
Transcriptomics and Proteomics Analyses Reveal JAK Signaling and Inflammatory Phenotypes during Cellular Senescence in Blind Mole Rats: The Reflections of Superior Biology

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Fibroblast Isolation and Cell Culture
2.3. β-Galactosidase Staining
2.4. Cell Growth Curve
2.5. RNA Extraction and qPCR
2.6. Protein Extraction and Nano LC-ESI-MS/MS Analysis
2.7. Data Analysis of LC MS/MS
2.8. Reanalysis of RNA Sequencing and Proteomics Datasets
2.9. Statistical Analysis
3. Results
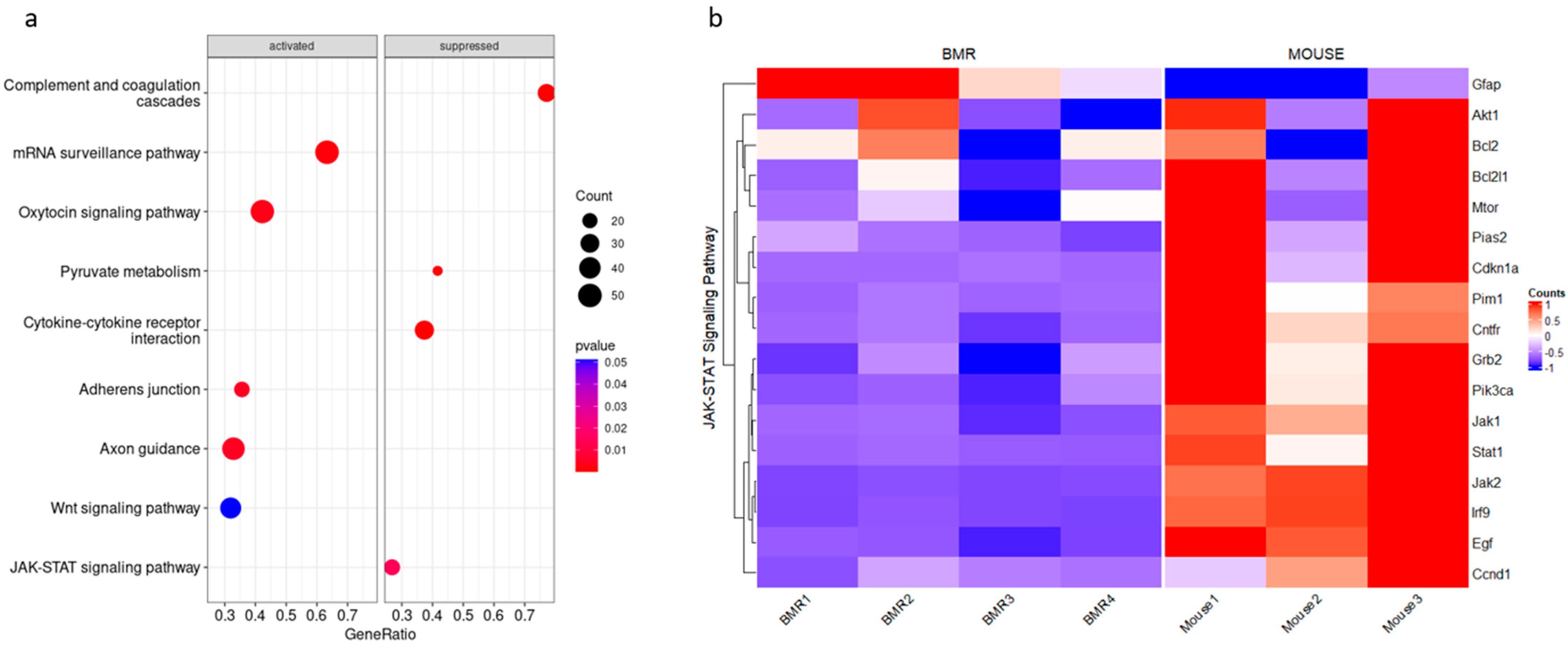
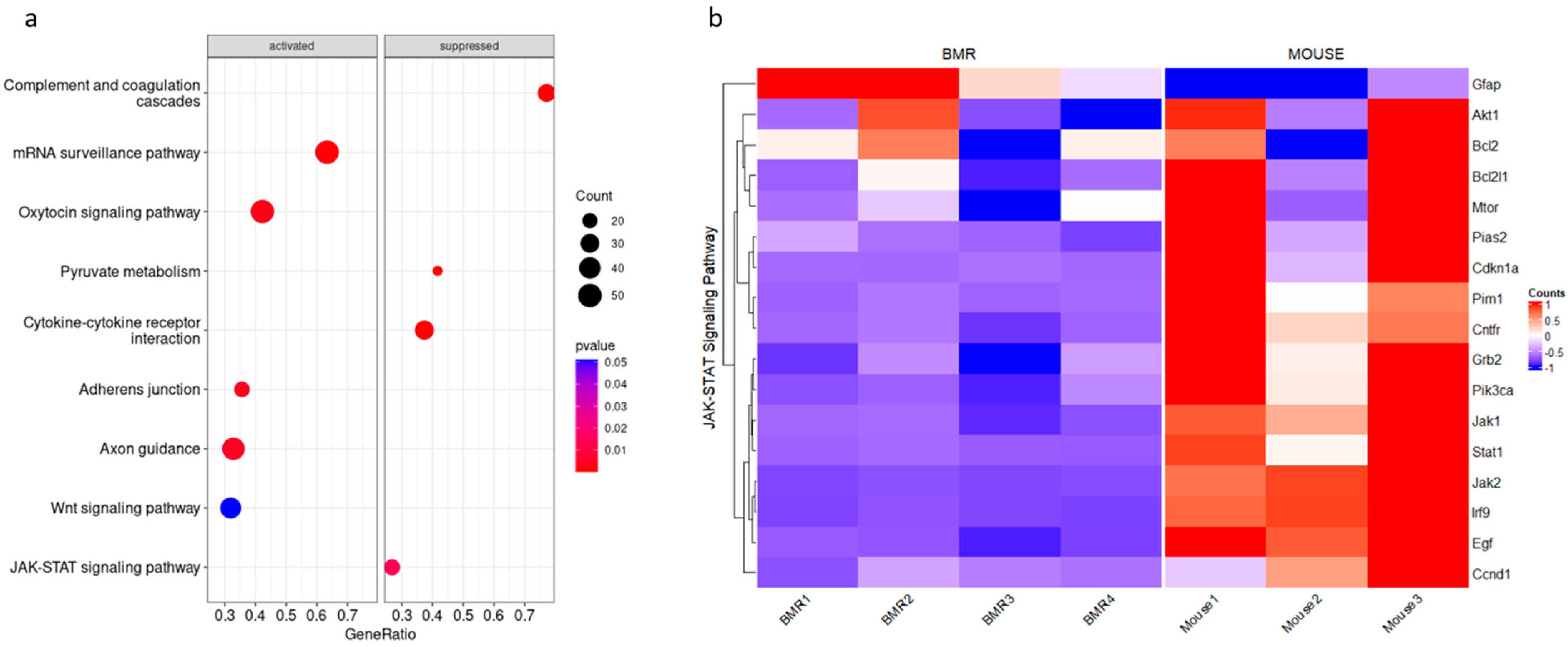
3.1. JAK–STAT Pathway Is Suppressed in the BMR Compared to the Mouse
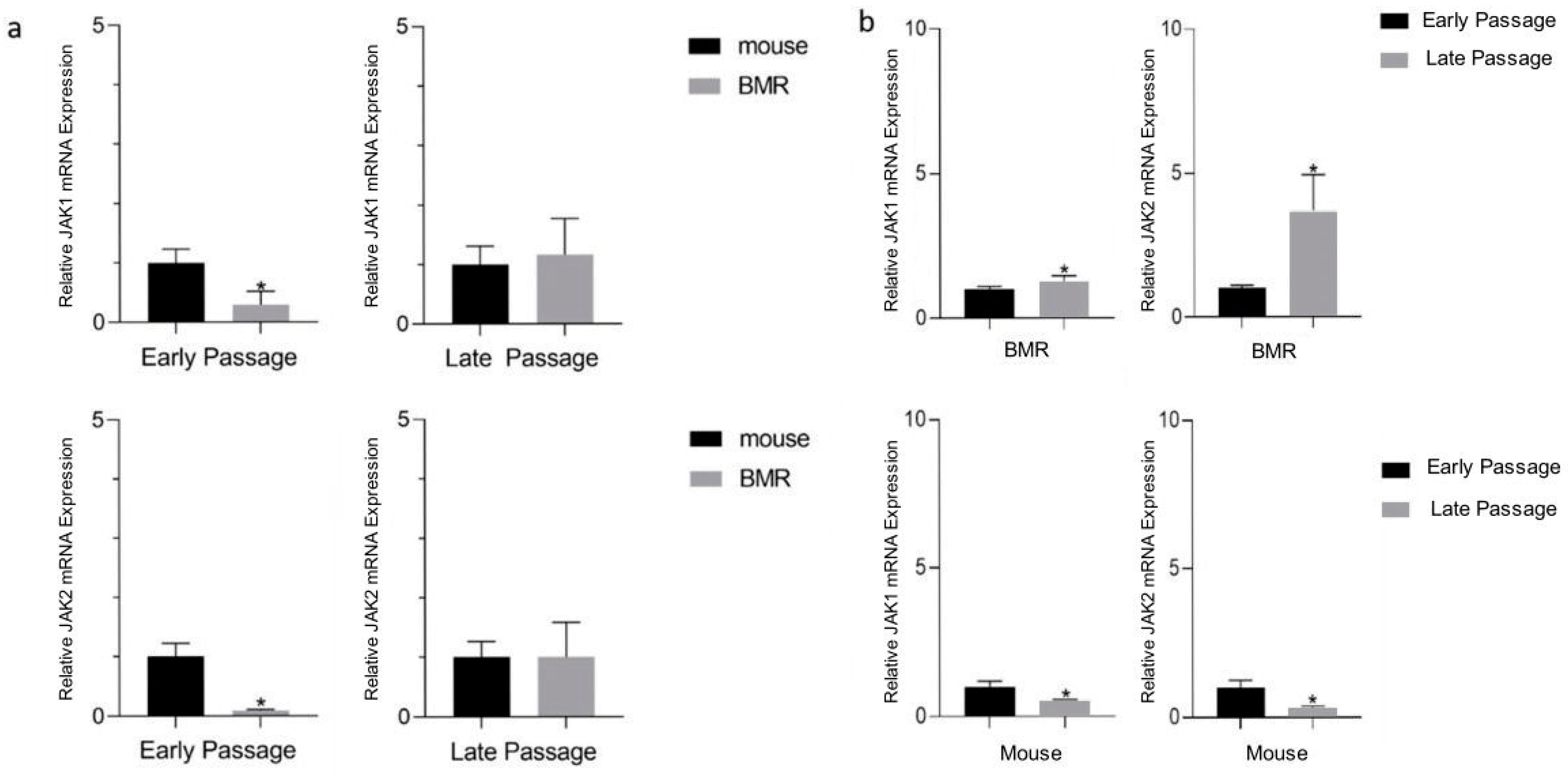
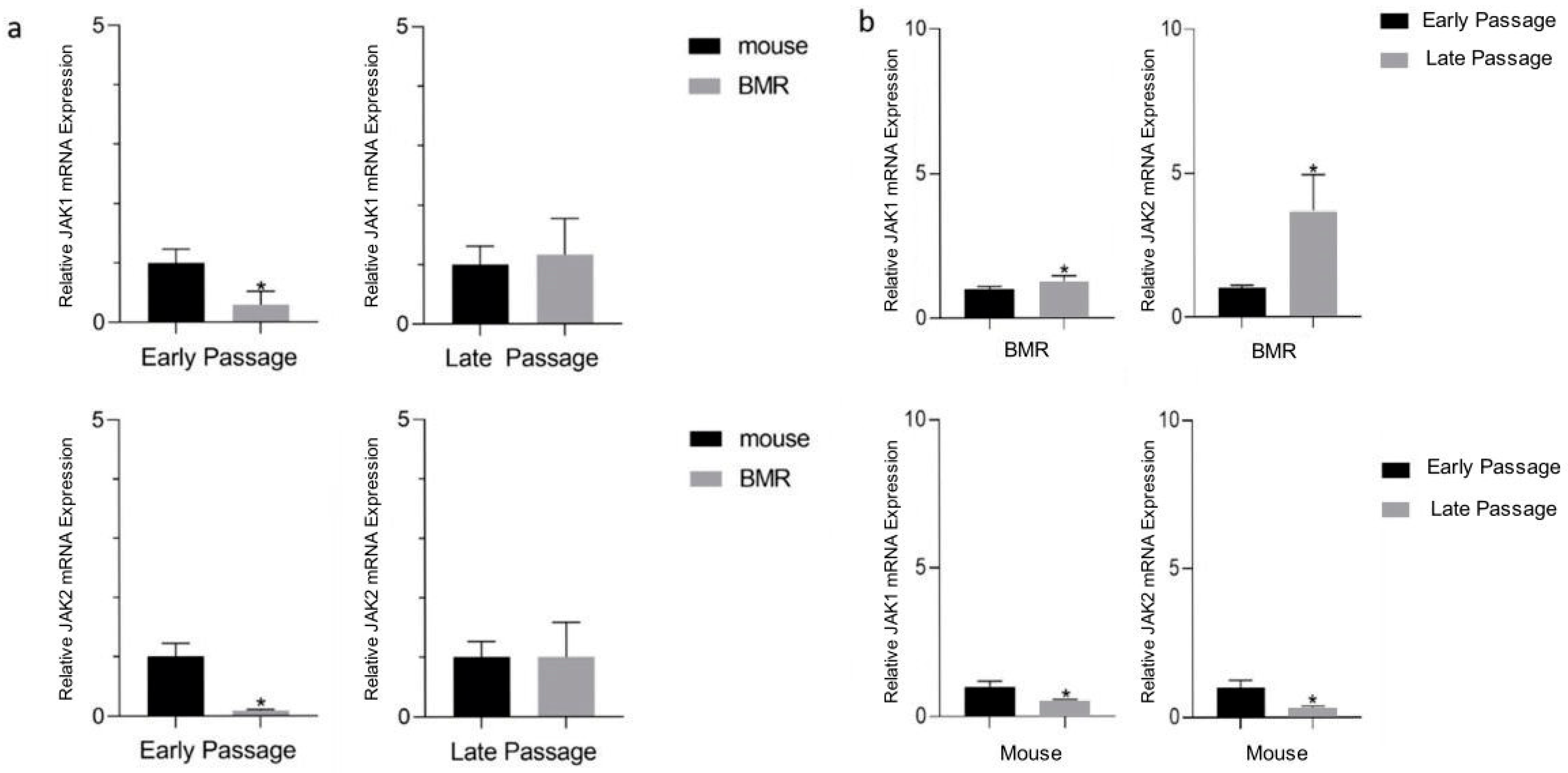
3.2. Jak1 and Jak2 Levels Are Elevated in the BMR during Replicative Senescence
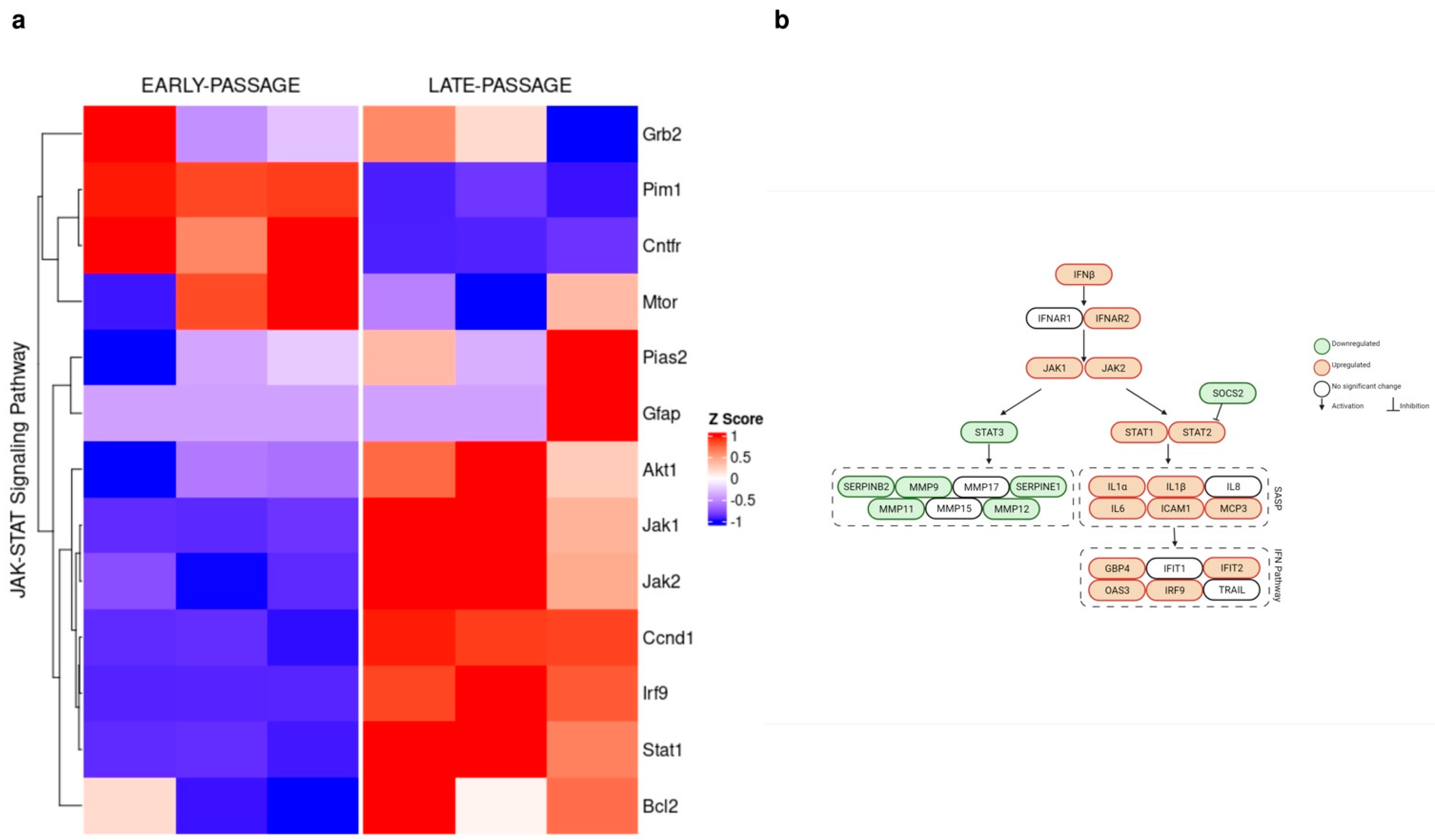
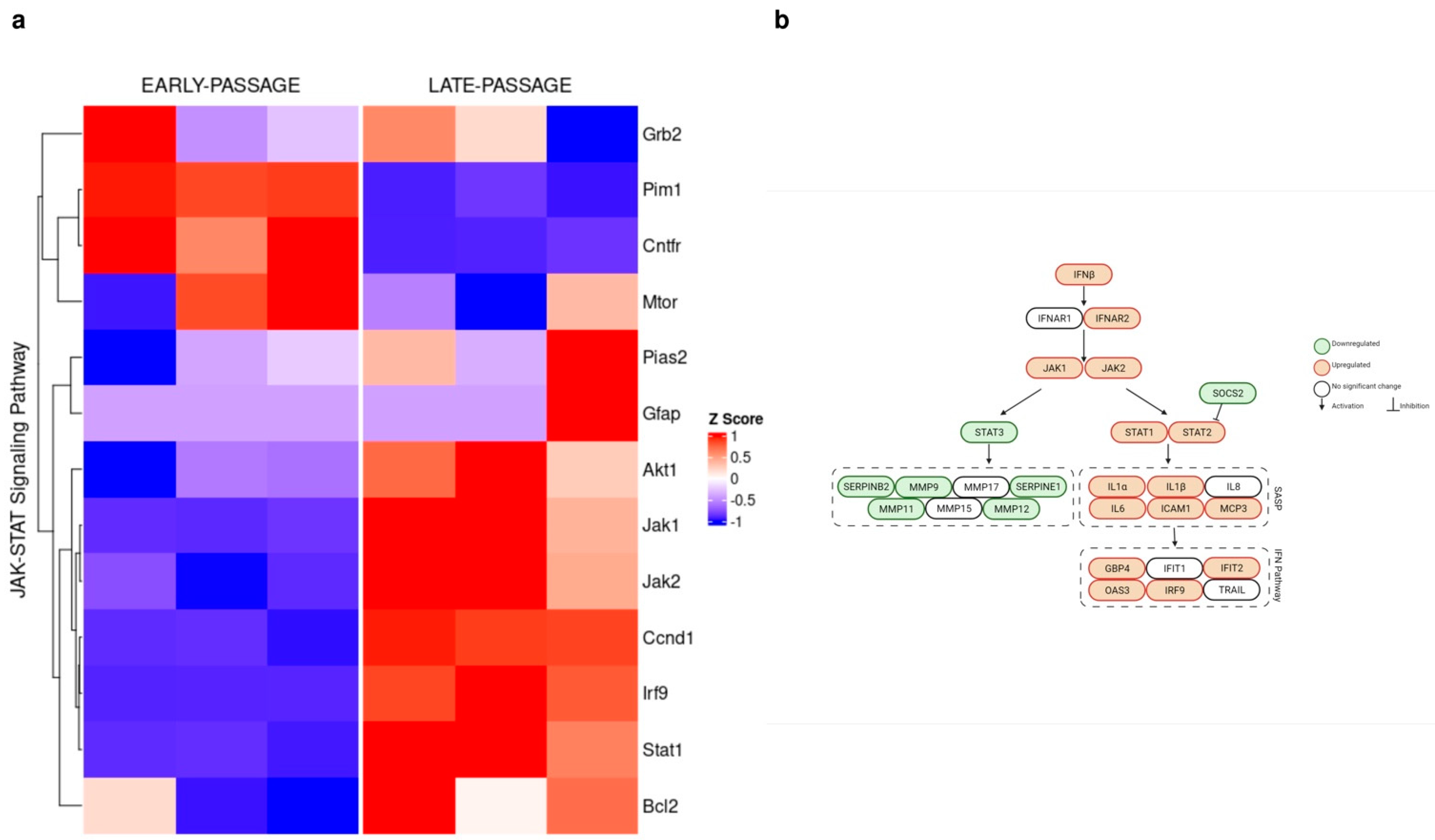
3.3. Divergent Action of Inflammatory Factors during Cellular Senescence
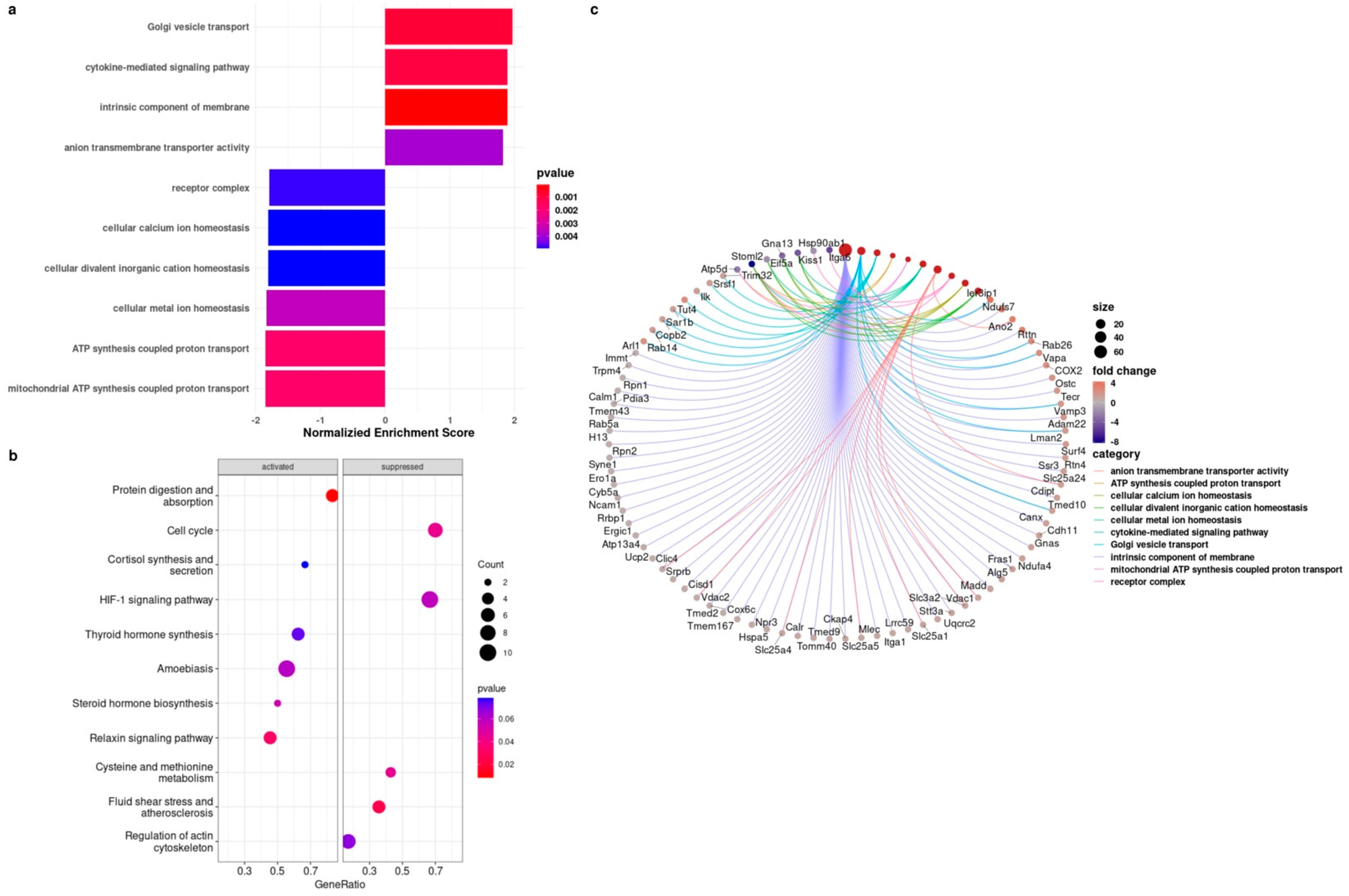
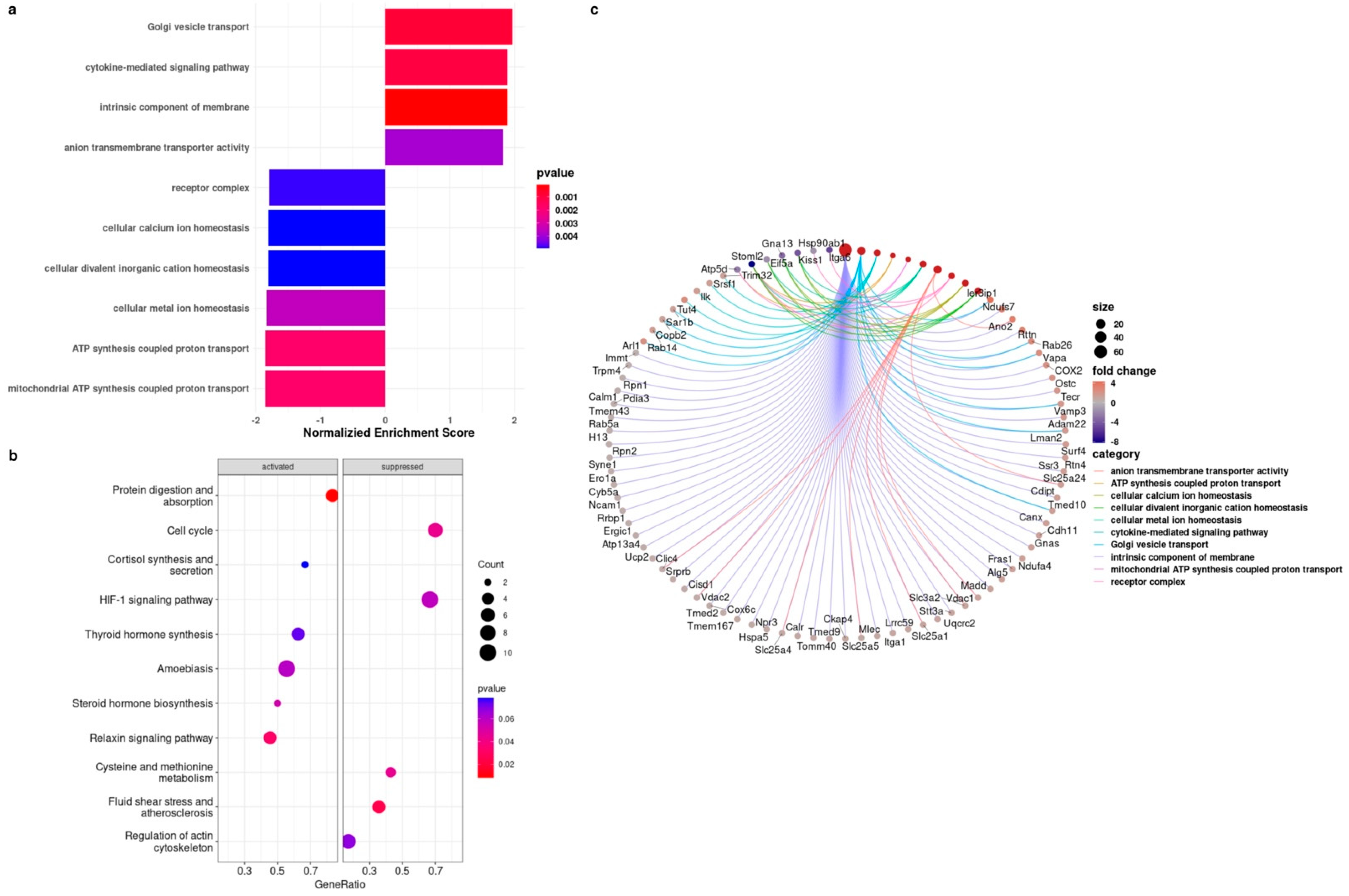
3.4. Proteomics Analysis Showed the Upregulation of the Cytokine-Mediated Signaling Pathway in BMRs during Replicative Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMR | Blind mole rat |
| CCD | Concerted cell death |
| CDKN1a | Cyclin-dependent kinase inhibitor 1A |
| DEG | Differential expression gene |
| DMEM | Dulbecco’s modified Eagle’s medium |
| ECM | Extracellular matrix |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| GSEA | Gene set enrichment analysis |
| HIF | Hypoxia-inducible factor |
| ICAM1 | Intercellular adhesion molecule 1 |
| IFNβ | Interferon β |
| IL17 | Interleukin 17 |
| Il1α | Interleukin 1 alpha |
| Il1β | Interleukin 1 beta |
| IL6 | Interleukin 6 |
| ILK | Integrin-linked protein kinase |
| IRF9 | Interferon regulatory factor 9 |
| JAK | Janus kinase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC-ESI-MS/MS | Liquid chromatography/electrospray ionization tandem mass spectrometry |
| MCP | Monocyte chemotactic protein |
| MMP | Matrix metallo-proteinases |
| mRNA | Messenger ribonucleic acid |
| NF-κB | Nuclear factor kappa B |
| p38 MAPK | p38 mitogen-activated protein kinases |
| PBS | Phosphate-buffered saline |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| qRT-PCR | Quantitative real-time PCR |
| SASP | Senescence-associated secretory phenotype |
| SA-β-Gal | Senescence-associated β-galactosidase |
| SD | Standard deviation |
| SERPINs | Serine protease inhibitors |
| SRSF1 | Serine-/arginine-rich splicing factor 1 |
| STAT | Signal transducer and activator of transcription |
| TRIM32 | E3 ubiquitin-protein ligase |
| TUT4 | Terminal uridylyltransferase 4 |
References
- Bozaykut, P.; Sozen, E.; Kaga, E.; Ece, A.; Ozaltin, E.; Bergquist, J.; Ozer, N.K.; Yilmaz, B.K. HSP70 Inhibition Leads to the Activation of Proteasomal System under Mild Hyperthermia Conditions in Young and Senescent Fibroblasts. Oxidative Med. Cell. Longev. 2020, 2020, 9369524. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Hine, C.; Tian, X.; Ablaeva, J.; Gudkov, A.V.; Nevo, E.; Seluanov, A. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 19392–19396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manov, I.; Hirsh, M.; Iancu, T.C.; Malik, A.; Sotnichenko, N.; Band, M.; Avivi, A.; Shams, I. Pronounced cancer resistance in a subterranean rodent, the blind mole-rat, Spalax: In vivo and in vitroevidence. BMC Biol. 2013, 11, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Oreskovic, E.; Zhang, Q.; Lu, Q.; Gilman, A.; Lin, Y.S.; He, J.; Zheng, Z.; Lu, J.Y.; Lee, J.; et al. Transposon-triggered innate immune response confers cancer resistance to the blind mole rat. Nat. Immunol. 2021, 22, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory Cytokines, Aging, and Age-Related Diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Duraisingham, S.S.; Rouphael, N.; Cavanagh, M.M.; Nakaya, H.I.; Goronzy, J.J.; Pulendran, B. Systems Biology of Vaccination in the Elderly. In Systems Biology. Current Topics in Microbiology and Immunology; Katze, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 363, pp. 117–142. [Google Scholar] [CrossRef]
- Chakraborti, S.; Nirmal, D.; Dikshit, M. Role of INOS in Insulin Resistance and Endothelial Dysfunction. In Oxidative Stress in Heart Diseases; Chakraborti, S., Dhalla, N., Ganguly, N., Dikshit, M., Eds.; Springer: Singapore, 2019. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular Pathways: Molecular Basis for Sensitivity and Resistance to JAK Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Odeh, A.; Dronina, M.; Domankevich, V.; Shams, I.; Manov, I. Downregulation of the inflammatory network in senescent fibroblasts and aging tissues of the long-lived and cancer-resistant subterranean wild rodent. Spalax. Aging Cell 2020, 19, e13045. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-G.; Mikhalchenko, A.E.; Yim, S.H.; Gladyshev, V.N. A naked mole rat iPSC line expressing drug-inducible mouse pluripotency factors developed from embryonic fibroblasts. Stem Cell Res. 2018, 31, 197–200. [Google Scholar] [CrossRef]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; Volume 445, pp. 51–56. [Google Scholar]
- Emmert-Streib, F.; Glazko, G.V. Pathway Analysis of Expression Data: Deciphering Functional Building Blocks of Complex Diseases. PLoS Comput. Biol. 2011, 7, e1002053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, P.; Singh, K.; Krug, L.; Koroma, A.; Hainzl, A.; Bloch, W.; Kochanek, S.; Wlaschek, M.; Schorpp-Kistner, M.; Angel, P.; et al. Persistent JunB activation in fibroblasts disrupts stem cell niche interactions enforcing skin aging. Cell Rep. 2021, 36. [Google Scholar] [CrossRef]
- Altwasser, R.; Paz, A.; Korol, A.; Manov, I.; Avivi, A.; Shams, I. The transcriptome landscape of the carcinogenic treatment response in the blind mole rat: Insights into cancer resistance mechanisms. BMC Genom. 2019, 20, 17. [Google Scholar] [CrossRef]
- Marthandan, S.; Priebe, S.; Baumgart, M.; Groth, M.; Cellerino, A.; Guthke, R.; Hemmerich, P.; Diekmann, S. Similarities in Gene Expression Profiles during In Vitro Aging of Primary Human Embryonic Lung and Foreskin Fibroblasts. BioMed Res. Int. 2015, 2015, 731938. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, R.; Sugawara, H.; Shumway, M.; On behalf of the International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.; Irizarry, R.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; E Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Biragyn, A.; Aliseychik, M.; Rogaev, E. Potential importance of B cells in aging and aging-associated neurodegenerative diseases. Semin. Immunopathol. 2017, 39, 283–294. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Seluanov, A.; Gladyshev, V.N.; Vijg, J.; Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Cancer 2018, 18, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Jojic, V.; Kidd, B.; Shen-Orr, S.; Price, J.; Jarrell, J.; Tse, T.; Huang, H.; Lund, P.; Maecker, H.T.; et al. Apoptosis and other immune biomarkers predict influenza vaccine responsiveness. Mol. Syst. Biol. 2013, 9, 659. [Google Scholar] [CrossRef]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef]
- Zeng, G.; Millis, A.J. Differential Regulation of Collagenase and Stromelysin mRNA in Late Passage Cultures of Human Fibroblasts. Exp. Cell Res. 1996, 222, 150–156. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Novotny, M.; Bulek, K.; Liu, C.; Li, X.; A Hamilton, T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 2011, 12, 853–860. [Google Scholar] [CrossRef]
- Herjan, T.; Hong, L.; Bubenik, J.; Bulek, K.; Qian, W.; Liu, C.; Li, X.; Chen, X.; Yang, H.; Ouyang, S.; et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat. Immunol. 2018, 19, 354–365. [Google Scholar] [CrossRef]
- Liu, Y.; Lagowski, J.P.; Gao, S.; Raymond, J.H.; White, C.R.; Kulesz-Martin, M.F. Regulation of the Psoriatic Chemokine CCL20 by E3 Ligases Trim32 and Piasy in Keratinocytes. J. Investig. Dermatol. 2010, 130, 1384–1390. [Google Scholar] [CrossRef] [Green Version]
- Albor, A.; El-Hizawi, S.; Horn, E.J.; Laederich, M.; Frosk, P.; Wrogemann, K.; Kulesz-Martin, M. The Interaction of Piasy with Trim32, an E3-Ubiquitin Ligase Mutated in Limb-girdle Muscular Dystrophy Type 2H, Promotes Piasy Degradation and Regulates UVB-induced Keratinocyte Apoptosis through NFκB. J. Biol. Chem. 2006, 281, 25850–25866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Gao, Y.; Wang, L.; Liu, S.; Han, B.; Ma, L.; Ling, Y.; Mao, S.; Wang, X. Overexpression of Integrin-linked Kinase Promotes Lung Cancer Cell Migration and Invasion via NF-κB-mediated Upregulation of Matrix Metalloproteinase-9. Int. J. Med. Sci. 2013, 10, 995–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, I.; Joo, C.; Kim, Y.-K.; Ha, M.; Yoon, M.-J.; Cho, J.; Yeom, K.-H.; Han, J.; Kim, V.N. TUT4 in Concert with Lin28 Suppresses MicroRNA Biogenesis through Pre-MicroRNA Uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, Z.; Feng, Z.; Wang, J.; Ouyang, C.; Liu, W.; Fu, B.; Cai, G.; Wu, C.; Wei, R.; et al. Integrin-Linked Kinase Induces Both Senescence-Associated Alterations and Extracellular Fibronectin Assembly in Aging Cardiac Fibroblasts. J. Gerontol. Ser. A 2006, 61, 1232–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | BMR | Mouse | p-Value of BMRs | p-Value of Mice |
|---|---|---|---|---|
| Mmp12 | ↓ | − | 2.09 × 10−3 | 7.93 × 10−1 |
| Serpine1 | ↓ | ↑ | 9.18 × 10−29 | 1.29 × 10−3 |
| Mmp11 | ↓ | ↑ | 8.69 × 10−1 | 2.24 × 10−2 |
| Mmp9 | ↓ | ↓ | 3.32 × 10−124 | 2.09 × 10−2 |
| Serpinb2 | ↓ | ↓ | 3.96 × 10−28 | 4.68 × 10−26 |
| Il1β | ↑ | − | 1.95 × 10−23 | 7.64 × 10−1 |
| Il1α | ↑ | ↓ | 1.33 × 10−236 | 6.78 × 10−8 |
| Icam1 | ↑ | − | 1.42 × 10−11 | 4.88 × 10−1 |
| Ifnβ | ↑ | − | 8.11 × 10−3 | − |
| Il10 | ↓ | − | 8.64 × 10−11 | 2.78 × 10−1 |
| Jak1 | ↑ | − | 1.31 × 10−2 | 2.83 × 10−1 |
| Jak2 | ↑ | − | 3.50 × 10−1 | 8.41 × 10−1 |
| Stat1 | ↑ | ↑ | 2.23 × 10−1 | 6.84 × 10−3 |
| Stat2 | ↑ | − | 2.02 × 10−12 | 2.28 × 10−1 |
| Stat3 | ↓ | − | 5.96 × 10−7 | 3.44 × 10−1 |
| Stat4 | ↓ | ↑ | 8.64 × 10−11 | 4.94 × 10−3 |
| Stat6 | ↓ | ↓ | 2.40 × 10−5 | 3.40 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inci, N.; Akyildiz, E.O.; Bulbul, A.A.; Turanli, E.T.; Akgun, E.; Baykal, A.T.; Colak, F.; Bozaykut, P. Transcriptomics and Proteomics Analyses Reveal JAK Signaling and Inflammatory Phenotypes during Cellular Senescence in Blind Mole Rats: The Reflections of Superior Biology. Biology 2022, 11, 1253. https://doi.org/10.3390/biology11091253
Inci N, Akyildiz EO, Bulbul AA, Turanli ET, Akgun E, Baykal AT, Colak F, Bozaykut P. Transcriptomics and Proteomics Analyses Reveal JAK Signaling and Inflammatory Phenotypes during Cellular Senescence in Blind Mole Rats: The Reflections of Superior Biology. Biology. 2022; 11(9):1253. https://doi.org/10.3390/biology11091253
Chicago/Turabian StyleInci, Nurcan, Erdogan Oguzhan Akyildiz, Abdullah Alper Bulbul, Eda Tahir Turanli, Emel Akgun, Ahmet Tarik Baykal, Faruk Colak, and Perinur Bozaykut. 2022. "Transcriptomics and Proteomics Analyses Reveal JAK Signaling and Inflammatory Phenotypes during Cellular Senescence in Blind Mole Rats: The Reflections of Superior Biology" Biology 11, no. 9: 1253. https://doi.org/10.3390/biology11091253
APA StyleInci, N., Akyildiz, E. O., Bulbul, A. A., Turanli, E. T., Akgun, E., Baykal, A. T., Colak, F., & Bozaykut, P. (2022). Transcriptomics and Proteomics Analyses Reveal JAK Signaling and Inflammatory Phenotypes during Cellular Senescence in Blind Mole Rats: The Reflections of Superior Biology. Biology, 11(9), 1253. https://doi.org/10.3390/biology11091253