
Intrinsically Fluorescent Anti-Cancer Drugs




Abstract
:Simple Summary
Abstract
1. Introduction
2. Protein Kinase Inhibitors
3. Fluorescence, Tyrosine-Kinase Inhibitors and Beyond
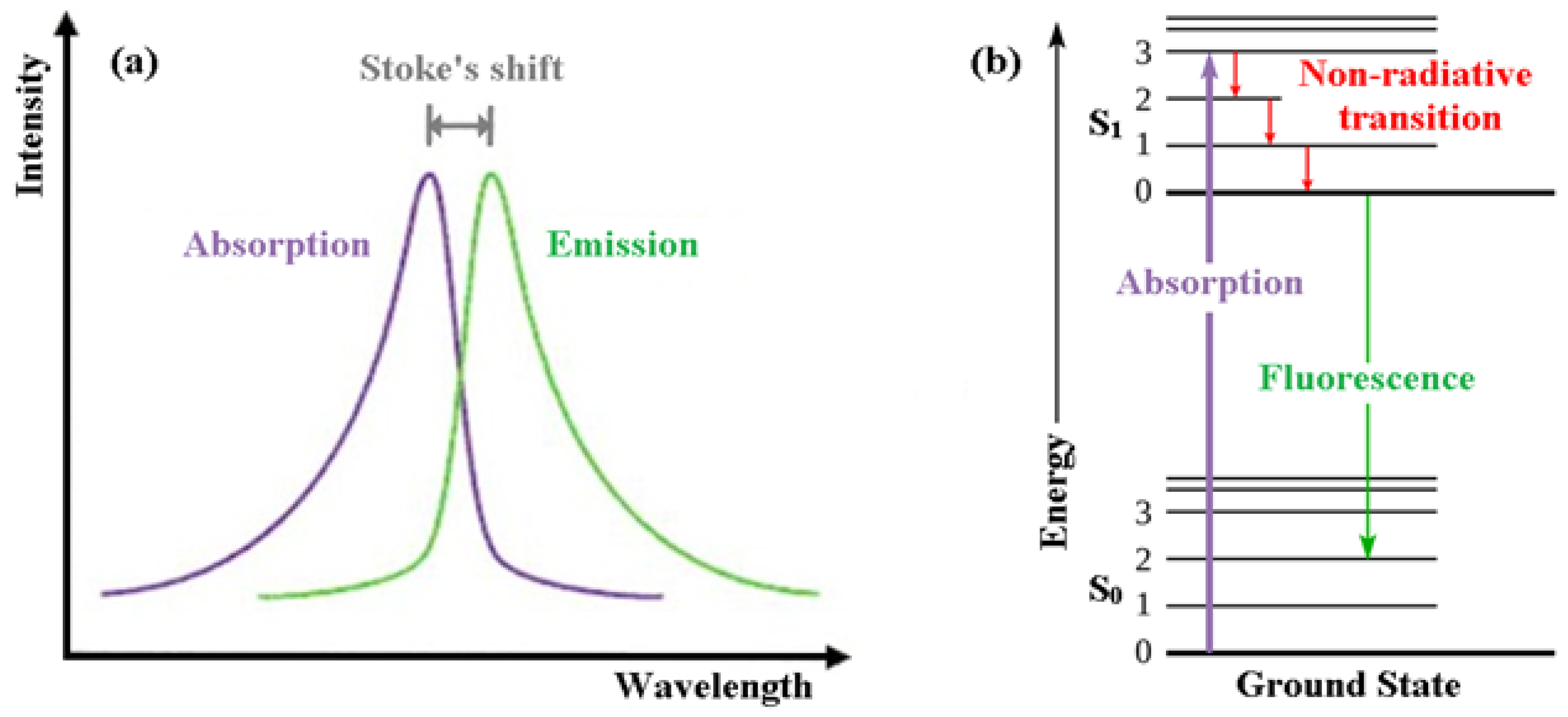
3.1. What Is Fluorescence?
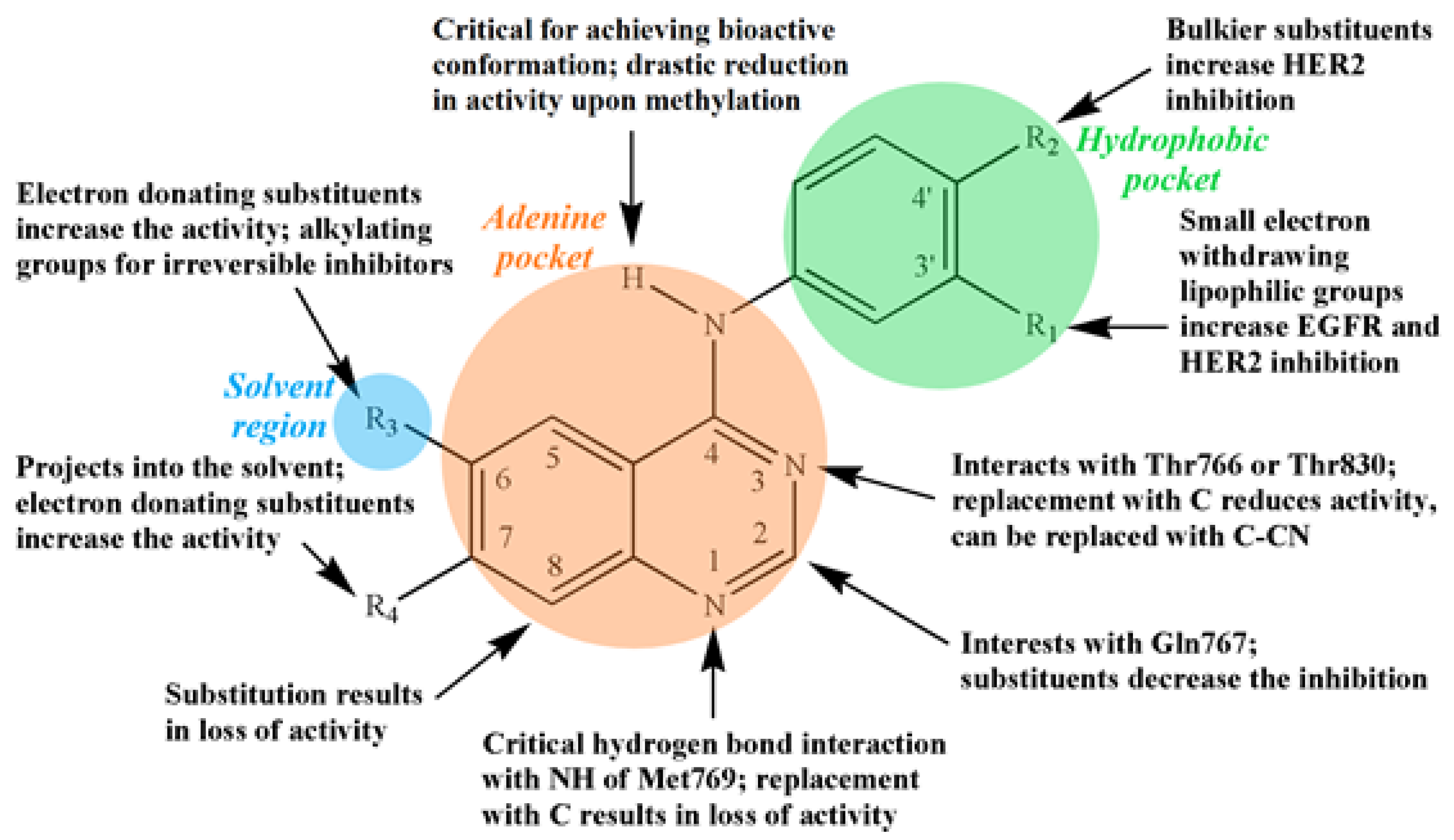
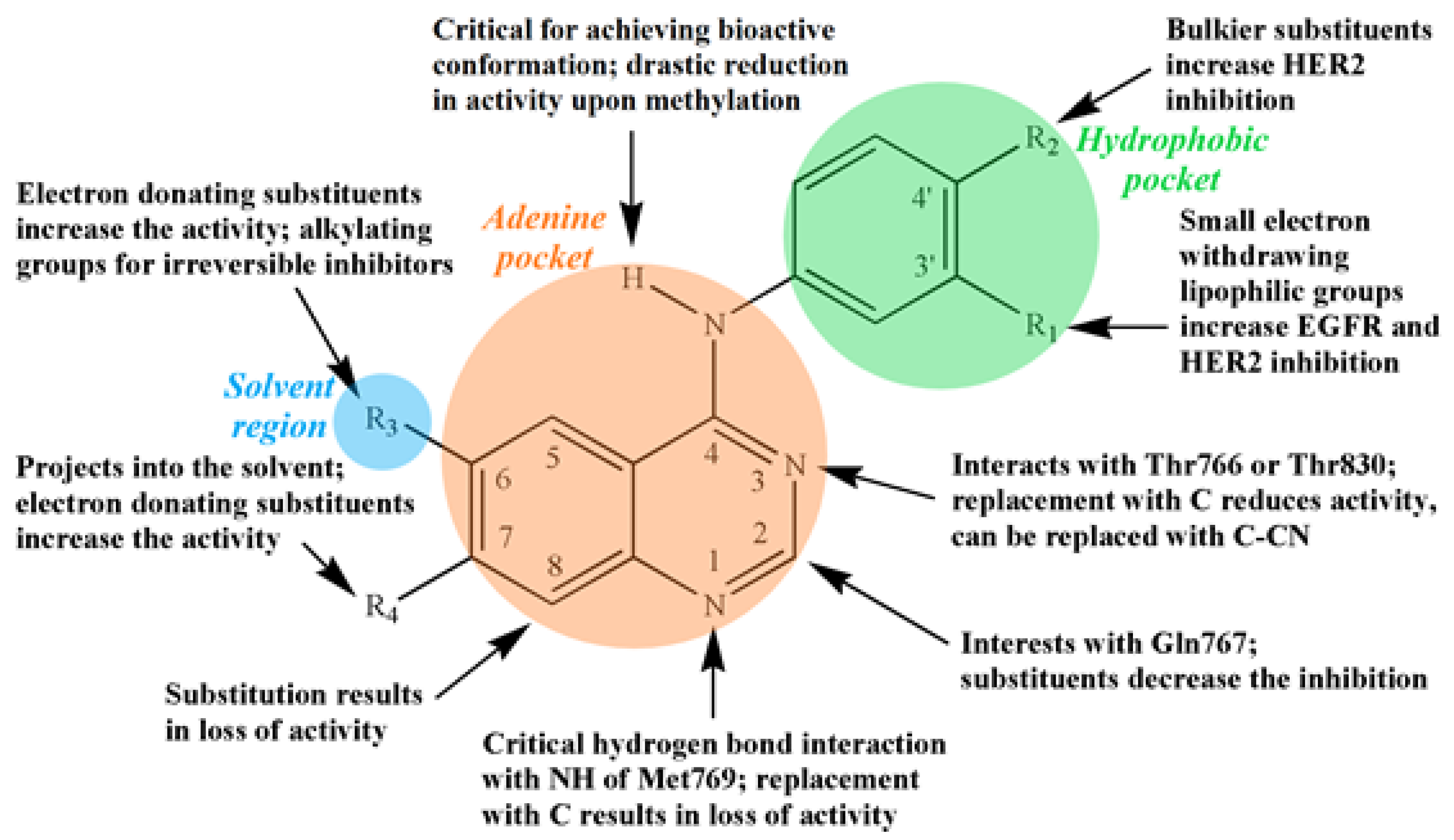
3.2. Quinazoline-Based TKI
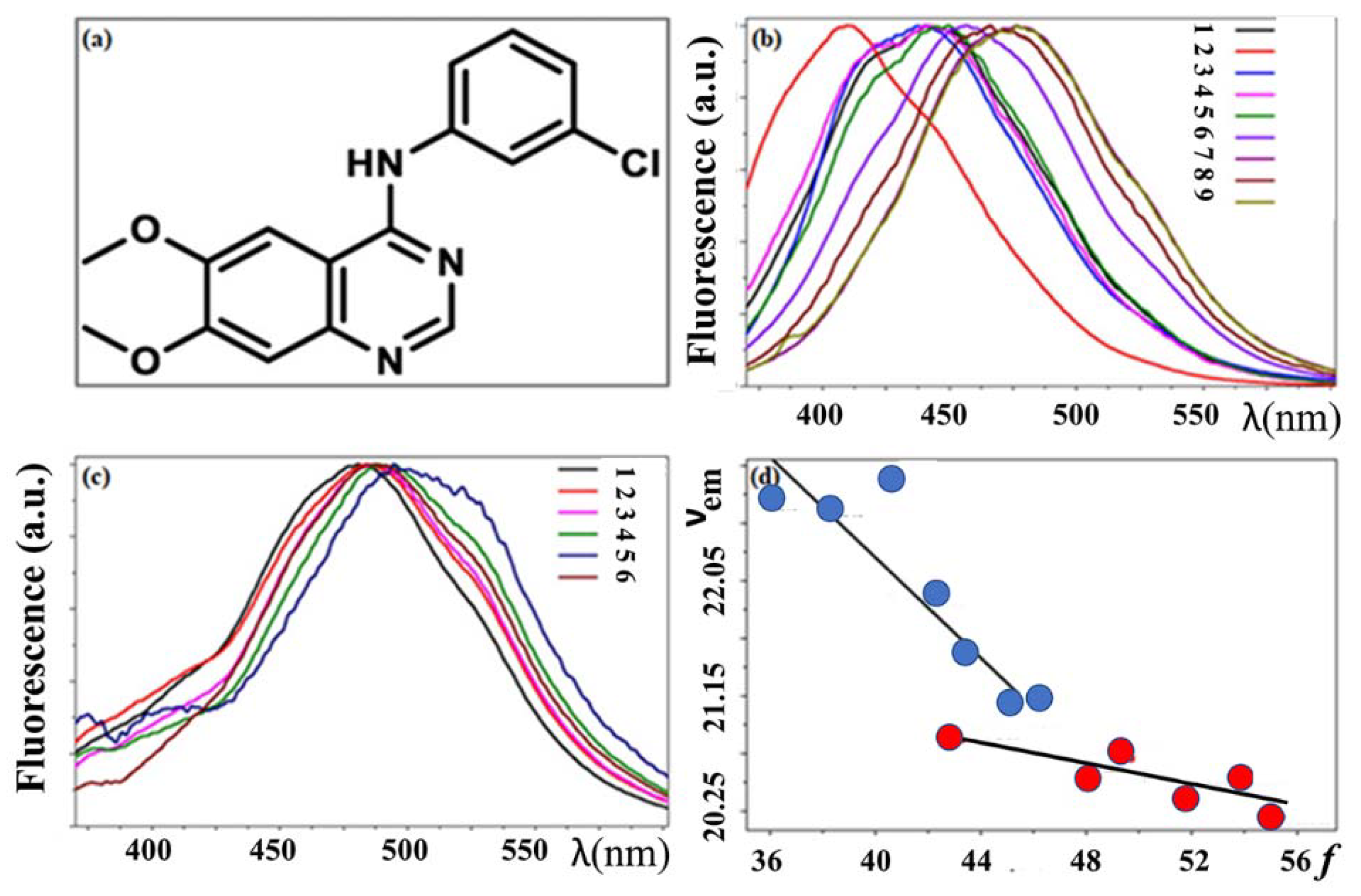
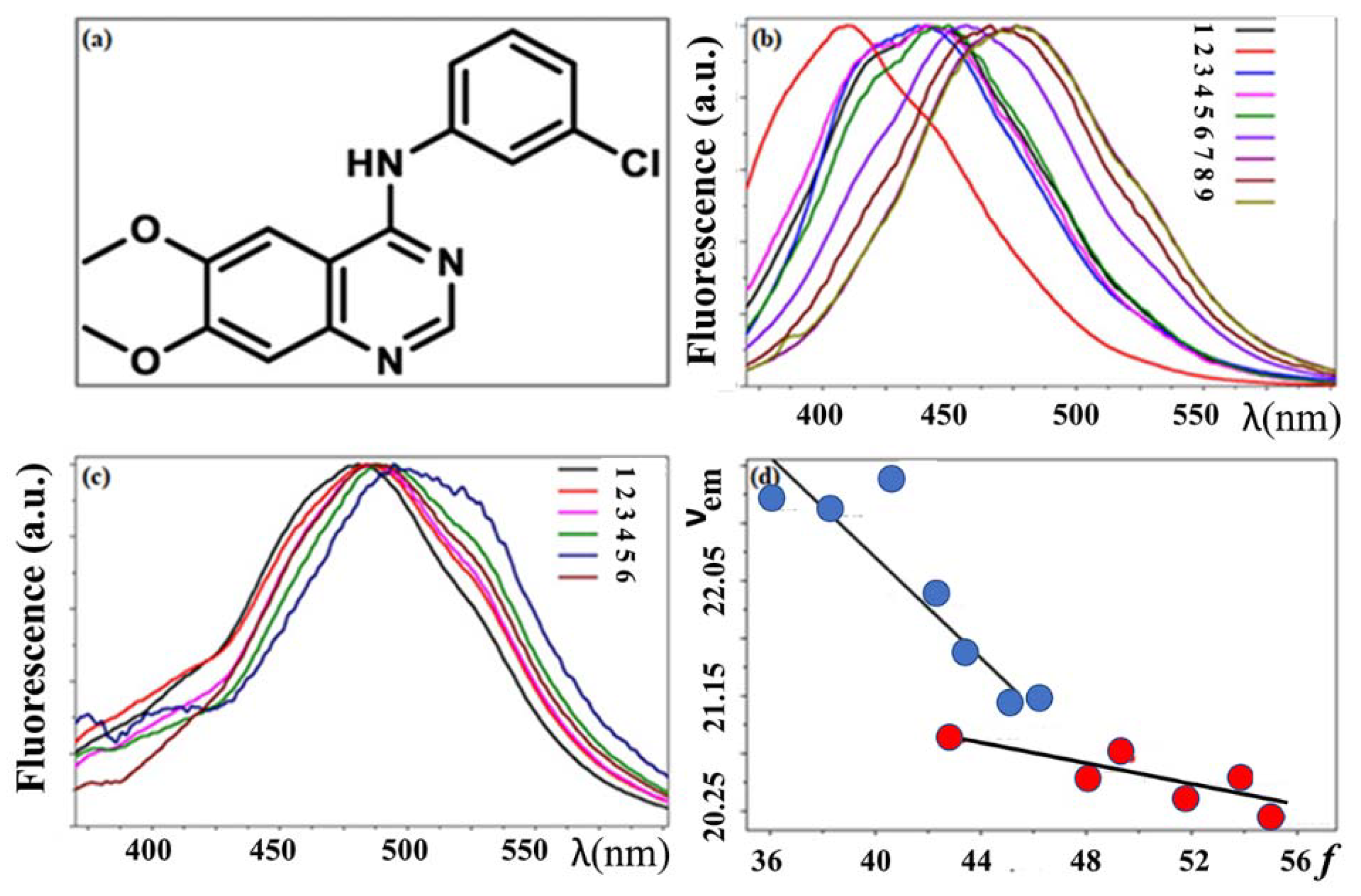
3.3. Basic Fluorescence Spectroscopy of TKIs
3.4. Fluorescence Binding Assay
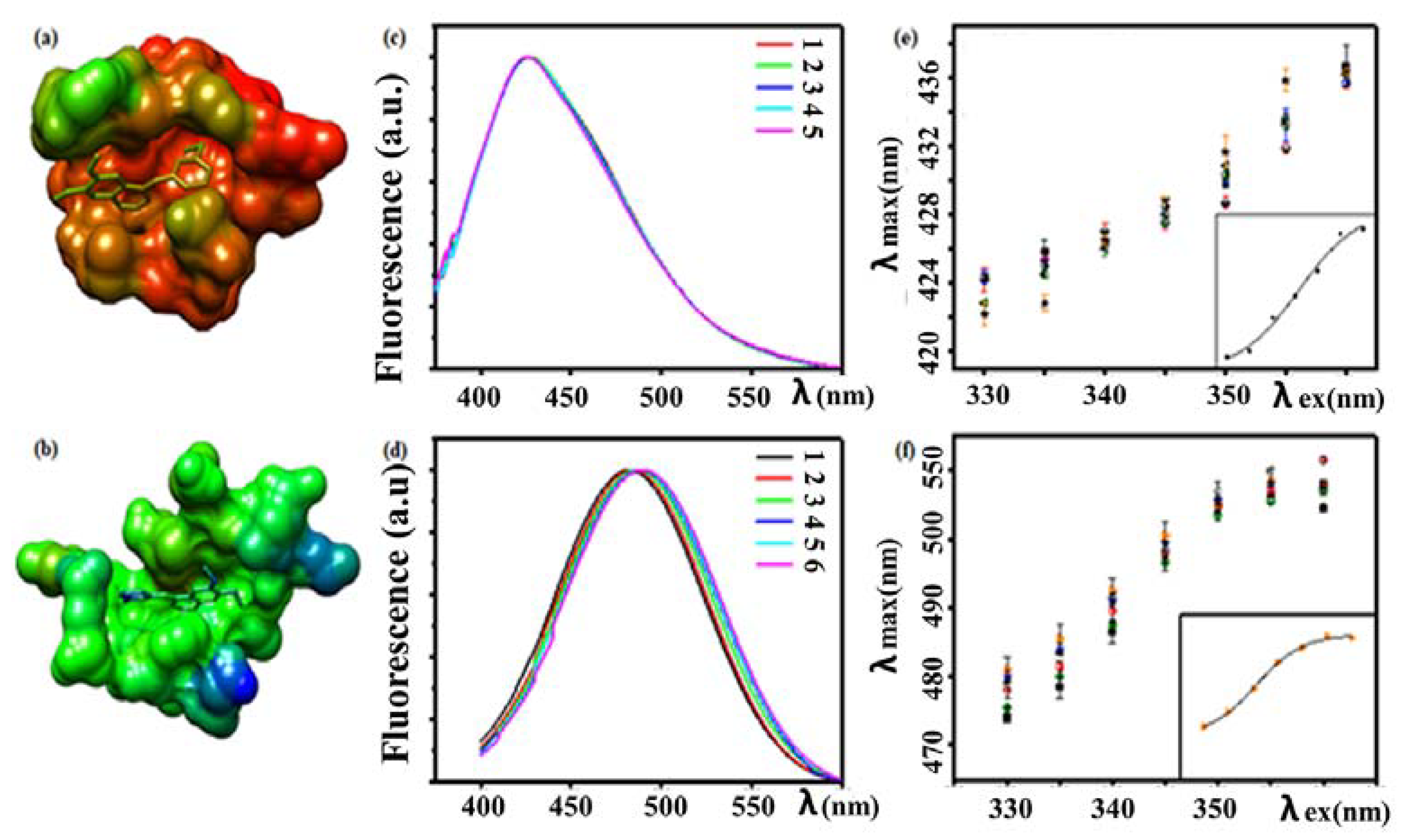
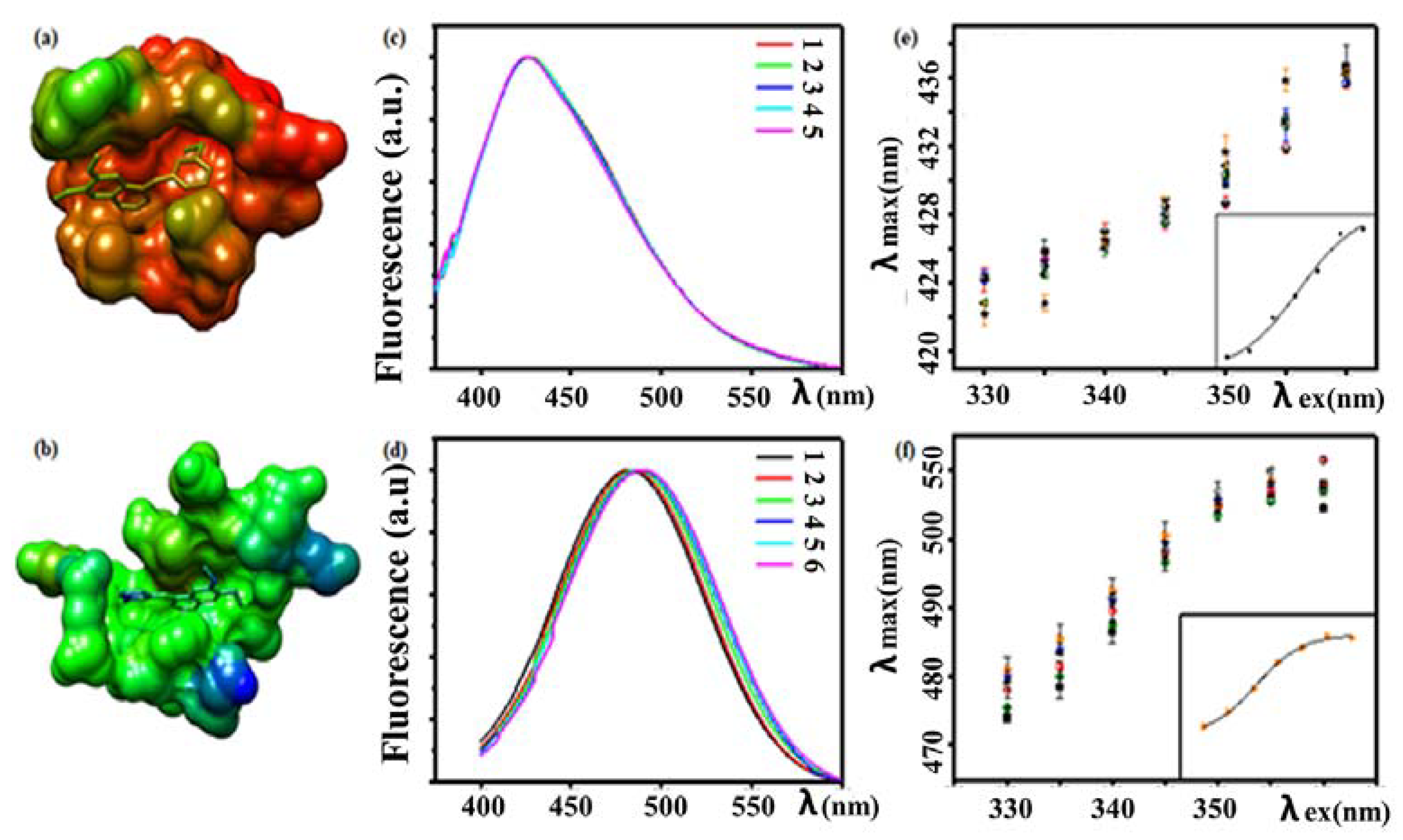
3.4.1. Fluorescence Analysis of Drug–Protein Interactions
3.4.2. Fluorescence Analysis of Drug–DNA Interactions
3.4.3. Fluorescence Analysis of Drug–Lipid Interactions
3.4.4. Fluorescence Analysis of Drug–Cell Interactions
3.4.5. Towards Pre-Clinical In Vivo Models for Cancer Research
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanese, S.K.; Parton, D.L.; Işlk, M.; Rodríguez-Laureano, L.; Hanson, S.M.; Behr, J.M.; Gradia, S.; Jeans, C.; Levinson, N.M.; Seeliger, M.A.; et al. An Open Library of Human Kinase Domain Constructs for Automated Bacterial Expression. Biochemistry 2018, 57, 4675–4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E. Chemistry and pharmacology of anticancer drugs. Chem. Pharmacol. Anticancer Drugs 2007, 97, 1713. [Google Scholar] [CrossRef] [Green Version]
- Juan, O.; Popat, S. Treatment choice in epidermal growth factor receptor mutation-positive non-small cell lung carcinoma: Latest evidence and clinical implications. Ther. Adv. Med. Oncol. 2017, 9, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Control. Release 2012, 157, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Product Monograph Including Patient Medication Information Gefitinib Tablets (IRESSA), (n.d.). Available online: https://www.astrazeneca.ca/content/dam/az-ca/downloads/productinformation/iressa-product-monograph-en.pdf (accessed on 6 January 2022).
- Product Monograph Erlotinib Hydrochloride Tablets (TARCEVA), (n.d.). Available online: http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tarceva/Tarceva_PM_E.pdf (accessed on 6 January 2022).
- Product Monograph Afatinib Tablets (Giotrif), (n.d.). Available online: https://www.boehringer-ingelheim.ca/sites/ca/files/documents/giotrifpmen.pdf (accessed on 6 January 2022).
- Product Monograph Including Patient Medication Information Osimertinib Tablets (TAGRISSO), (n.d.). Available online: https://www.astrazeneca.ca/content/dam/az-ca/downloads/productinformation/tagrisso-product-monograph-en.pdf (accessed on 6 January 2022).
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006; 954p. [Google Scholar] [CrossRef]
- Rye, H.S.; Dabora, J.M.; Quesada, M.A.; Mathies, R.A.; Glazer, A.N. Fluorometric assay using dimeric dyes for double- and single-stranded DNA and RNA with picogram sensitivity. Anal. Biochem. 1993, 208, 144–150. [Google Scholar] [CrossRef]
- Ungnade, H.E. The Effect of Solvents on the Absorption Spectra of Aromatic Compounds. J. Am. Chem. Soc. 1953, 75, 432–434. [Google Scholar] [CrossRef]
- Novaki, L.P.; El Seoud, O.A. Solvatochromism in aqueous micellar solutions: Effects of the molecular structures of solvatochromic probes and cationic surfactants. Phys. Chem. Chem. Phys. 1999, 1, 1957–1964. [Google Scholar] [CrossRef]
- Matsui, M.; Yamamoto, T.; Kakitani, K.; Biradar, S.; Kubota, Y.; Funabiki, K. UV–vis absorption and fluorescence spectra, solvatochromism, and application to pH sensors of novel xanthene dyes having thienyl and thieno[3,2-b]thienyl rings as auxochrome. Dye. Pigment. 2017, 139, 533–540. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Dempcy, R.O.; Skibo, E.B. Rational Design of Quinazoline-Based Irreversible Inhibitors of Human Erythrocyte Purine Nucleoside Phosphorylase. Biochemistry 1991, 30, 8480–8487. [Google Scholar] [CrossRef]
- Davoll, J.; Johnson, A.M. Quinazoline Analogues of Folic Acid. J. Chem. Soc. 1970, 8, 997–1002. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Medchemcomm 2017, 8, 871–885. [Google Scholar] [CrossRef]
- Shagufta, S.; Ahmad, I. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef]
- Al-Obeidi, F.A.; Lam, K.S. Development of inhibitors for protein tyrosine kinases. Oncogene 2000, 19, 5690–5701. [Google Scholar] [CrossRef] [Green Version]
- Dhuguru, J.; Liu, W.; Gonzalez, W.G.; Babinchak, W.M.; Miksovska, J.; Landgraf, R.; Wilson, J.N. Emission tuning of fluorescent kinase inhibitors: Conjugation length and substituent effects. J. Org. Chem. 2014, 79, 4940–4947. [Google Scholar] [CrossRef]
- Ismail, R.S.; Ismail, N.S.; Abuserii, S.; Abou El Ella, D.A. Recent advances in 4-aminoquinazoline based scaffold derivatives targeting EGFR kinases as anticancer agents. Futur. J. Pharm. Sci. 2016, 2, 9–19. [Google Scholar] [CrossRef]
- Johansson, M.H. Reversible Michael Additions: Covalent Inhibitors and Prodrugs. Mini-Rev. Med. Chem. 2012, 12, 1330–1344. [Google Scholar] [CrossRef]
- Marini, A.; Muñoz-Losa, A.; Biancardi, A.; Mennucci, B. What is solvatochromism? J. Phys. Chem. B 2010, 17128–17135. [Google Scholar] [CrossRef]
- Bilot, V.L.; Kawski, A. Zur Theorie des Einflusses von Lösungsmitteln auf die Elektronenspektren der Moleküle. Z. Naturforsch. A 1962, 17, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Lippert, E. Habilitationsschrift Zur Erlangung Der Lehrberichtigung (Venia Legendi) Fur Das Fach Physikalische Chemie Ander Techischen-Hochschule-Stuttgart—Spektroskopische Bestimmung Des Dipolmomentes Aromatischer Verbindungen Im Ersten Angeregten Singulettzustand. Z. Fur Elektrochem. 1957, 61, 962–975. [Google Scholar]
- Mataga, N.; Kaifu, Y.; Koizumi, M. Solvent Effects upon Fluorescence Spectra and the Dipole moments of Excited Molecules. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Bakhshiev, N.G. Universal Intermolecular Interactions and Their Effect on the Position of the Electronic Spectra of Molecules in Two-Component Solutions. VII. Theory (General Case of an Isotropic Solution). Opt. Spectrosc. 1964, 16, 446. [Google Scholar]
- Kawski, A. On the Estimation of Excited-State Dipole Moments from Solvatochromic Shifts of Absorption and Fluorescence Spectra. Z. Nat. A 2002, 57, 255–262. [Google Scholar] [CrossRef]
- Levinson, N.M.; Boxer, S.G. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS ONE 2012, 7, e29828. [Google Scholar] [CrossRef]
- Assessment Report Gefitinib Mylan; EMA/587444/2018; The European Medicines Agency: Amsterdam, The Netherlands, 2018; p. 44.
- Dömötör, O.; Pelivan, K.; Borics, A.; Keppler, B.K.; Kowol, C.R.; Enyedy, É.A. Comparative studies on the human serum albumin binding of the clinically approved EGFR inhibitors gefitinib, erlotinib, afatinib, osimertinib and the investigational inhibitor KP2187. J. Pharm. Biomed. Anal. 2018, 154, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Morales-Vilchis, M.G.; Flores-Sánchez, P.; Escalante, J. Identification, synthesis and structure assignment of two impurities of Erlotinib, a drug used for the treatment of some types of cancer. J. Mex. Chem. Soc. 2019, 63, 43–49. [Google Scholar] [CrossRef]
- Wilson, J.N.; Liu, W.; Brown, A.S.; Landgraf, R. Binding-induced, turn-on fluorescence of the EGFR/ERBB kinase inhibitor, lapatinib. Org. Biomol. Chem. 2015, 13, 5006–5011. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Zhang, L. Analysis of Positional Isomers of Lapatinib with Agilent Poroshell 120 PFP Columns, Agilent Technical Note. Available online: https://www.agilent.com/cs/library/applications/5991-4373EN.pdf (accessed on 6 January 2022).
- Patil Smita, J.; Doijad Rajendra, C.; Dhumal Priya, P. Development of UV-spectrophotometric method for the determination of Imatinib Mesylate (ITM) in bulk and formulation. Asian J. Pharm. Clin. Res. 2013, 6, 54–57. [Google Scholar]
- Zidan, D.W.; Hassan, W.S.; Elmasry, M.S.; Shalaby, A.A. A novel spectrofluorimetric method for determination of imatinib in pure, pharmaceutical preparation, human plasma, and human urine. Luminescence 2018, 33, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Liang, X. Fragmentation pathways and differentiation of positional isomers of sorafenib and structural analogues by ESI-IT-MSn and ESI-Q-TOF-MS/MS coupled with DFT calculations. J. Mass Spectrom. 2018, 53, 579–589. [Google Scholar] [CrossRef]
- Bondì, M.L.; Scala, A.; Sortino, G.; Amore, E.; Botto, C.; Azzolina, A.; Balasus, D.; Cervello, M.; Mazzaglia, A. Nanoassemblies Based on Supramolecular Complexes of Nonionic Amphiphilic Cyclodextrin and Sorafenib as Effective Weapons to Kill Human HCC Cells. Biomacromolecules 2015, 16, 3784–3791. [Google Scholar] [CrossRef]
- Kabir, M.L.; Backler, F.; Clayton, A.H.A.; Wang, F. Deducing the Conformational Properties of a Tyrosine Kinase Inhibitor in Solution by Optical Spectroscopy and Computational Chemistry. Front. Chem. 2020, 8, 596. [Google Scholar] [CrossRef]
- Fan, Y.F.; Zhang, W.; Zeng, L.; Lei, Z.N.; Cai, C.Y.; Gupta, P.; Yang, D.H.; Cui, Q.; Qin, Z.D.; Chen, Z.S.; et al. Dacomitinib antagonizes multidrug resistance (MDR) in cancer cells by inhibiting the efflux activity of ABCB1 and ABCG2 transporters. Cancer Lett. 2018, 421, 186–198. [Google Scholar] [CrossRef]
- Illendula, S.; Sumanjali, T.; Sandhya, D.; Lavanya, N.; Rao, G.K.; Rao, K.N.V.; Dutt, K.R. Method development and validation of AFATINIB in bulk and pharmaceutical dosage form by UV spectrophotometry. INDO Am. J. Pharm. Sci. 2018, 5, 1169–1173. [Google Scholar] [CrossRef]
- Alanazi, A.M.; Abdelhameed, A.S. A spectroscopic approach to investigate the molecular interactions between the newly approved irreversible ErbB blocker “Afatinib” and bovine serum albumin. PLoS ONE 2016, 11, e0146297. [Google Scholar] [CrossRef] [Green Version]
- Togami, K.; Fukuda, K.; Yamaguchi, K.; Chono, S.; Tada, H. Facile and sensitive HPLC-UV method for determination of nintedanib in rat plasma. Int. J. Pharm. Pharm. Sci. 2018, 10, 133–137. [Google Scholar] [CrossRef]
- Englinger, B.; Kallus, S.; Senkiv, J.; Heilos, D.; Gabler, L.; van Schoonhoven, S.; Terenzi, A.; Moser, P.; Pirker, C.; Timelthaler, G.; et al. Intrinsic fluorescence of the clinically approved multikinase inhibitor nintedanib reveals lysosomal sequestration as resistance mechanism in FGFR-driven lung cancer. J. Exp. Clin. Cancer Res. 2017, 36, 122. [Google Scholar] [CrossRef] [PubMed]
- Khattab, M.; Wang, F.; Clayton, A.H.A. UV-Vis spectroscopy and solvatochromism of the tyrosine kinase inhibitor AG-1478. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2016, 164, 128–132. [Google Scholar] [CrossRef] [PubMed]
- CArteaga, L.; Ramsey, T.T.; Shawver, L.K.; Guyer, C.A. Unliganded epidermal growth factor receptor dimerization induced by direct interaction of quinazolines with the ATP binding site. J. Biol. Chem. 1997, 272, 23247–23254. [Google Scholar] [CrossRef] [Green Version]
- Levitzki, A.; Gazit, A. Tyrosine kinase inhibition: An approach to drug development. Science 1995, 267, 1782–1788. [Google Scholar] [CrossRef]
- Khattab, M.; Wang, F.; Clayton, A.H.A. Micro-solvation of tyrosine-kinase inhibitor AG1478 explored with fluorescence spectroscopy and computational chemistry. RSC Adv. 2017, 7, 31725–31735. [Google Scholar] [CrossRef] [Green Version]
- Jameson, D.M.; Ross, J.A. Fluorescence polarization/anisotropy in diagnostics and imaging. Chem. Rev. 2010, 110, 2685–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Aulabaugh, A. Application of Fluorescence Polarization in HTS Assays. In High Throughput Screening; Humana Press: New York, NY, USA, 2016; Volume 1439, pp. 115–130. [Google Scholar] [CrossRef]
- Vayá, I.; Lhiaubet-Vallet, V.; Jiménez, M.C.; Miranda, M.A. Photoactive assemblies of organic compounds and biomolecules: Drug-protein supramolecular systems. Chem. Soc. Rev. 2014, 43, 4102–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Ruíz, R.; Lence, E.; Andreu, I.; Limones-Herrero, D.; González-Bello, C.; Miranda, M.A.; Jiménez, M.C. A New Pathway for Protein Haptenation by β-Lactams. Chem.-A Eur. J. 2017, 23, 13986–13994. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, S.; Curutchet, C. Can Förster Theory Describe Stereoselective Energy Transfer Dynamics in a Protein-Ligand Complex? J. Phys. Chem. B 2017, 121, 2265–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molins-Molina, O.; Pérez-Ruiz, R.; Lence, E.; González-Bello, C.; Miranda, M.A.; Consuelo Jiménez, M. Photobinding of triflusal to human serum albumin investigated by fluorescence, proteomic analysis, and computational studies. Front. Pharmacol. 2019, 10, 1028. [Google Scholar] [CrossRef]
- Spitaleri, A.; Rocchia, W. Molecular Dynamics-Based Approaches Describing Protein Binding. In Biomolecular Simulations in Structure-Based Drug Discovery; Wiley VCH: Weinheim, Germany, 2019; pp. 29–42. [Google Scholar] [CrossRef]
- Vayá, I.; Andreu, I.; Lence, E.; González-Bello, C.; Consuelo Cuquerella, M.; Navarrete-Miguel, M.; Roca-Sanjuán, D.; Miranda, M.A. Characterization of Locally Excited and Charge-Transfer States of the Anticancer Drug Lapatinib by Ultrafast Spectroscopy and Computational Studies. Chem.-A Eur. J. 2020, 26, 15922–15930. [Google Scholar] [CrossRef]
- Andreu, I.; Lence, E.; González-Bello, C.; Mayorga, C.; Cuquerella, M.C.; Vayá, I.; Miranda, M.A. Protein Binding of Lapatinib and Its N- and O-Dealkylated Metabolites Interrogated by Fluorescence, Ultrafast Spectroscopy and Molecular Dynamics Simulations. Front. Pharmacol. 2020, 11, 576495. [Google Scholar] [CrossRef]
- Khattab, M.; Wang, F.; Clayton, A.H.A. Conformational Plasticity in Tyrosine Kinase Inhibitor-Kinase Interactions Revealed with Fluorescence Spectroscopy and Theoretical Calculations. J. Phys. Chem. B 2018, 122, 4667–4679. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Chaudhuri, A.; Chattopadhyay, A. Organization and dynamics of membrane probes and proteins utilizing the red edge excitation shift. J. Phys. Chem. B 2011, 115, 5693–5706. [Google Scholar] [CrossRef]
- Catici, D.A.M.; Amos, H.E.; Yang, Y.; van den Elsen, J.M.H.; Pudney, C.R. The red edge excitation shift phenomenon can be used to unmask protein structural ensembles: Implications for NEMO–ubiquitin interactions. FEBS J. 2016, 283, 2272–2284. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, A.; Haldar, S. Dynamic insight into protein structure utilizing red edge excitation shift. Acc. Chem. Res. 2014, 47, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.L.; Wang, F.; Clayton, A.H.A. Red-edge excitation shift spectroscopy (Rees): Application to hidden bound states of ligands in protein–ligand complexes. Int. J. Mol. Sci. 2021, 22, 2582. [Google Scholar] [CrossRef] [PubMed]
- Levinson, N.M.; Boxer, S.G. A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat. Chem. Biol. 2014, 10, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.H.A.; Perugini, M.A.; Weinstock, J.; Rothacker, J.; Watson, K.G.; Burgess, A.W.; Nice, E.C. Fluorescence and analytical ultracentrifugation analyses of the interaction of the tyrosine kinase inhibitor, tyrphostin AG1478-mesylate, with albumin. Anal. Biochem. 2005, 342, 292–299. [Google Scholar] [CrossRef]
- Rasoulzadeh, F.; Asgari, D.; Naseri, A.; Rashidi, M.R. Spectroscopic studies on the interaction between erlotinib hydrochloride and bovine serum albumin. DARU J. Pharm. Sci. 2010, 18, 179–184. [Google Scholar]
- Trummer, B.J.; Iyer, V.; Balu-Iyer, S.V.; O’Connor, R.; Straubinger, R.M. Physicochemical properties of epidermal growth factor receptor inhibitors and development of a nanoliposomal formulation of gefitinib. J. Pharm. Sci. 2012, 101, 2763–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.Y.; Zhou, K.L.; Lou, Y.Y.; Shi, J.H. Exploring the binding interaction of calf thymus DNA with lapatinib, a tyrosine kinase inhibitor: Multi-spectroscopic techniques combined with molecular docking. J. Biomol. Struct. Dyn. 2019, 37, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.J.; Wang, B.L.; Kou, S.B.; Lin, Z.Y.; Zhou, K.L.; Lou, Y.Y.; Shi, J.H. Assessment on the binding characteristics of dasatinib, a tyrosine kinase inhibitor to calf thymus DNA: Insights from multi-spectroscopic methodologies and molecular docking as well as DFT calculation. J. Biomol. Struct. Dyn. 2020, 38, 4210–4220. [Google Scholar] [CrossRef]
- Chen, C.B.; Chen, J.; Wang, J.; Zhu, Y.Y.; Shi, J.H. Combined spectroscopic and molecular docking approach to probing binding interactions between lovastatin and calf thymus DNA. Luminescence 2015, 30, 1004–1010. [Google Scholar] [CrossRef]
- Karapetyan, N.H.; Torosyan, A.L.; Malakyan, M.; Bajinyan, S.A.; Haroutiunian, S.G. Investigation of irradiated rats DNA in the presence of Cu(II) chelates of amino acids Schiff bases. J. Biomol. Struct. Dyn. 2016, 34, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Palmajumder, E.; Sepay, N.; Mukherjea, K.K. Development of oxidovanadium and oxido-peroxido vanadium-based artificial DNA nucleases via multi spectroscopic investigations and theoretical simulation of DNA binding. J. Biomol. Struct. Dyn. 2018, 36, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Chen, J.; Wang, J.; Zhu, Y.Y. Binding interaction between sorafenib and calf thymus DNA: Spectroscopic methodology, viscosity measurement and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Chen, J.; Wang, J.; Zhu, Y.Y.; Wang, Q. Binding interaction of sorafenib with bovine serum albumin: Spectroscopic methodologies and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Liu, T.T.; Jiang, M.; Chen, J.; Wang, Q. Characterization of interaction of calf thymus DNA with gefitinib: Spectroscopic methods and molecular docking. J. Photochem. Photobiol. B Biol. 2015, 147, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Bi, S.; Wang, Y.; Wu, J. Characterization of the binding of neomycin/paromomycin sulfate with DNA using acridine orange as fluorescence probe and molecular docking technique. J. Biomol. Struct. Dyn. 2017, 35, 2077–2089. [Google Scholar] [CrossRef]
- Hegde, A.H.; Seetharamappa, J. Fluorescence and circular dichroism studies on binding and conformational aspects of an anti-leukemic drug with DNA. Mol. Biol. Rep. 2014, 41, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltammetry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Bhattacharya, P.; Chakravorti, S. Quest for mode of binding of 2-(4-(dimethylamino)styryl)-1-methylpyridinium iodide with calf thymus DNA. J. Phys. Chem. B 2010, 114, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Shahabadi, N.; Hadidi, S. Spectroscopic studies on the interaction of calf thymus DNA with the drug levetiracetam. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 96, 278–283. [Google Scholar] [CrossRef]
- Dogan-Topal, B.; Bozal-Palabiyik, B.; Ozkan, S.A.; Uslu, B. Investigation of anticancer drug lapatinib and its interaction with dsDNA by electrochemical and spectroscopic techniques. Sens. Actuators B Chem. 2014, 194, 185–194. [Google Scholar] [CrossRef]
- Goossens, J.F.; Bouey-Bencteux, E.; Houssin, R.; Hénichart, J.P.; Colson, P.; Houssier, C.; Laine, W.; Baldeyrou, B.; Bailly, C. Dna interaction of the tyrosine protein kinase inhibitor PD153035 and its N-methyl analogue. Biochemistry 2001, 40, 4663–4671. [Google Scholar] [CrossRef]
- Gallois, L.; Fiallo, M.; Laigle, A.; Priebe, W.; Garnier-Suillerot, A. The overall partitioning of anthracyclines into phosphatidyl-containing model membranes depends neither on the drug charge nor the presence of anionic phospholipids. Eur. J. Biochem. 1996, 241, 879–887. [Google Scholar] [CrossRef]
- de Wolf, F.A.; Demel, R.A.; Bets, D.; van Kats, C.; de Kruijff, B. Characterization of the interaction of doxorubicin with (poly)phosphoinositides in model systems Evidence for specific interaction with phosphatidylinositol-monophosphate and -diphosphate. FEBS Lett. 1991, 288, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.L.; Barceló-Coblijn, G.; De Almeida, R.F.M.; Noguera-Salvà, M.A.; Terés, S.; Higuera, M.; Liebisch, G.; Schmitz, G.; Busquets, X.; Escribá, P.V. The role of membrane fatty acid remodeling in the antitumor mechanism of action of 2-hydroxyoleic acid. Biochim. Biophys. Acta 2013, 1828, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Khosa, A.; Saha, R.N.; Singhvi, G. Drug delivery to the brain. In Nanomaterials for Drug Delivery and Therapy; William Andrew Applied Science Publishing: Norwich, NY, USA, 2019; pp. 461–514. [Google Scholar]
- Usama, S.M.; Zhao, B.; Burgess, K. Fluorescent kinase inhibitors as probes in cancer. Chem. Soc. Rev. 2021, 50, 9794–9816. [Google Scholar] [CrossRef]
- Solanki, A.; Wang, L.; Korber, J.; McMahon, N.; Tichauer, K.; Samkoe, K.S.; Gibbs, S.L. Intracellular paired agent imaging enables improved evaluation of tyrosine kinase inhibitor target engagement. Proc. SPIE Int. Soc. Opt. Eng. 2020, 11219, 23–28. [Google Scholar]
- Xu, C.; Zipfel, W.; Shear, J.B.; Williams, R.M.; Webb, W.W. Multiphoton fluorescence excitation: New spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 10763–10768. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (Intended Target) | Absorbance Max (nm) | Fluorescence Max (nm) |
|---|---|---|
| Sorafenib (Raf kinase) | (DCM/ACN) 1 264 nm | 487 nm [42,43] |
| Gefitinib (EGFR) | n-hexane (332 nm, 344 nm) | 368 nm, RI 7 = 1 [35,36] |
| Gefitinib (EGFR) | Benzene (332 nm, 340 nm) | 446 nm RI = 0.3 [35,36] |
| Gefitinib (EGFR) | n-octanol (334 nm, 344 nm) | 390–450 nm RI = 0.07 [35,36] |
| Gefitinib (EGFR) | Water (330 nm) | Non-fluorescent |
| Gefitinib (EGFR) | HSA (not reported) | 378 nm [35,36] |
| Erlotinib (EGFR) | n-hexane (332 nm, 344 nm) | 372 nm RI = 1 [36,37] |
| Erlotinib (EGFR) | Benzene (336 nm, 346 nm) | 423 nm RI = 0.38 [36,37] |
| Erlotinib (EGFR) | n-octanol (336 nm, 346 nm) | 400–470 nm RI = 0.07 [36,37] |
| Erlotinib (EGFR) | Water (332 nm) | Non-fluorescent [36,37] |
| Erlotinib (EGFR) | HAS 2 (336 nm, 347 nm) | 380–400 nm [36,37] |
| Lapatinib (EGFR, HER2) | THF 3 (380 nm) | 475 nm [38] |
| Lapatinib (EGFR, HER2) | Methanol (367 nm) | Non-fluorescent |
| Lapatinib (EGFR, HER2) | BSA 4 (361 nm) | 423 nm QY 8 = 0.07 [38,39] |
| Lapatinib (EGFR, HER2) | ErBB2 (368 nm) | 445 nm QY = 0.30 [38,39] |
| Lapatinib (EGFR, HER2) | Aggregates (371 nm) | 464 nm QY = 0.04 [38,39] |
| Afatinib | Methanol (246 nm, 340nm) | Not reported [46,47] |
| Imatinib (Abl kinase) | Water (281 nm) | 307nm [40,41] |
| Dacomitinib (mutated EGFR) | DMSO 5 (343 nm) | 500 nm 9 [44,45] |
| Nintedanib | PBS 6/DMSO (390 nm) | 482 nm [48,49] |
| Bosutinib (Abl kinase) | 350 nm | 480 nm [34] |
| Drug (Intended Target) | DNA Binding Mode | Binding Affinity (Ref.) |
|---|---|---|
| Sorafenib (Raf kinase) | Minor groove | 5.6 × 103 M−1 [77] |
| Gefitinib (EGFR) | Minor groove | 1.0 × 104 M−1 [79] |
| Lapatinib (EGFR, HER2) | Minor groove | 1.0 × 104 M−1 [72] |
| Imatinib (Abl kinase) | Inter-collator | 7 × 103 M−1 [81] |
| PD153035 (EGFR) | Inter-collator | Weak [86] |
| EBE-A22 (cytotoxic) | Inter-collator | 2 × 104 M−1 [86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabir, M.L.; Wang, F.; Clayton, A.H.A. Intrinsically Fluorescent Anti-Cancer Drugs. Biology 2022, 11, 1135. https://doi.org/10.3390/biology11081135
Kabir ML, Wang F, Clayton AHA. Intrinsically Fluorescent Anti-Cancer Drugs. Biology. 2022; 11(8):1135. https://doi.org/10.3390/biology11081135
Chicago/Turabian StyleKabir, Md. Lutful, Feng Wang, and Andrew H. A. Clayton. 2022. "Intrinsically Fluorescent Anti-Cancer Drugs" Biology 11, no. 8: 1135. https://doi.org/10.3390/biology11081135
APA StyleKabir, M. L., Wang, F., & Clayton, A. H. A. (2022). Intrinsically Fluorescent Anti-Cancer Drugs. Biology, 11(8), 1135. https://doi.org/10.3390/biology11081135