
Dissecting the Activity of Catechins as Incomplete Aldose Reductase Differential Inhibitors through Kinetic and Computational Approaches




 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of AKR1B1 Activity
2.3. Purification of Human Recombinant AKR1B1
2.4. Kinetic Analysis
2.5. Molecular Docking
2.6. Molecular Dynamics Simulations
2.7. Binding Energy Evaluations
2.8. Other Methods
3. Results
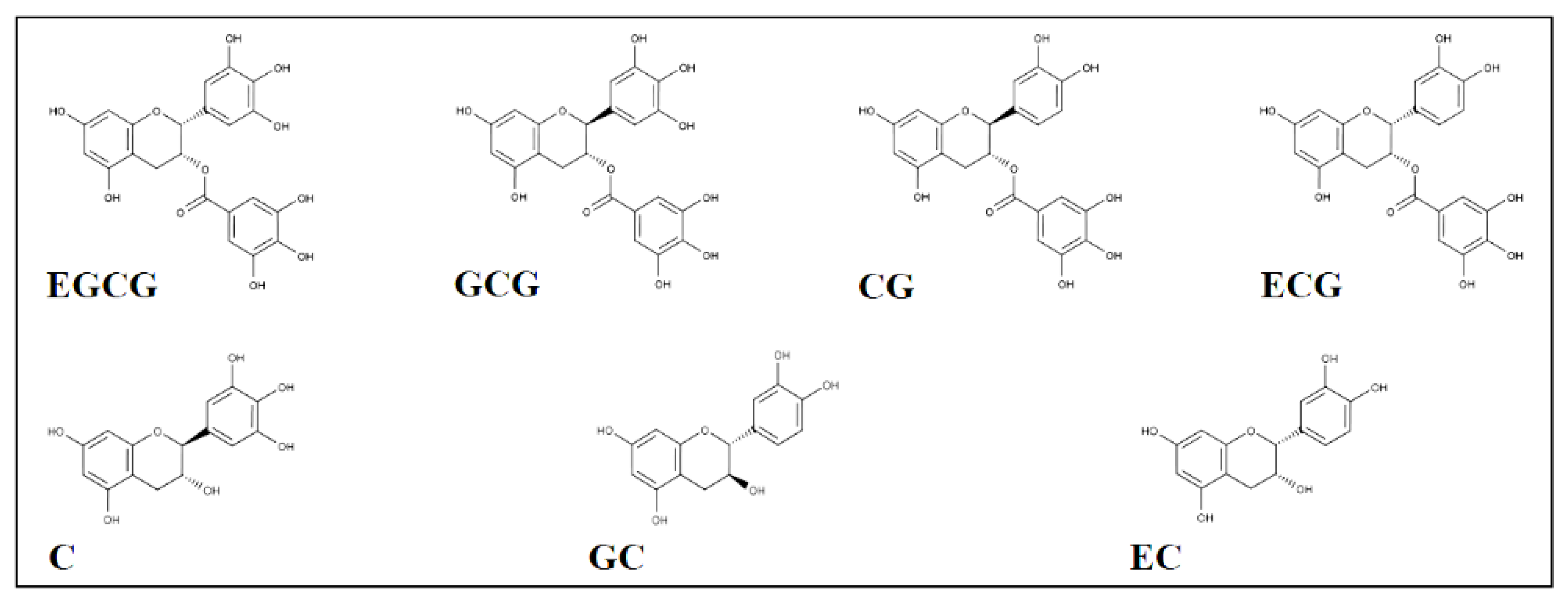
3.1. AKR1B1 Inhibition by Catechins: Evidence of Differential Inhibition
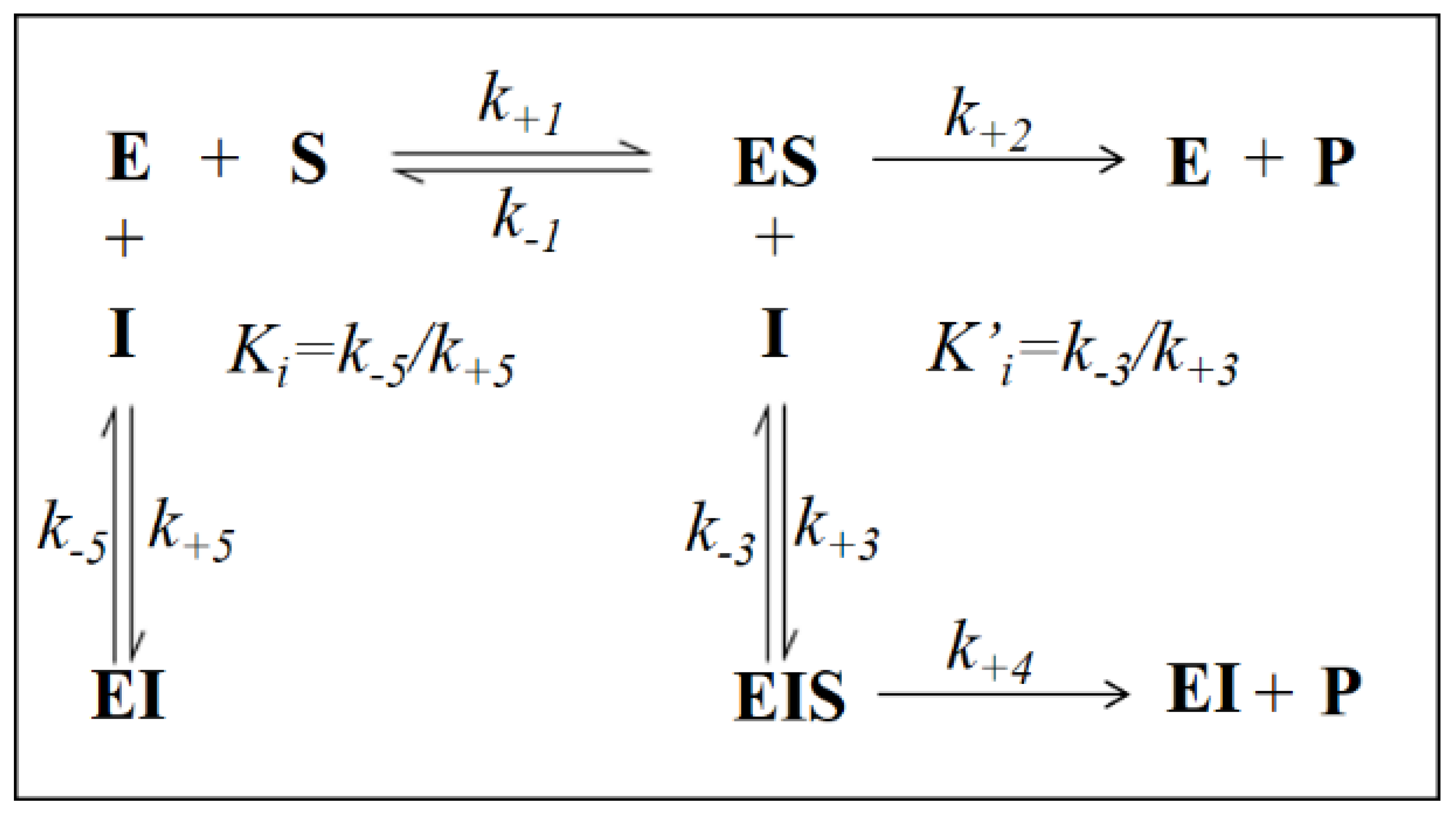
3.2. Inhibition Kinetic Analysis
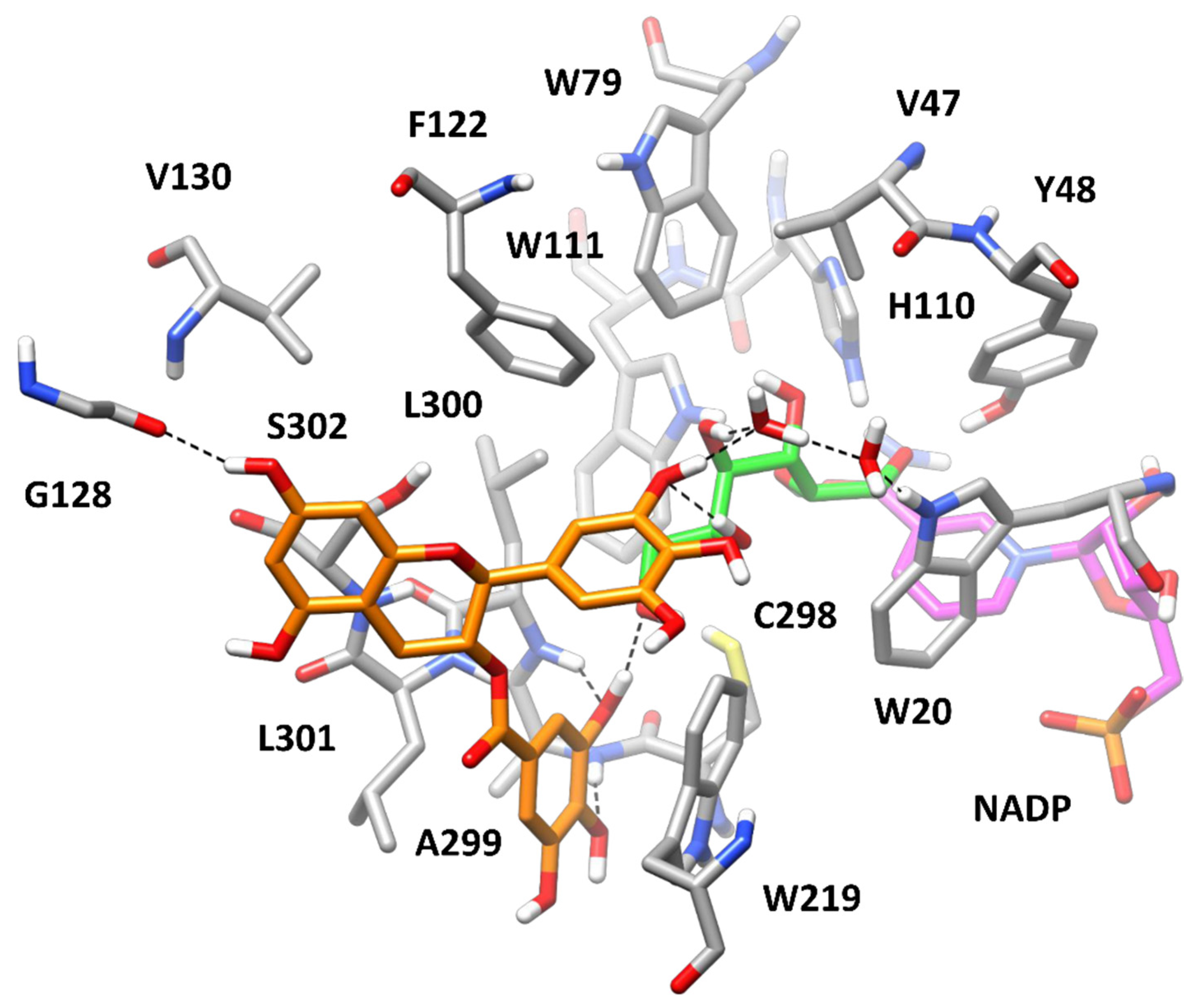
3.3. Computational Study of AKR1B1 Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Biol. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Gupta, S.K.; Ali, V.; Singh, P.; Verma, M. Aldose reductase: A cause and a potential target for the treatment of diabetic complications. Arch. Pharm. Res. 2021, 44, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Jannapureddy, S.; Sharma, M.; Yepuri, G.; Schmidt, A.M.; Ramasamy, R. Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 2021, 12, 636267. [Google Scholar] [CrossRef] [PubMed]
- Grewal, A.S.; Thapa, K.; Kanojia, N.; Sharma, N.; Singh, S. Natural Compounds as Source of Aldose Reductase (AR) Inhibitors for the Treatment of Diabetic Complications: A Mini Review. Curr. Drug Metab. 2020, 21, 1091–1116. [Google Scholar] [CrossRef]
- Kovacikova, L.; Prnova, M.S.; Majekova, M.; Bohac, A.; Karasu, C.; Stefek, M. Development of Novel Indole-Based Bifunctional Aldose Reductase Inhibitors/Antioxidants as Promising Drugs for the Treatment of Diabetic Complications. Molecules 2021, 26, 2867. [Google Scholar] [CrossRef]
- Kumar, M.; Choudhary, S.; Singh, P.K.; Silakari, O. Addressing selectivity issues of aldose reductase 2 inhibitors for the management of diabetic complications. Future. Med. Chem. 2020, 12, 1327–1358. [Google Scholar] [CrossRef]
- Choudhary, S.; Kalra, V.; Kumar, M.; Tiwary, A.K.; Sood, J.; Silakari, O. Bio-Inspired Strategies against Diabetes and Associated Complications: A Review. Recent Pat. Drug Deliv. Formul. 2019, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Grewal, A.S.; Bhardwaj, S.; Pandita, D.; Lather, V.; Sekhon, B.S. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-diabetic Diseases. Mini Rev. Med. Chem. 2016, 16, 120–162. [Google Scholar] [CrossRef]
- Srivastava, S.; Watowich, S.J.; Petrash, J.M.; Srivastava, S.K.; Bhatnagar, A. Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry 1999, 38, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Dixit, B.L.; Balendiran, G.K.; Watowich, S.J.; Srivastava, S.; Ramana, K.V.; Petrash, J.M.; Bhatnagar, A.; Srivastava, S.K. Kinetic and structural characterization of the glutathione-binding site of aldose reductase. J. Biol. Chem. 2000, 275, 21587–21595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestri, F.; Sorce, C.; Moschini, R.; Cappiello, M.; Misuri, L.; Del Corso, A.; Mura, U. Edible vegetables as a source of aldose reductase differential inhibitors. Chem. Biol. Interact. 2017, 276, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, C.E. Aldose reductase: Model for a new paradigm of enzymatic perfection in detoxification catalysts. Biochemistry 1992, 31, 10139–10145. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Bhatnagar, A.; Srivastava, S.; Yadav, U.C.; Awasthi, S.; Awasthi, Y.C.; Srivastava, S.K. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy- trans-2-nonenal (HNE): Role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 2006, 281, 17652–17660. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Long, E.K.; Hahn, W.S.; Bernlohr, D.A. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 2014, 63, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Elmazoglu, Z.; Prnova, M.S.; Santamaria, A.; Stefek, M.; Karasu, C. Combatting Nitrosative Stress and Inflammation with Novel Substituted Triazinoindole Inhibitors of Aldose Reductase in PC12 Cells Exposed to 6-Hydroxydopamine Plus High Glucose. Neurotox. Res. 2021, 39, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Sonowal, H.; Ramana, K.V. Development of Aldose Reductase Inhibitors for the Treatment of Inflammatory Disorders and Cancer: Current Drug Design Strategies and Future Directions. Curr. Med. Chem. 2021, 28, 3683–3712. [Google Scholar] [CrossRef]
- Wang, C.; La, L.; Feng, H.; Yang, Q.; Wu, F.; Wang, C.; Wu, J.; Hou, L.; Hou, C.; Liu, W. Aldose Reductase Inhibitor Engeletin Suppresses Pelvic Inflammatory Disease by Blocking the Phospholipase C/Protein Kinase C-Dependent/NF-κB and MAPK Cascades. J. Agric. Food Chem. 2020, 68, 11747–11757. [Google Scholar] [CrossRef] [PubMed]
- Chatzopoulou, M.; Pegklidou, K.; Papastavrou, N.; Demopoulos, V.J. Development of aldose reductase inhibitors for the treatment of inflammatory disorders. Expert Opin. Drug Discov. 2013, 8, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Del Corso, A.; Balestri, F.; Di Bugno, E.; Moschini, R.; Cappiello, M.; Sartini, S.; La Motta, C.; Da Settimo, F.; Mura, U. A new approach to control the enigmatic activity of aldose reductase. PLoS ONE 2013, 8, e74076. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Moschini, R.; Mura, U.; Cappiello, M.; Del Corso, A. In Search of Differential Inhibitors of Aldose Reductase. Biomolecules 2022, 12, 485. [Google Scholar] [CrossRef]
- Balestri, F.; Poli, G.; Pineschi, C.; Moschini, R.; Cappiello, M.; Mura, U.; Tuccinardi, T.; Del Corso, A. Aldose Reductase Differential Inhibitors in Green Tea. Biomolecules 2020, 10, 1003. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Quattrini, L.; Coviello, V.; Sartini, S.; Da Settimo, F.; Cappiello, M.; Moschini, R.; Del Corso, A.; Mura, U.; La Motta, C. Acid Derivatives of Pyrazolo[1,5-a]pyrimidine as Aldose Reductase Differential Inhibitors. Cell Chem. Biol. 2018, 25, 1414–1418. [Google Scholar] [CrossRef]
- Balestri, F.; De Leo, M.; Sorce, C.; Cappiello, M.; Quattrini, L.; Moschini, R.; Pineschi, C.; Braca, A.; La Motta, C.; Da Settimo, F.; et al. Soyasaponins from Zolfino bean as aldose reductase differential inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food Nutr Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. L-Idose: An attractive substrate alternative to D-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef]
- Mavrevski, R.; Traykov, M.; Trenchev, I.; Trencheva, M. Approaches to modeling of biological experimental data with GraphPad prism software. WSEAS Trans. Syst Control. 2018, 13, 242–247. [Google Scholar]
- Balestri, F.; Cappiello, M.; Moschini, R.; Mura, U.; Del Corso, A. Models of enzyme inhibition and apparent dissociation constants from kinetic analysis to study the differential inhibition of aldose reductase. J. Enzym. Inhib. Med. Chem. 2022, 37, 1426–1436. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, L.; Chen, W.; Chen, Y.; Xie, W.; Hu, X. Partial inhibition of aldose reductase by nitazoxanide and its molecular basis. Chem. Med. Chem. 2012, 7, 1921–1923. [Google Scholar] [CrossRef] [PubMed]
- Steuber, H.; Heine, A.; Klebe, G. Structural and thermodynamic study on aldose reductase: Nitro-substituted inhibitors with strong enthalpic binding contribution. J. Mol. Biol. 2007, 368, 618–638. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2013, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER, Version 20; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Poli, G.; Lapillo, M.; Jha, V.; Mouawad, N.; Caligiuri, I.; Macchia, M.; Rizzolio, F.; Tuccinardi, T.; Granchi, C. Computationally driven discovery of phenyl(piperazin-1-yl)methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 589–596. [Google Scholar] [CrossRef]
- Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.R.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules. 2018, 23, 1506. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Manetti, F.; Schenone, S.; Martinelli, A.; Botta, M. Construction and validation of a RET TK catalytic domain by homology modeling. J. Chem. Inf. Model. 2007, 47, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Lapillo, M.; Granchi, C.; Caciolla, J.; Mouawad, N.; Caligiuri, I.; Rizzolio, F.; Langer, T.; Minutolo, F.; Tuccinardi, T. Binding investigation and preliminary optimisation of the 3-amino-1,2,4-triazin-5(2H)-one core for the development of new Fyn inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Balestri, F.; Barracco, V.; Renzone, G.; Tuccinardi, T.; Pomelli, C.S.; Cappiello, M.; Lessi, M.; Rotondo, R.; Bellina, F.; Scaloni, A.; et al. Stereoselectivity of Aldose Reductase in the Reduction of Glutathionyl-Hydroxynonanal Adduct. Antioxidants 2019, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, M.; Peroni, E.; Lepore, A.; Moschini, R.; Del Corso, A.; Balestri, F.; Mura, U. Rapid colorimetric determination of reduced and oxidized glutathione using an end point coupled enzymatic assay. Anal. Bioanal. Chem. 2013, 405, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Urzhumtsev, A.; Tête-Favier, F.; Mitschler, A.; Barbanton, J.; Barth, P.; Urzhumtseva, L.; Biellmann, J.F.; Podjarny, A.; Moras, D. A ‘specificity’ pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1197, 5, 601–612. [Google Scholar] [CrossRef]
- Shah, A.; Shinde, R.; Kare, P.; Hymavathi, V.; Chavan, S.; Elizabeth Sobhia, M. Induced fit binding of aldose reductase inhibitors to AKR1B10. Med. Chem. Res. 2012, 21, 1245–1252. [Google Scholar] [CrossRef]
- Rechlin, C.; Scheer, F.; Terwesten, F.; Wulsdorf, T.; Pol, E.; Fridh, V.; Toth, P.; Diederich, W.E.; Heine, A.; Klebe, G. Price for Opening the Transient Specificity Pocket in Human Aldose Reductase upon Ligand Binding: Structural, Thermodynamic, Kinetic, and Computational Analysis. ACS Chem. Biol. 2017, 12, 1397–1415. [Google Scholar] [CrossRef] [PubMed]
- Sandner, A.; Ngo, K.; Sager, C.P.; Scheer, F.; Daude, M.; Diederich, W.E.; Heine, A.; Klebe, G. Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase? Biomolecules 2021, 11, 1837. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, C.E.; Shahbaz, M.; Putney, C.G. Mechanistic basis for nonlinear kinetics of aldehyde reduction catalyzed by aldose reductase. Biochemistry 1990, 29, 9947–9955. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Abate, M.; Del Corso, A.; Mura, U. Modulation of aldose reductase activity by aldose hemiacetals. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 2329–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catechin | L-Idose | HNE | GSHNE |
|---|---|---|---|
| CG | |||
| Ki | 246 ± 170 | ND > 1021 | ND > 1022 |
| Ki* | 4.1 ± 0.2 | 4.5 ± 0.4 | 5.0 ± 0.2 |
| k+4 | 1.4 ± 0.6 | 4.7 ± 0.7 | 4.1 ± 0.8 |
| k+4/k+2 (%) a | 0.7 ± 0.3 | 6.0 ± 0.9 | 3.8 ± 0.8 |
| GCG | |||
| Ki | 71 ± 9 | ND > 1015 | ND > 1023 |
| Ki* | 7.1 ± 0.4 | 9.0 ± 0.5 | 17 ± 2 |
| k+4 | 5.6 ± 0.5 | 9.5 ± 0.9 | 6.0 ± 1.0 |
| k+4/k+2 (%) a | 2.9 ± 0.25 | 12.2 ± 1.1 | 5.5 ± 0.9 |
| ECG | |||
| Ki | 116 ± 8 | ND > 104 | 91 ± 10 |
| Ki* | 46 ± 5 | 54 ± 9 | 46 ± 11 |
| k+4 | 5.2 ± 3.2 | 6.7 ± 2.7 | 15.6 ± 4.3 |
| k+4/k+2 (%) a | 2.7 ± 1.7 | 8.6 ± 0.4 | 14.3 ± 3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balestri, F.; Poli, G.; Piazza, L.; Cappiello, M.; Moschini, R.; Signore, G.; Tuccinardi, T.; Mura, U.; Del Corso, A. Dissecting the Activity of Catechins as Incomplete Aldose Reductase Differential Inhibitors through Kinetic and Computational Approaches. Biology 2022, 11, 1324. https://doi.org/10.3390/biology11091324
Balestri F, Poli G, Piazza L, Cappiello M, Moschini R, Signore G, Tuccinardi T, Mura U, Del Corso A. Dissecting the Activity of Catechins as Incomplete Aldose Reductase Differential Inhibitors through Kinetic and Computational Approaches. Biology. 2022; 11(9):1324. https://doi.org/10.3390/biology11091324
Chicago/Turabian StyleBalestri, Francesco, Giulio Poli, Lucia Piazza, Mario Cappiello, Roberta Moschini, Giovanni Signore, Tiziano Tuccinardi, Umberto Mura, and Antonella Del Corso. 2022. "Dissecting the Activity of Catechins as Incomplete Aldose Reductase Differential Inhibitors through Kinetic and Computational Approaches" Biology 11, no. 9: 1324. https://doi.org/10.3390/biology11091324
APA StyleBalestri, F., Poli, G., Piazza, L., Cappiello, M., Moschini, R., Signore, G., Tuccinardi, T., Mura, U., & Del Corso, A. (2022). Dissecting the Activity of Catechins as Incomplete Aldose Reductase Differential Inhibitors through Kinetic and Computational Approaches. Biology, 11(9), 1324. https://doi.org/10.3390/biology11091324