1. Introduction
As the demand for medical care increases, the cost of drug development is growing and unacceptable [
1]. The main reason for the relatively low productivity in the pharmaceutical industry is attributed to the high cost of searching for new drug targets. However, finding appropriate drug targets from the numerous and disorderly informatics is one of the important purposes of bioinformatics. For a long period, many studies on therapeutic targets have focused on protein and have spent much time and effort exploring the drug response of proteins. However, about 80% of approved drugs target protein and 99% of them target only specific proteins [
2]. This means that there are still vast proteins that are “undruggable”. Therefore, some researchers have shifted their focus in target selection to other biological entities such as microRNA (miRNA).
MicroRNA is a kind of endogenous non-coding RNA with a length of about 20 nucleotides, existing in humans, plants, animals and viruses [
3]. To date, about 2600 human mature miRNAs have been discovered [
4]. A considerable amount of literature has been published on miRNAs regarding their biogenesis, mechanic of action and function [
5,
6,
7]. Research in this area has shown that the abnormal expression of miRNAs is involved in plenty of diseases including cancer, neurologic disorders, autoimmune diseases and cardiovascular diseases [
8,
9,
10,
11,
12]. Furthermore, from post-transcriptional regulation, miRNAs can affect the gene to produce specific proteins, including the aforementioned “undruggable” proteins. Thus, miRNAs are considered to be potential high-value therapeutic targets and identifying the underlying drug–miRNA associations has major implications for the pharmaceutical industry [
13,
14].
Many researchers believe that miRNA pharmacogenomics would promote the development of personalized medicine [
15,
16]. However, there are two main challenges for miRNA-target therapeutics: the effective means of delivering the therapeutic agents to the target tissues and the safety evaluation of the potential drug response [
17]. In the first challenge, the problem of poor cell permeability and pharmacokinetics can be solved by Lipinski’s Rule of Five [
18]. In the other challenge, it is inevitable to study the situation of the association between drugs and miRNAs. For most drugs, it is relatively difficult to completely identify their association with different miRNA profiling through wet experiments because it is an intricate problem concerning a series of factors, and it is also labor-intensive and time-consuming work. [
19,
20]. Even though much effort has been invested in identifying DMA by wet experiment, the existing knowledge about drugs and miRNAs is not sufficient for guiding miRNA-targeted drug research. For improving the research and development of miRNA-target therapeutics, we need to accelerate the identification of DMA for future research. Compared with wet experiments, the computational method is the better choice for completing this mission, since it is lower in cost and higher in efficiency [
21]. In particular, machine learning has made great contributions in the field of bioinformatics [
22,
23,
24,
25]. In molecular biology research, novel datasets and innovated concepts are being generated [
26,
27,
28,
29]. Thus, it is important to adopt techniques that can handle these data efficiently. Machine learning can process the vast amount of data generated by new high-throughput devices to extract undiscovered relationships that exist and are imperceptible to experts [
30,
31,
32,
33].
After years of efforts, several computational methods for predicting DMA have emerged. One category of these methods was based on the self-similarity network and the association network. For example, Lv et al. developed a model based on the drug–miRNA network to identify DMA. They constructed the drug–miRNA integrated network and applied a random walk with restart (RWR) algorithm to predict the underlying miRNA targets of drugs. This model can predict related miRNAs for drugs in the absence of known drug–miRNA associations, but it is sophisticated and contains too many adjusted parameters [
34]. Furthermore, Qu et al. presented an in silico method for DMA prediction called HSDMA, which was also based on the drug–miRNA similarity network. [
35]. They introduced the path-based relevance measurement method of HeteSim. In the HeteSim method, considering different search paths between the miRNAs and drugs is the most predominant issue, because the path in the heterogeneous network has semantics [
36]. It can predict potential DMA by calculating the association score of each drug–miRNA pair based on the given search path, but the function for integrating different patterns of the search path is relatively simple. Moreover, Guan et al. proposed a prediction model called GIDMA. Inspired by the concept of graphlet interactions, they defined 28 types of graphlet interaction isomers that contained 1 to 4 vertexes and various connection patterns for describing the different relationships between 2 nodes [
37]. Thereafter, the association score between the drugs and miRNAs was calculated based on the number of each isomer on the self-similarity network [
38]. Furthermore, Wang et al. designed an DMA prediction model called RFDMA. This model combined the integrated similarity of miRNA and the drugs, and predicted DMA using the random forest algorithm [
39]. Qu et al. presented a new method called TLHNDMA based on a triple-layer heterogeneous network. This network not only used data on drug self-similarity and miRNA self-similarity but also considered disease similarity. An iterative updating algorithm was also developed to propagate information in the network and complete the prediction task [
40]. Additionally, Zhan et al. proposed a model called SNMFDMA, which did not directly use the similarity matrixes of drugs and miRNAs. They first used symmetric non-negative matrix factorization to process the similarity matrixes to generate new similarity matrixes. The Kronecker product of the new similarities matrixes was then regarded as the similarity of the drugs and miRNAs. Finally, regularized least squares were implemented to predict the potential associations between drugs and miRNAs [
41].
Another category of prediction methods leverages other features to represent the drugs and miRNAs instead of self-similarity. An example of this is the study carried out by Huang et al. in which they constructed an end-to-end model named GCMDR to discover associations between miRNA and drug resistance. These authors combined the side information such as the miRNA expression profile, drug substructure fingerprints, gene ontology and disease ontology as attribute features of the miRNAs and drugs. This model used GCN to learn low-dimensional embedding vectors for each biological entity and predicted the association between the drugs and miRNAs [
42]. Yu et al. built a web server for predicting the effects of drugs on miRNAs. They utilize k-mer, sequence information and the MACCS fingerprints to represent the miRNAs and drugs. The regulation of the miRNA expression of the drugs was then predicted using random forests [
43].
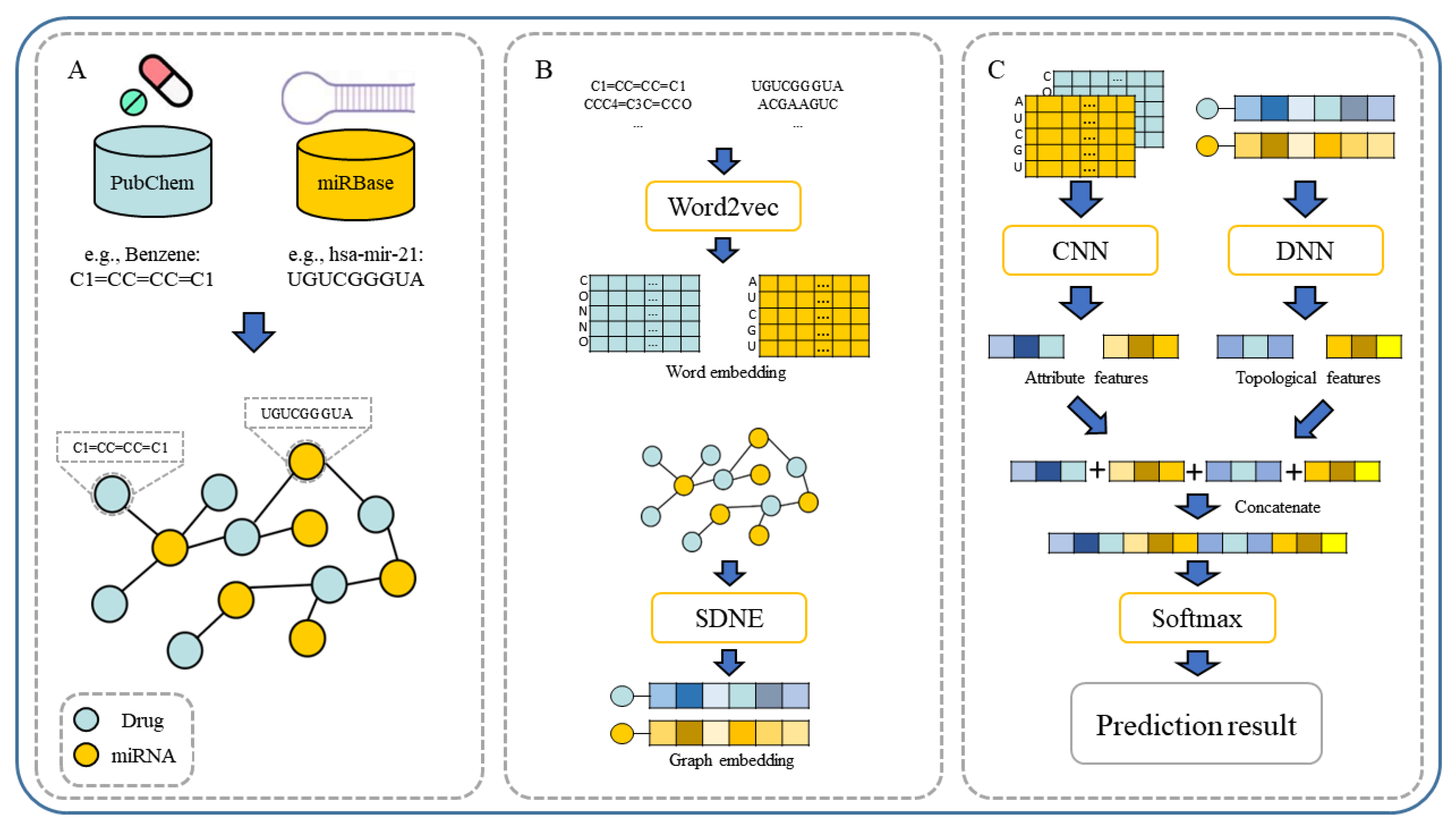
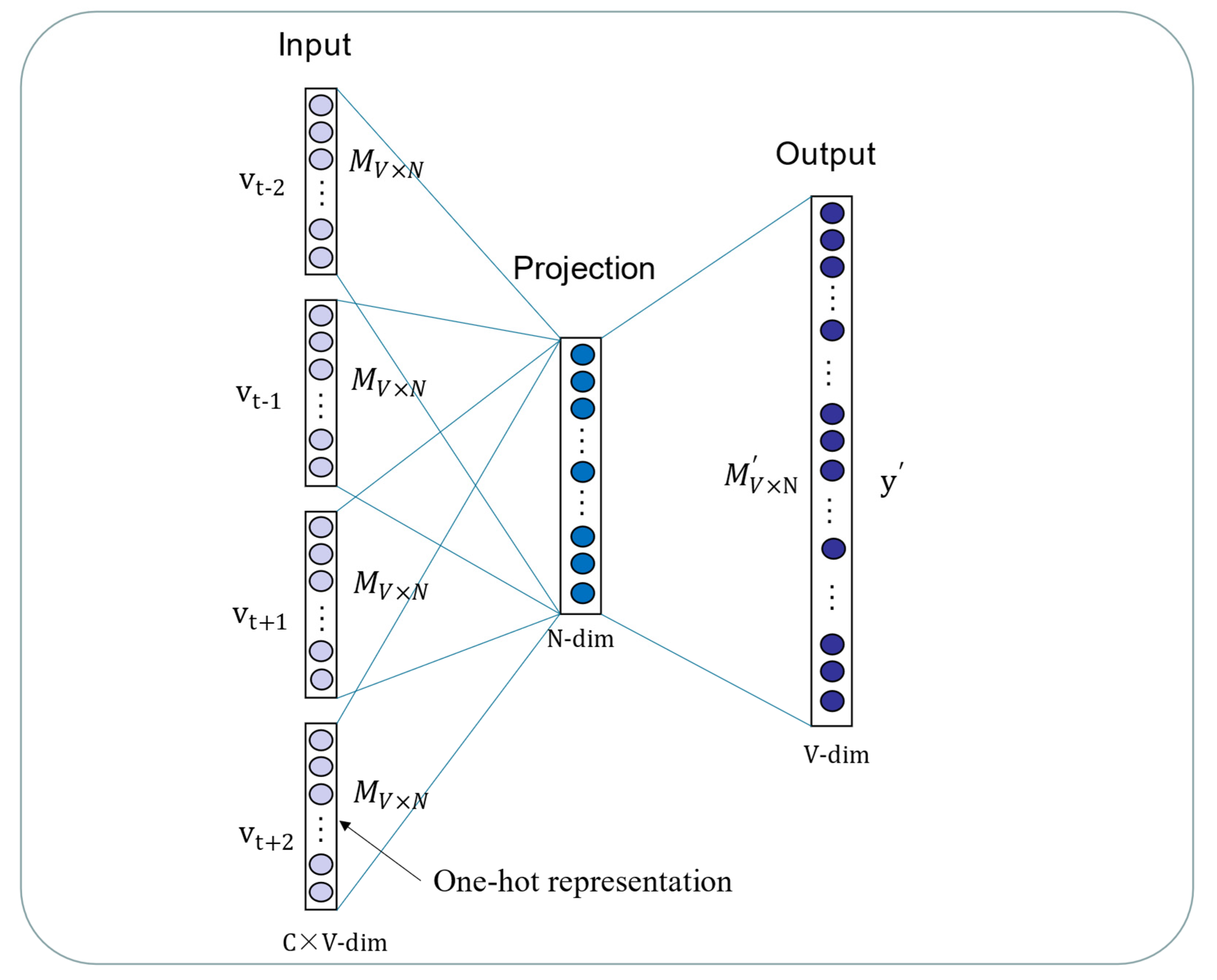
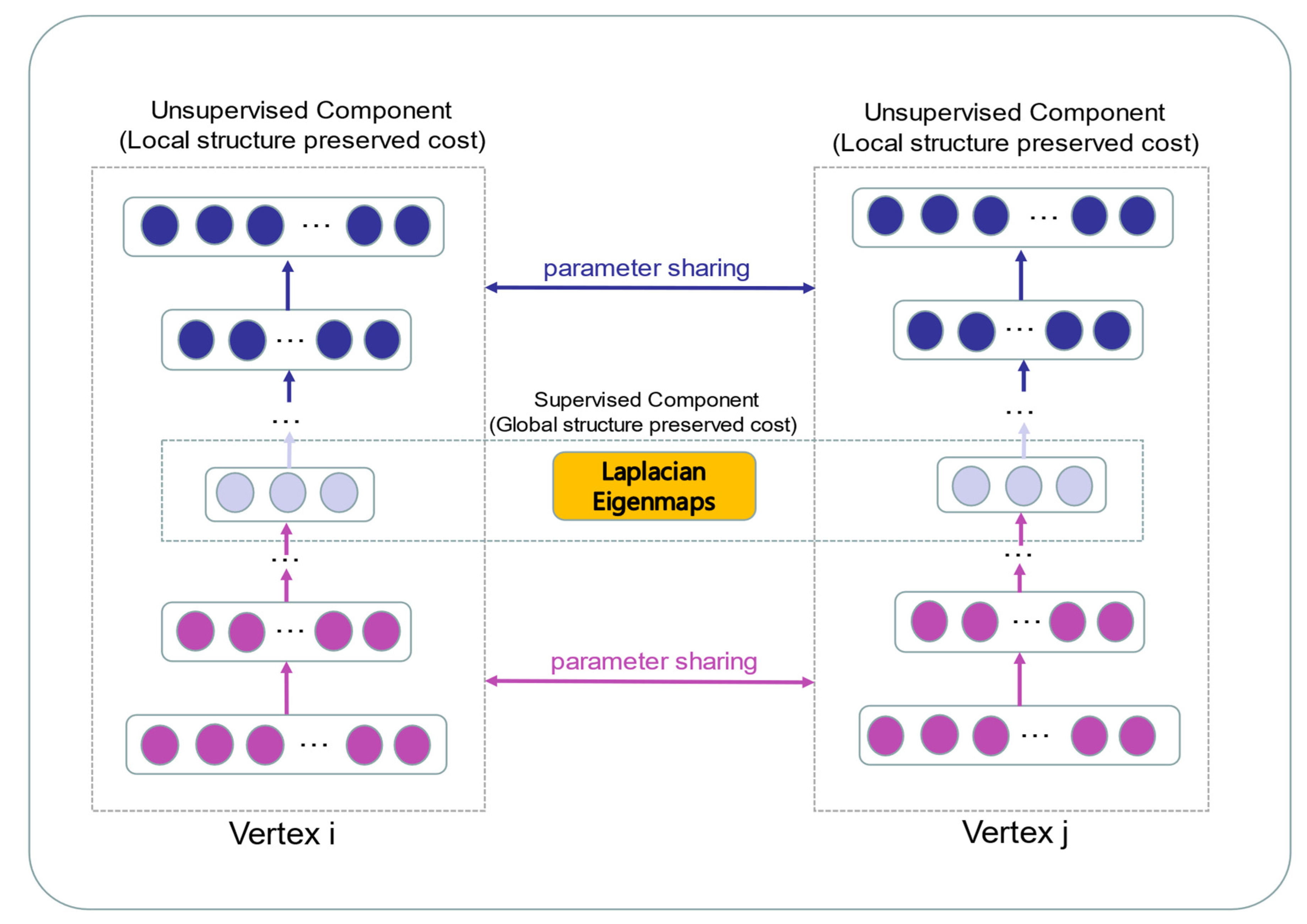
In our paper, we propose a novel multiple features integration method based on the integration of multiple features, named MFIDMA. First, a bipartite network was established to represent the relationship between drugs and miRNAs. Second, the structural deep network embedding (SDNE) algorithm was implemented to extract topology information and generate the embedding vectors of each node in the network. Third, the miRNAs were directly represented using sequences and the drugs were indicated by simplified molecular input line entry specification (SMILES). The Word2vec algorithm was then adopted to extract attribute features. Finally, two kinds of features were separately entered into the convolutional neural network (CNN) and the deep neural network (DNN) for deep learning feature extraction and classification.
Figure 1 provides the flowchart of the MFIDMA model.
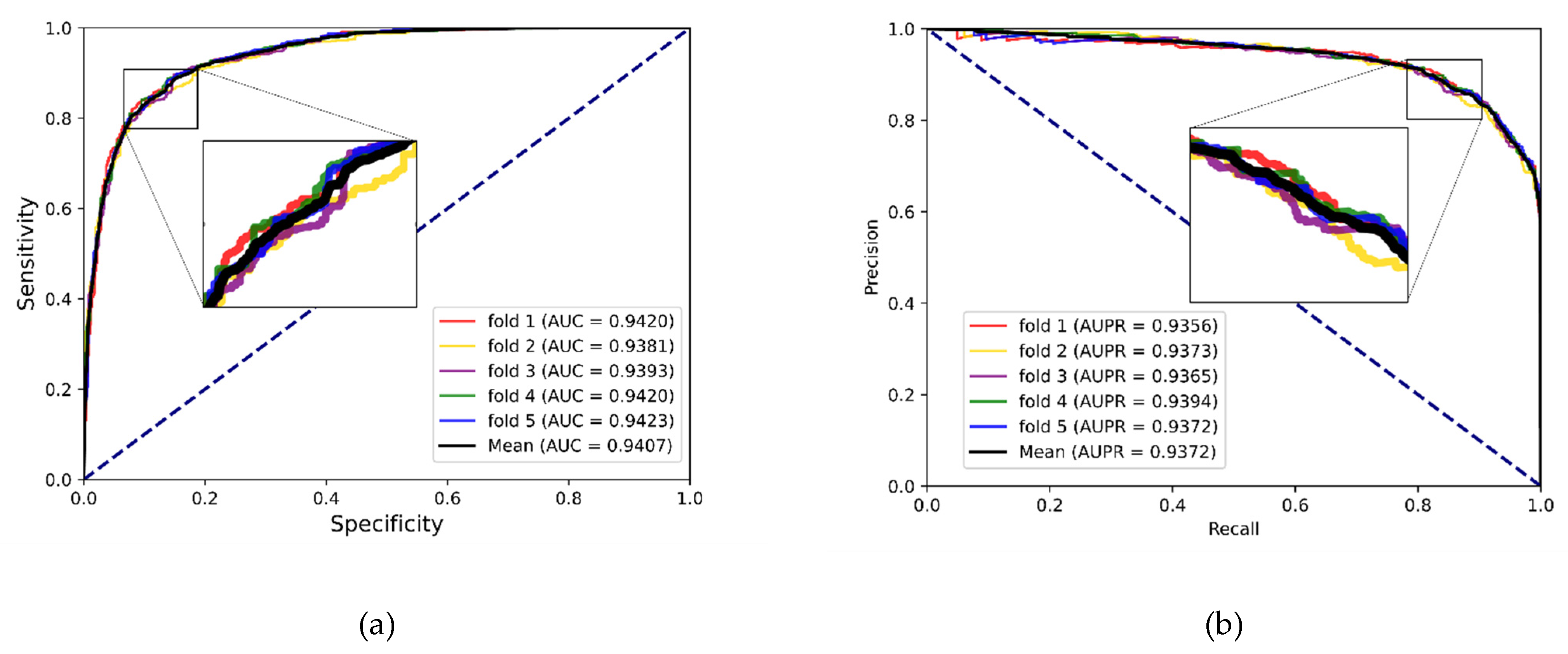
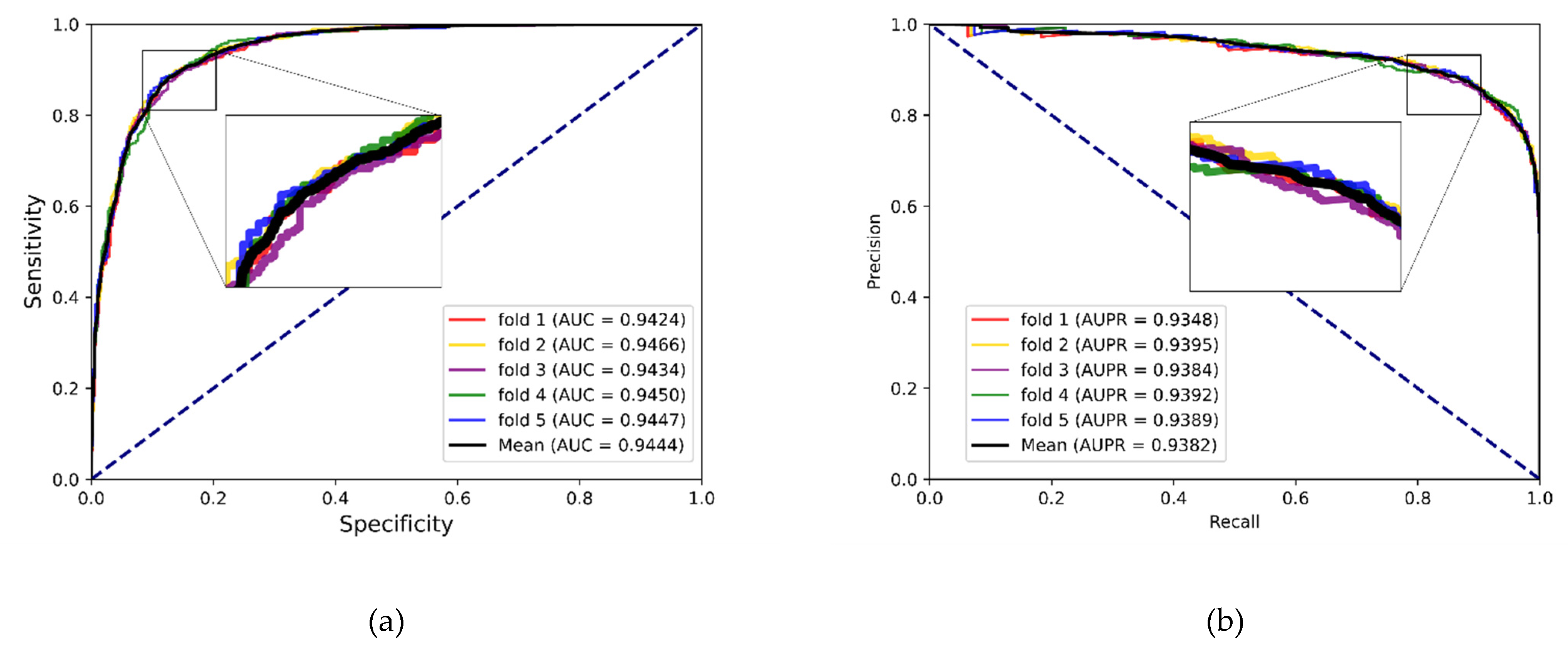
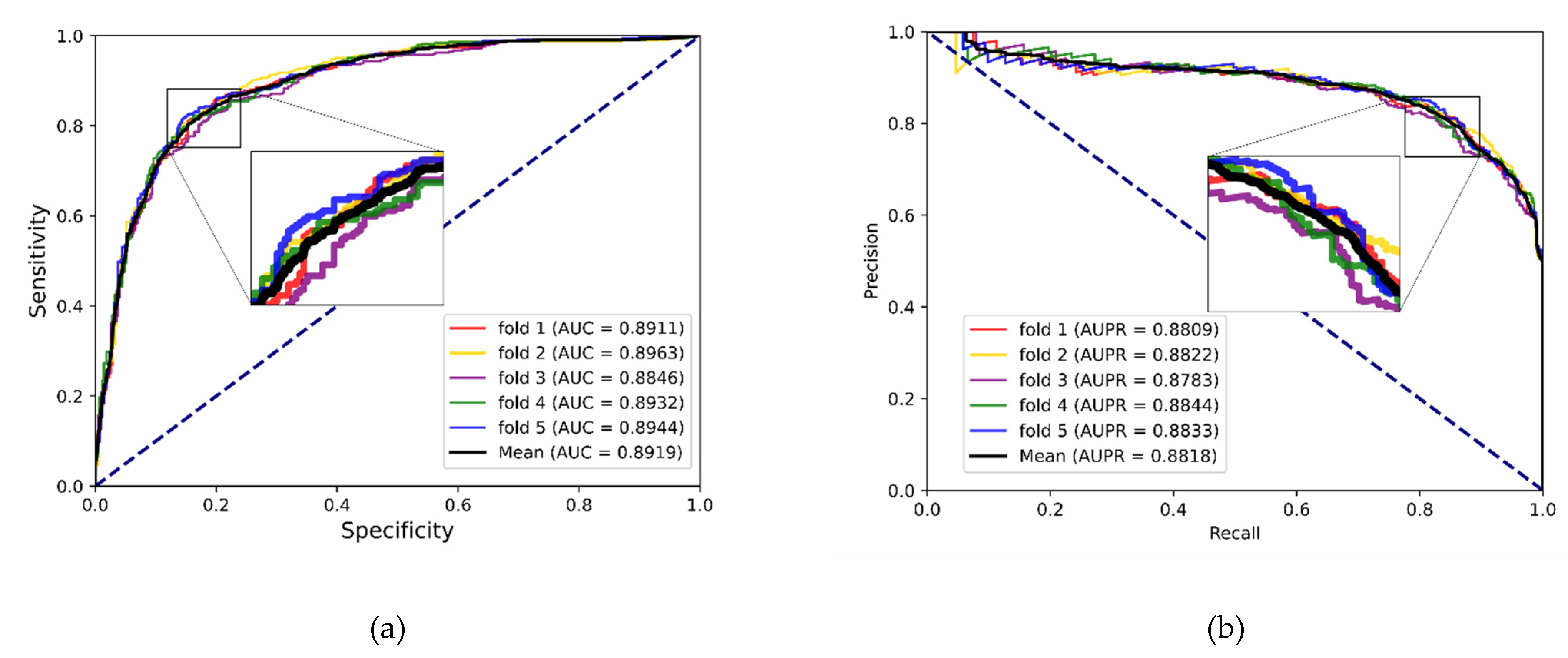
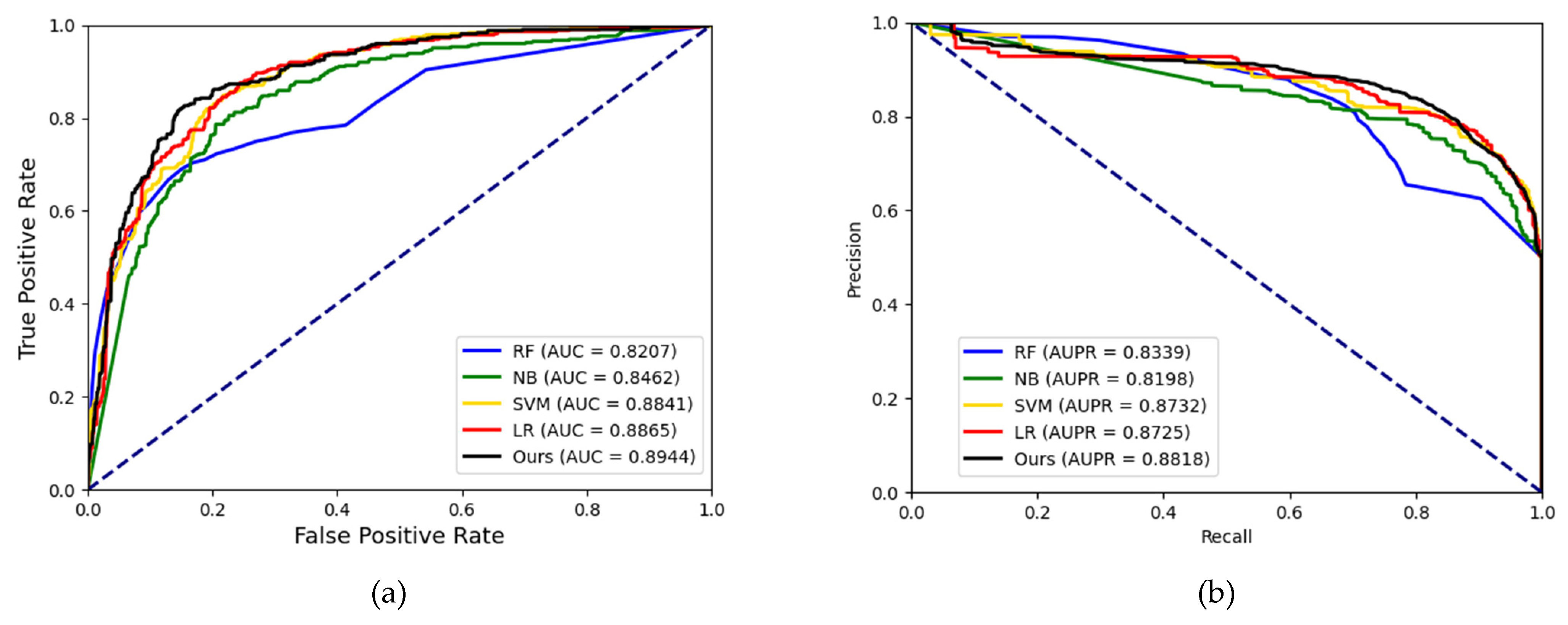
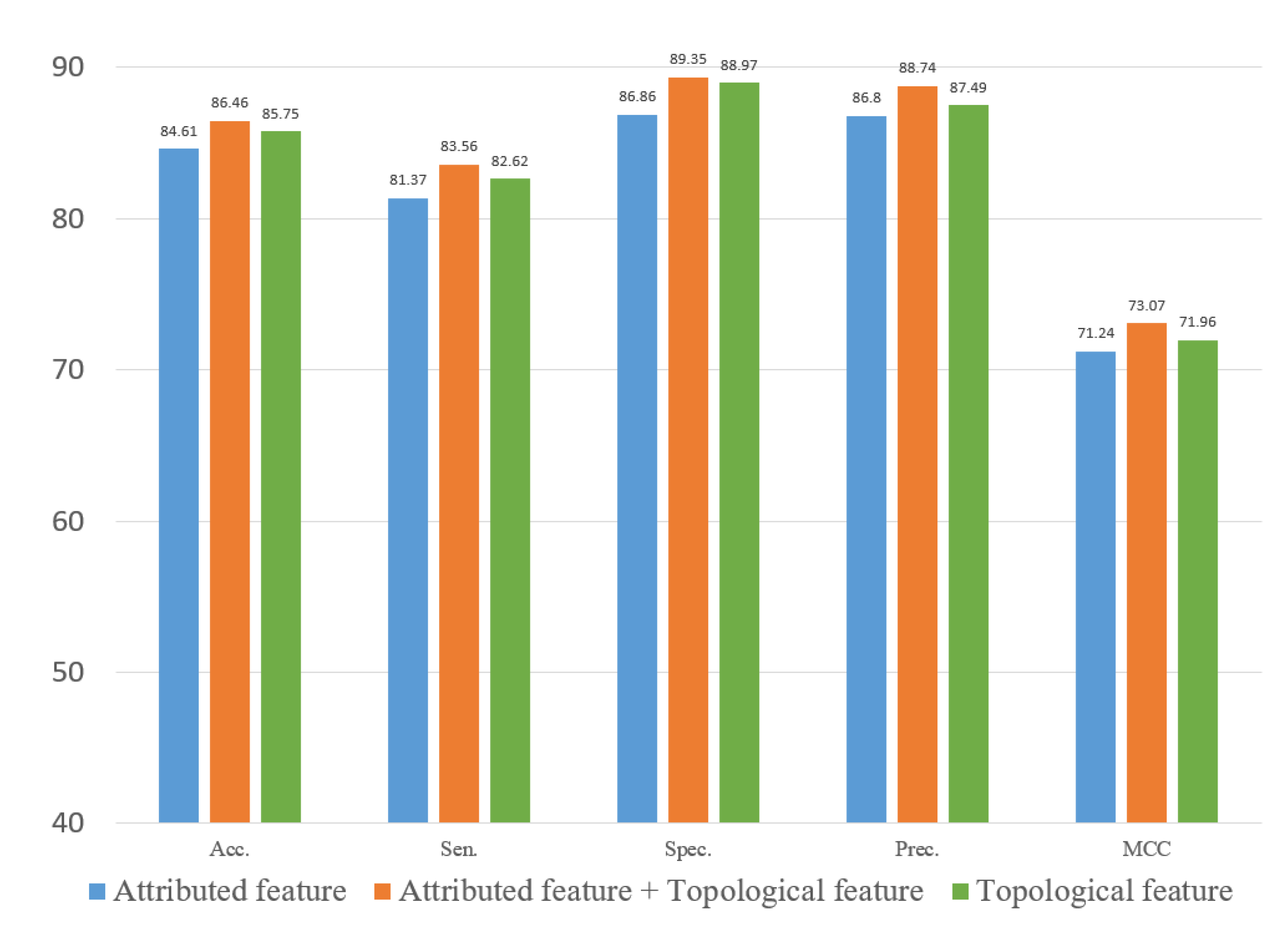
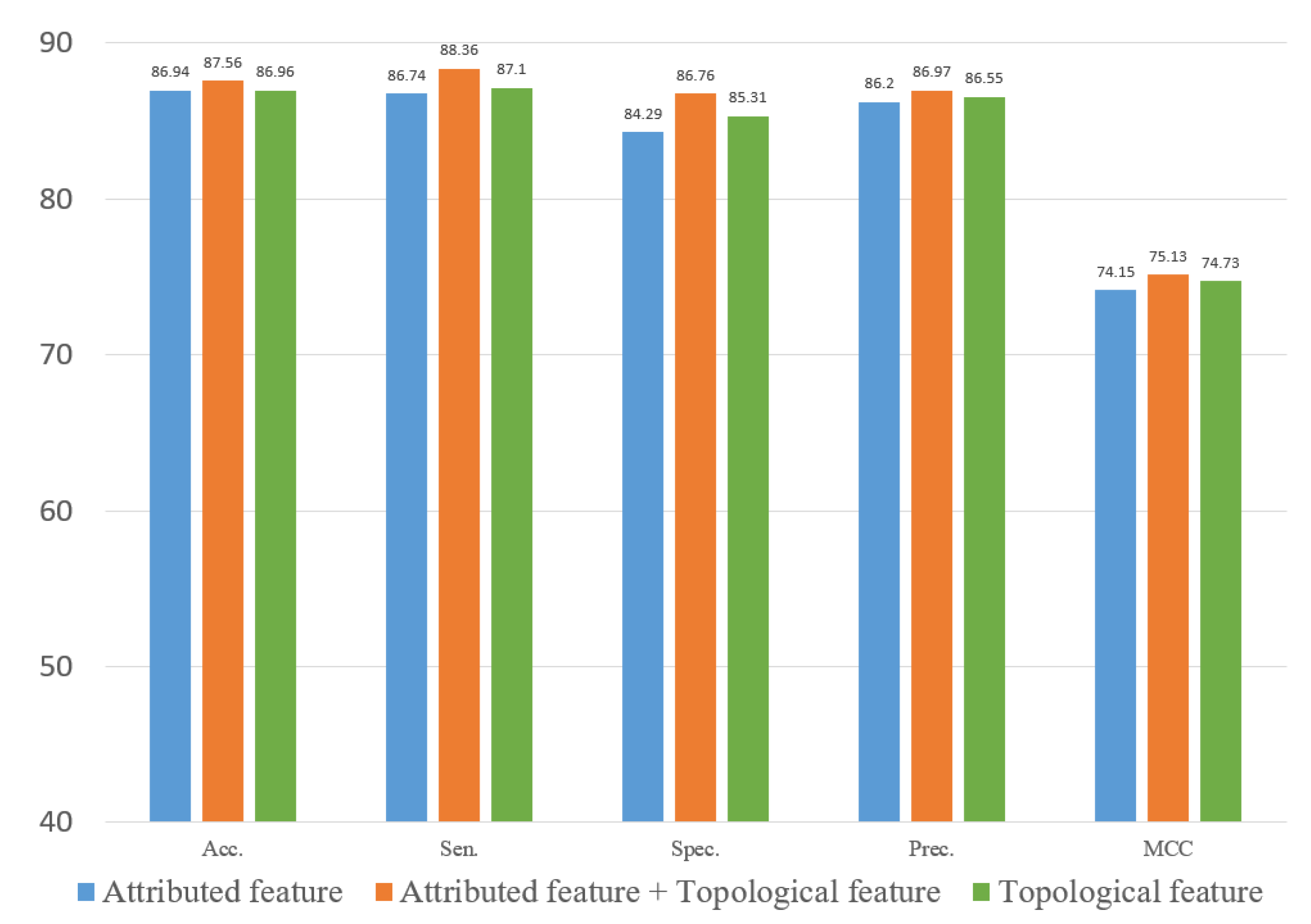
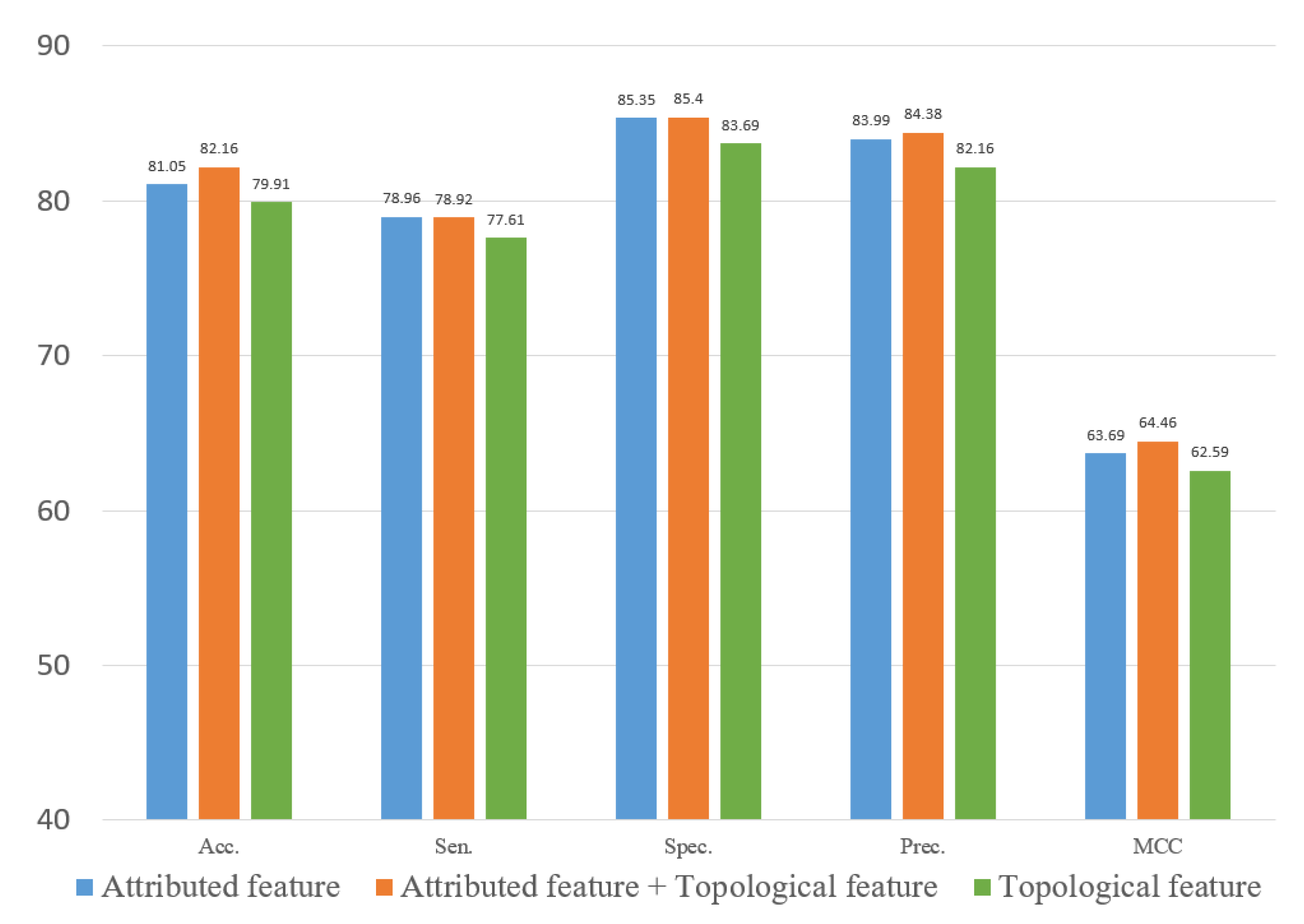
In experiments, the known drug–miRNA pairs were collected from three databases including ncDR [
44], RNAInter [
45] and SM2miR [
46]. It is worth noting that the SM2miR database was divided into three datasets according to its versions. After preprocessing these databases, there were three datasets available: ncDR, RNAInter and SM2miR. For evaluating the prediction ability of the MFIDMA, we implemented the proposed model on those three datasets and obtained average accuracies of 86.46%, 87.56% and 82.16% under a five-fold cross-validation. The average AUC values achieved 0.9407, 0.9444 and 0.8919 on ncDR, RNAInter and SM2miR, respectively. In addition, serval experiments were conducted for performance comparisons with respect to the choices of features and prediction methods. Furthermore, we carried out case studies using hsa-let-7c-5p and Verapamil to prove the prediction ability of the proposed method. There are 9 of the top 15 predicted drugs and 10 of the top 15 predicted miRNAs confirmed by the PubMed database, respectively. The results of the cross-validations and case studies demonstrated that the MFIDMA model could predict DMA accurately and robustly. This study may be helpful for predicting drug response and overcoming drug resistance for subsequent treatment and improving the situation for drug-target discovery.
4. Case Study
To further evaluate the prediction capability of the MFIDMA method, we selected the miRNA hsa-let-7c-5p and the drug Verapamil as objects to implement the proposed method as case studies based on the SM2miR v1.0 database. For Verapamil, we removed 167 known DMA related to Verapamil from the dataset; the remaining association were regarded as positive samples. Negative samples were randomly selected from the non-association pairs in the dataset and on the same scale as the positive samples. The combination of the positive samples and negative samples was treated as the training set to train the model. We then connected hsa-let-7c-5pl with the other drugs for validation. After sorting the results of the prediction scores in descending order, 9 of the top 15 candidate drugs were verified by the PubMed literature. The result of the validation is shown in
Table 7, and some supporting evidence was found. For example, the expression level of hsa-let-7c-5p reduced in cells resistant to gemcitabine [
77]. Through inactivating the IL-6/STAT3 pathway, transfection of hsa-let-7c-5p recovered the sensitivity to cisplatin [
78]. The sensitivity of 5-Fluorouracil was influenced by Akt2, which declined due to the over-activating of hsa-let-7c-5p [
79]. Fulvestrant regulated the expression of hsa-let-7c-5p to affect Gefitinib [
80]. Moreover, the same approach was implemented on Verapamil with 5573 positive samples.
Table 8 shows 10 of the top 15 candidate miRNAs that were verified from the RNAInter database, and we have evidence to support them. For example, hsa-miR-34a-5p was down-regulated in Verapamil-resistant MCF-7 breast cancer cells [
81]. Hsa-miR-21-5p and hsa-miR-15a-5p played regulatory roles in MCF7/AdrVp [
82]. The results of the case studies indicated that the proposed method could predict the drug–miRNA association with high efficiency and robustness.
5. Conclusions
In general, it seems that as the understanding of molecular mechanisms improve, it is suggested that the abnormal expression level of miRNA is associated with diseases. Micro-RNA also offers a new insight into drug-target selection. Discovering DMA is crucial to developing miRNA therapeutics and miRNA-target drugs. Consequently, several studies have investigated the computational model to identify DMA. Herein, our study has offered a multiple feature integrated model, MFIDMA, to identify the potential association between drugs and miRNAs. In MFIDMA, we formulated the drug–miRNA network and utilized SDNE to obtain the topological features. The miRNA sequences and drug SMILES were regarded as a biological sentence and generated attribute features using the Word2vec algorithm. The DNN and CNN models were then used to extract deep learning information. Finally, the predicted results of DMA were obtained using a full connection layer with integrated features. To assess the MFIDMA model, this was implemented on three datasets with a five-fold cross-validation. Our model achieved average AUC values of 0.9407, 0.9444 and 0.8919 on three of the datasets we collected. In addition, we carried out case studies and comparative experiments with other existing methods. Comprehensively, the results of the abovementioned experiments illustrated that the proposed model can predict DMA precisely and robustly. Moreover, in MFIDMA, we used miRNA sequence information and drug SMILES instead of self-similarity, which allowed our model to process new miRNAs and drugs. Future research will attempt to use side information about miRNAs and drugs such as miRNA family information, drug fingerprints and miRNA-gene information.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}