574–5586. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective Loss of Glial Glutamate Transporter GLT-1 in Amyotrophic Lateral Sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lai, L.; Butchbach, M.E.R.; Stockinger, M.P.; Shan, X.; Bishop, G.A.; Lin, C.G. Increased Expression of the Glial Glutamate Transporter EAAT2 Modulates Excitotoxicity and Delays the Onset but Not the Outcome of ALS in Mice. Hum. Mol. Genet. 2003, 12, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic Reactive Astrocytes Induce Cell Death via Saturated Lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of Reactive Astrocyte Activating Factors Slows Disease Progression in an ALS Mouse Model. Nat. Commun. 2020, 11, 3753. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T Cells Support Glial Neuroprotection, Slow Disease Progression, and Modify Glial Morphology in an Animal Model of Inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef]
- Garofalo, S.; Cocozza, G.; Porzia, A.; Inghilleri, M.; Raspa, M.; Scavizzi, F.; Aronica, E.; Bernardini, G.; Peng, L.; Ransohoff, R.M.; et al. Natural Killer Cells Modulate Motor Neuron-Immune Cell Cross Talk in Models of Amyotrophic Lateral Sclerosis. Nat. Commun. 2020, 11, 1773. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, C.-F.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. TDP-43-Immunoreactive Neuronal and Glial Inclusions in the Neostriatum in Amyotrophic Lateral Sclerosis with and without Dementia. Acta Neuropathol. 2008, 115, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1 Mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-F.; Eguchi, H.; Tagawa, A.; Onodera, O.; Iwasaki, T.; Tsujino, A.; Nishizawa, M.; Kakita, A.; Takahashi, H. TDP-43 Immunoreactivity in Neuronal Inclusions in Familial Amyotrophic Lateral Sclerosis with or without SOD1 Gene Mutation. Acta Neuropathol. 2007, 113, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-Mediated Recovery from ALS-Relevant Motor Neuron Degeneration in a Mouse Model of TDP-43 Proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- Peng, A.Y.T.; Agrawal, I.; Ho, W.Y.; Yen, Y.-C.; Pinter, A.J.; Liu, J.; Phua, Q.X.C.; Koh, K.B.; Chang, J.-C.; Sanford, E.; et al. Loss of TDP-43 in Astrocytes Leads to Motor Deficits by Triggering A1-like Reactive Phenotype and Triglial Dysfunction. Proc. Natl. Acad. Sci. USA 2020, 117, 29101–29112. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Mariani, A.; Watkins, L.R.; Link, C.D. TDP-43 Knockdown Causes Innate Immune Activation via Protein Kinase R in Astrocytes. Neurobiol. Dis. 2019, 132, 104514. [Google Scholar] [CrossRef]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.-G. Expression of ALS-Linked TDP-43 Mutant in Astrocytes Causes Non-Cell-Autonomous Motor Neuron Death in Rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Petrozziello, T.; Mills, A.N.; Farhan, S.M.K.; Mueller, K.A.; Granucci, E.J.; Glajch, K.E.; Chan, J.; Chew, S.; Berry, J.D.; Sadri-Vakili, G. Lipocalin-2 Is Increased in Amyotrophic Lateral Sclerosis. Muscle Nerve 2020, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Huang, C.; Tong, J.; Qiu, G.; Huang, B.; Wu, Q.; Li, F.; Xu, Z.; Bowser, R.; Xia, X.-G.; et al. Reactive Astrocytes Secrete Lcn2 to Promote Neuron Death. Proc. Natl. Acad. Sci. USA 2013, 110, 4069–4074. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.; Kang, H.-Y.; Lim, H.R.; Kwon, Y.; Jo, M.; Jeon, Y.-M.; Kim, S.R.; Kim, K.; Ha, C.M.; et al. The Overexpression of TDP-43 in Astrocytes Causes Neurodegeneration via a PTP1B-Mediated Inflammatory Response. J. Neuroinflamm. 2020, 17, 299. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in Neuroinflammation and ALS and Frontotemporal Dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to C9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 Is Required for Proper Macrophage and Microglial Function in Mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.-J.; Terpstra, M.L.; Zundel, C.A.C.; Vieira de Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 Ablation in Mice Does Not Cause Motor Neuron Degeneration or Motor Deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Lorenzini, I.; Mota, T.A.; Bell, S.; Mahan, T.E.; Ulrich, J.D.; Davtyan, H.; Rexach, J.E.; Muhammad, A.K.M.G.; Shelest, O.; et al. C9orf72 Deficiency Promotes Microglial-Mediated Synaptic Loss in Aging and Amyloid Accumulation. Neuron 2021, 109, 2275–2291.e8. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- Nguyen, L.; Montrasio, F.; Pattamatta, A.; Tusi, S.K.; Bardhi, O.; Meyer, K.D.; Hayes, L.; Nakamura, K.; Banez-Coronel, M.; Coyne, A.; et al. Antibody Therapy Targeting RAN Proteins Rescues C9 ALS/FTD Phenotypes in C9orf72 Mouse Model. Neuron 2020, 105, 645–662.e11. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.A.; Arthur, K.C.; Tienari, P.J.; Houlden, H.; Chiò, A.; Traynor, B.J. Age-Related Penetrance of the C9orf72 Repeat Expansion. Sci. Rep. 2017, 7, 2116. [Google Scholar] [CrossRef] [PubMed]
- Mordes, D.A.; Morrison, B.M.; Ament, X.H.; Cantrell, C.; Mok, J.; Eggan, P.; Xue, C.; Wang, J.-Y.; Eggan, K.; Rothstein, J.D. Absence of Survival and Motor Deficits in 500 Repeat C9ORF72 BAC Mice. Neuron 2020, 108, 775–783.e4. [Google Scholar] [CrossRef]
- Nguyen, L.; Laboissonniere, L.A.; Guo, S.; Pilotto, F.; Scheidegger, O.; Oestmann, A.; Hammond, J.W.; Li, H.; Hyysalo, A.; Peltola, R.; et al. Survival and Motor Phenotypes in FVB C9-500 ALS/FTD BAC Transgenic Mice Reproduced by Multiple Labs. Neuron 2020, 108, 784–796.e3. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.-F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 Poly(GA) Aggregates Sequester and Impair HR23 and Nucleocytoplasmic Transport Proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef]
- Schludi, M.H.; Becker, L.; Garrett, L.; Gendron, T.F.; Zhou, Q.; Schreiber, F.; Popper, B.; Dimou, L.; Strom, T.M.; Winkelmann, J.; et al. Spinal Poly-GA Inclusions in a C9orf72 Mouse Model Trigger Motor Deficits and Inflammation without Neuron Loss. Acta Neuropathol. 2017, 134, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) Impairs Protein Translation and Stress Granule Dynamics in C9orf72-Associated Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- Hao, Z.; Liu, L.; Tao, Z.; Wang, R.; Ren, H.; Sun, H.; Lin, Z.; Zhang, Z.; Mu, C.; Zhou, J.; et al. Motor Dysfunction and Neurodegeneration in a C9orf72 Mouse Line Expressing Poly-PR. Nat. Commun. 2019, 10, 2906. [Google Scholar] [CrossRef] [PubMed]
- Verdone, B.M.; Cicardi, M.E.; Wen, X.; Sriramoji, S.; Russell, K.; Markandaiah, S.S.; Jensen, B.K.; Krishnamurthy, K.; Haeusler, A.R.; Pasinelli, P.; et al. A Mouse Model with Widespread Expression of the C9orf72-Linked Glycine-Arginine Dipeptide Displays Non-Lethal ALS/FTD-like Phenotypes. Sci. Rep. 2022, 12, 5644. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Mareljic, N.; Michaelsen, M.; Parhizkar, S.; Heindl, S.; Nuscher, B.; Farny, D.; Czuppa, M.; Schludi, C.; Graf, A.; et al. Active Poly-GA Vaccination Prevents Microglia Activation and Motor Deficits in a C9orf72 Mouse Model. EMBO Mol. Med. 2020, 12, e10919. [Google Scholar] [CrossRef] [PubMed]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.-A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2019, 179, 1609–1622.e16. [Google Scholar] [CrossRef]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.-W.; Stiles, L.; et al. Conservation and Divergence of Vulnerability and Responses to Stressors between Human and Mouse Astrocytes. Nat. Commun. 2021, 12, 3958. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient Derivation of Microglia-like Cells from Human Pluripotent Stem Cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Lorenzini, I.; Alsop, E.; Levy, J.; Gittings, L.M.; Lall, D.; Rabichow, B.E.; Moore, S.; Pevey, R.; Bustos, L.M.; Burciu, C.; et al. Moderate Intrinsic Phenotypic Alterations in C9orf72 ALS/FTD IPSC-Microglia despite the Presence of C9orf72 Pathological Features. Front. Cell. Neurosci. 2023, 17, 1179796. [Google Scholar] [CrossRef]
- Vahsen, B.F.; Nalluru, S.; Morgan, G.R.; Farrimond, L.; Carroll, E.; Xu, Y.; Cramb, K.M.L.; Amein, B.; Scaber, J.; Katsikoudi, A.; et al. C9orf72-ALS Human IPSC Microglia Are pro-Inflammatory and Toxic to Co-Cultured Motor Neurons via MMP9. Nat. Commun. 2023, 14, 5898. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Mehta, A.R.; Nirujogi, R.S.; Cooper, J.; James, O.G.; Nanda, J.; Longden, J.; Burr, K.; McDade, K.; Salzinger, A.; et al. Cell-Autonomous Immune Dysfunction Driven by Disrupted Autophagy in C9orf72-ALS IPSC-Derived Microglia Contributes to Neurodegeneration. Sci. Adv. 2023, 9, eabq0651. [Google Scholar] [CrossRef] [PubMed]
- Quek, H.; Cuní-López, C.; Stewart, R.; Colletti, T.; Notaro, A.; Nguyen, T.H.; Sun, Y.; Guo, C.C.; Lupton, M.K.; Roberts, T.L.; et al. ALS Monocyte-Derived Microglia-like Cells Reveal Cytoplasmic TDP-43 Accumulation, DNA Damage, and Cell-Specific Impairment of Phagocytosis Associated with Disease Progression. J. Neuroinflamm. 2022, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.-Y.; Kwon, M.-S.; Oh, K.-W.; Nahm, M.; Park, J.; Kim, Y.-E.; Ki, C.-S.; Jin, H.K.; Bae, J.-S.; Kim, S.H. Role of NCKAP1 in the Defective Phagocytic Function of Microglia-Like Cells Derived from Rapidly Progressing Sporadic ALS. Mol. Neurobiol. 2023, 60, 4761–4777. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.F.; Amponsah, A.E.; Guo, R.; Liu, X.; Zhang, J.; Du, X.; Zhou, Z.; He, J.; Ma, J.; Cui, H. Autophagy-Mediated Inflammatory Cytokine Secretion in Sporadic ALS Patient IPSC-Derived Astrocytes. Oxid. Med. Cell. Longev. 2022, 2022, 6483582. [Google Scholar] [CrossRef]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct Conversion of Patient Fibroblasts Demonstrates Non-Cell Autonomous Toxicity of Astrocytes to Motor Neurons in Familial and Sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs Secreted through Astrocyte-Derived Extracellular Vesicles Cause Neuronal Network Degeneration in C9orf72 ALS. EBioMedicine 2019, 40, 626–635. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte Adenosine Deaminase Loss Increases Motor Neuron Toxicity in Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2019, 142, 586–605. [Google Scholar] [CrossRef]
- Zhao, C.; Devlin, A.-C.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 Human IPSC-Derived Astrocytes Cause Non-Cell Autonomous Motor Neuron Pathophysiology. Glia 2020, 68, 1046–1064. [Google Scholar] [CrossRef]
- Rojas, F.; Aguilar, R.; Almeida, S.; Fritz, E.; Corvalán, D.; Ampuero, E.; Abarzúa, S.; Garcés, P.; Amaro, A.; Diaz, I.; et al. Mature IPSC-Derived Astrocytes of an ALS/FTD Patient Carrying the TDP43A90V Mutation Display a Mild Reactive State and Release PolyP Toxic to Motoneurons. Front. Cell Dev. Biol. 2023, 11, 1226604. [Google Scholar] [CrossRef]
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD Brain Organoid Slice Cultures Display Distinct Early Astrocyte and Targetable Neuronal Pathology. Nat. Neurosci. 2021, 24, 1542–1554. [Google Scholar] [CrossRef]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic Lateral Sclerosis Patient IPSC-Derived Astrocytes Impair Autophagy via Non-Cell Autonomous Mechanisms. Mol. Brain 2017, 10, 22. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human IPSC-Derived Astrocytes from ALS Patients with Mutated C9ORF72 Show Increased Oxidative Stress and Neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 Expansion within Astrocytes Reduces Metabolic Flexibility in Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2019, 142, 3771–3790. [Google Scholar] [CrossRef]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.-R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct Responses of Neurons and Astrocytes to TDP-43 Proteinopathy in Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-Term Survival of Participants in the CENTAUR Trial of Sodium Phenylbutyrate-Taurursodiol in Amyotrophic Lateral Sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; et al. Effect of Sodium Phenylbutyrate/Taurursodiol on Tracheostomy/Ventilation-Free Survival and Hospitalisation in Amyotrophic Lateral Sclerosis: Long-Term Results from the CENTAUR Trial. J. Neurol. Neurosurg. Psychiatry 2022, 93, 871–875. [Google Scholar] [CrossRef]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic Lateral Sclerosis: A Neurodegenerative Disorder Poised for Successful Therapeutic Translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Deng, M. New Developments and Opportunities in Drugs Being Trialed for Amyotrophic Lateral Sclerosis from 2020 to 2022. Front. Pharmacol. 2022, 13, 1054006. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-Paralysis Tyrosine Kinase Inhibition with Masitinib Abrogates Neuroinflammation and Slows Disease Progression in Inherited Amyotrophic Lateral Sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for Mast Cells Contributing to Neuromuscular Pathology in an Inherited Model of ALS. JCI Insight 2017, 2, e95934. [Google Scholar] [CrossRef]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast Cells and Neutrophils Mediate Peripheral Motor Pathway Degeneration in ALS. JCI Insight 2018, 3, e123249. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an Add-on Therapy to Riluzole in Patients with Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 Mediates Axonal Degeneration by Promoting Inflammation and Necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Oskarsson, B.; Maragakis, N.; Bedlack, R.S.; Goyal, N.; Meyer, J.A.; Genge, A.; Bodkin, C.; Maiser, S.; Staff, N.; Zinman, L.; et al. MN-166 (Ibudilast) in Amyotrophic Lateral Sclerosis in a Phase IIb/III Study: COMBAT-ALS Study Design. Neurodegener. Dis. Manag. 2021, 11, 431–443. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Ying, Z.; Gao, Q. Ibudilast Enhances the Clearance of SOD1 and TDP-43 Aggregates through TFEB-Mediated Autophagy and Lysosomal Biogenesis: The New Molecular Mechanism of Ibudilast and Its Implication for Neuroprotective Therapy. Biochem. Biophys. Res. Commun. 2020, 526, 231–238. [Google Scholar] [CrossRef]
- Babu, S.; Hightower, B.G.; Chan, J.; Zürcher, N.R.; Kivisäkk, P.; Tseng, C.-E.J.; Sanders, D.L.; Robichaud, A.; Banno, H.; Evora, A.; et al. Ibudilast (MN-166) in Amyotrophic Lateral Sclerosis- an Open Label, Safety and Pharmacodynamic Trial. NeuroImage Clin. 2021, 30, 102672. [Google Scholar] [CrossRef]
- Parker, S.E.; Hanton, A.M.; Stefanou, S.N.; Noakes, P.G.; Woodruff, T.M.; Lee, J.D. Revisiting the Role of the Innate Immune Complement System in ALS. Neurobiol. Dis. 2019, 127, 223–232. [Google Scholar] [CrossRef]
- Baloh, R.H.; Johnson, J.P.; Avalos, P.; Allred, P.; Svendsen, S.; Gowing, G.; Roxas, K.; Wu, A.; Donahue, B.; Osborne, S.; et al. Transplantation of Human Neural Progenitor Cells Secreting GDNF into the Spinal Cord of Patients with ALS: A Phase 1/2a Trial. Nat. Med. 2022, 28, 1813–1822. [Google Scholar] [CrossRef]
- Henderson, C.E.; Phillips, H.S.; Pollock, R.A.; Davies, A.M.; Lemeulle, C.; Armanini, M.; Simmons, L.; Moffet, B.; Vandlen, R.A.; Simpson LC corrected to Simmons, L.; et al. GDNF: A Potent Survival Factor for Motoneurons Present in Peripheral Nerve and Muscle. Science 1994, 266, 1062–1064. [Google Scholar] [CrossRef]
- Alisky, J.M.; Davidson, B.L. Gene Therapy for Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. Hum. Gene Ther. 2000, 11, 2315–2329. [Google Scholar] [CrossRef]
- Klein, S.M.; Behrstock, S.; McHugh, J.; Hoffmann, K.; Wallace, K.; Suzuki, M.; Aebischer, P.; Svendsen, C.N. GDNF Delivery Using Human Neural Progenitor Cells in a Rat Model of ALS. Hum. Gene Ther. 2005, 16, 509–521. [Google Scholar] [CrossRef]
- Suzuki, M.; McHugh, J.; Tork, C.; Shelley, B.; Klein, S.M.; Aebischer, P.; Svendsen, C.N. GDNF Secreting Human Neural Progenitor Cells Protect Dying Motor Neurons, but Not Their Projection to Muscle, in a Rat Model of Familial ALS. PLoS ONE 2007, 2, e689. [Google Scholar] [CrossRef]
- Das, M.M.; Avalos, P.; Suezaki, P.; Godoy, M.; Garcia, L.; Chang, C.D.; Vit, J.-P.; Shelley, B.; Gowing, G.; Svendsen, C.N. Human Neural Progenitors Differentiate into Astrocytes and Protect Motor Neurons in Aging Rats. Exp. Neurol. 2016, 280, 41–49. [Google Scholar] [CrossRef]
- Gowing, G.; Shelley, B.; Staggenborg, K.; Hurley, A.; Avalos, P.; Victoroff, J.; Latter, J.; Garcia, L.; Svendsen, C.N. Glial Cell Line-Derived Neurotrophic Factor-Secreting Human Neural Progenitors Show Long-Term Survival, Maturation into Astrocytes, and No Tumor Formation Following Transplantation into the Spinal Cord of Immunocompromised Rats. Neuroreport 2014, 25, 367–372. [Google Scholar] [CrossRef]
- Izrael, M.; Slutsky, S.G.; Admoni, T.; Cohen, L.; Granit, A.; Hasson, A.; Itskovitz-Eldor, J.; Krush Paker, L.; Kuperstein, G.; Lavon, N.; et al. Safety and Efficacy of Human Embryonic Stem Cell-Derived Astrocytes Following Intrathecal Transplantation in SOD1G93A and NSG Animal Models. Stem Cell Res. Ther. 2018, 9, 152. [Google Scholar] [CrossRef]
- Gotkine, M.; Caraco, Y.; Lerner, Y.; Blotnick, S.; Wanounou, M.; Slutsky, S.G.; Chebath, J.; Kuperstein, G.; Estrin, E.; Ben-Hur, T.; et al. Safety and Efficacy of First-in-Man Intrathecal Injection of Human Astrocytes (AstroRx®) in ALS Patients: Phase I/IIa Clinical Trial Results. J. Transl. Med. 2023, 21, 122. [Google Scholar] [CrossRef]
- Lyczek, A.; Arnold, A.; Zhang, J.; Campanelli, J.T.; Janowski, M.; Bulte, J.W.M.; Walczak, P. Transplanted Human Glial-Restricted Progenitors Can Rescue the Survival of Dysmyelinated Mice Independent of the Production of Mature, Compact Myelin. Exp. Neurol. 2017, 291, 74–86. [Google Scholar] [CrossRef]
- Crespo-Castrillo, A.; Arevalo, M.-A. Microglial and Astrocytic Function in Physiological and Pathological Conditions: Estrogenic Modulation. Int. J. Mol. Sci. 2020, 21, 3219. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-F.; Wei, D.-N.; Tang, Y. Gut Microbiota Regulate Astrocytic Functions in the Brain: Possible Therapeutic Consequences. Curr. Neuropharmacol. 2021, 19, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Prinz, M. Regulation of Microglial Physiology by the Microbiota. Gut Microbes 2022, 14, 2125739. [Google Scholar] [CrossRef] [PubMed]
- Marcos, J.L.; Olivares-Barraza, R.; Ceballo, K.; Wastavino, M.; Ortiz, V.; Riquelme, J.; Martínez-Pinto, J.; Muñoz, P.; Cruz, G.; Sotomayor-Zárate, R. Obesogenic Diet-Induced Neuroinflammation: A Pathological Link between Hedonic and Homeostatic Control of Food Intake. Int. J. Mol. Sci. 2023, 24, 1468. [Google Scholar] [CrossRef]
- Obara-Michlewska, M. The Contribution of Astrocytes to Obesity-Associated Metabolic Disturbances. J. Biomed. Res. 2022, 36, 299–311. [Google Scholar] [CrossRef]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate Immune Memory in the Brain Shapes Neurological Disease Hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, J.; Youssef, M.M.M.; Santos, J.R.; Lee, J.; Park, J. Microglia and Astrocytes in Amyotrophic Lateral Sclerosis: Disease-Associated States, Pathological Roles, and Therapeutic Potential. Biology 2023, 12, 1307. https://doi.org/10.3390/biology12101307
You J, Youssef MMM, Santos JR, Lee J, Park J. Microglia and Astrocytes in Amyotrophic Lateral Sclerosis: Disease-Associated States, Pathological Roles, and Therapeutic Potential. Biology. 2023; 12(10):1307. https://doi.org/10.3390/biology12101307
Chicago/Turabian StyleYou, Justin, Mohieldin M. M. Youssef, Jhune Rizsan Santos, Jooyun Lee, and Jeehye Park. 2023. "Microglia and Astrocytes in Amyotrophic Lateral Sclerosis: Disease-Associated States, Pathological Roles, and Therapeutic Potential" Biology 12, no. 10: 1307. https://doi.org/10.3390/biology12101307
APA StyleYou, J., Youssef, M. M. M., Santos, J. R., Lee, J., & Park, J. (2023). Microglia and Astrocytes in Amyotrophic Lateral Sclerosis: Disease-Associated States, Pathological Roles, and Therapeutic Potential. Biology, 12(10), 1307. https://doi.org/10.3390/biology12101307