Enhanced Adipogenic Differentiation of Human Dental Pulp Stem Cells in Enzymatically Decellularized Adipose Tissue Solid Foams

 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
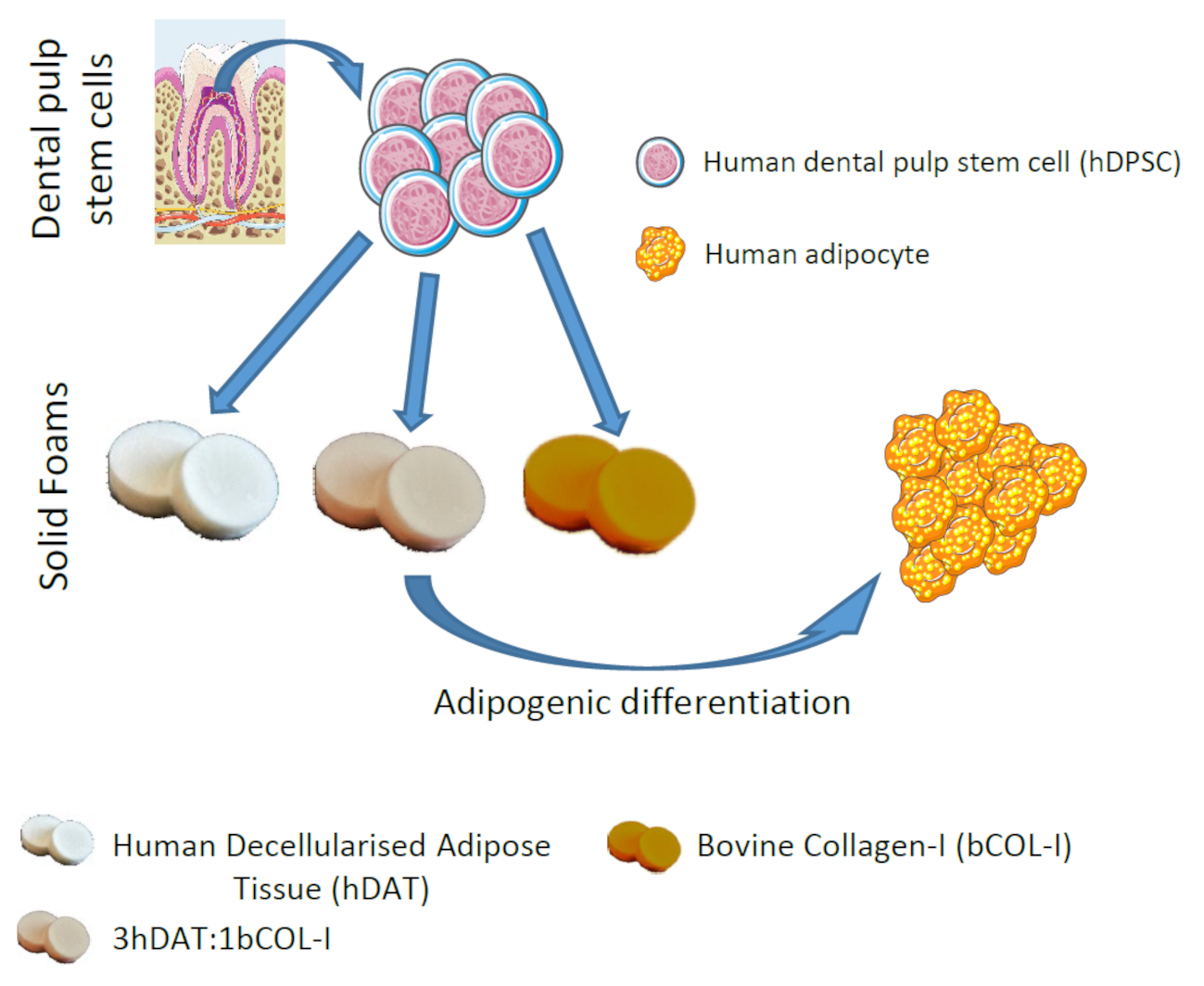
2.1. Human Adipose Tissue Decellularization
2.2. Characterization of hDAT
2.3. hDAT Processing as Solid Foam
2.4. Scanning Electron Microscopy (SEM)
2.5. Swelling Properties
2.6. Mechanical Test
2.7. Degradation Test
2.8. hDPSC In Vitro Culture
2.9. hDPSC Viability and Proliferation
2.10. Adipogenic Differentiation of hDPSC
2.11. RNA Extraction and qRT-PCR
2.12. PPAR-Gamma Immunofluorescence and Quantification
2.13. Oil Red Staining and Quantification
2.14. Statistical Analysis
3. Results
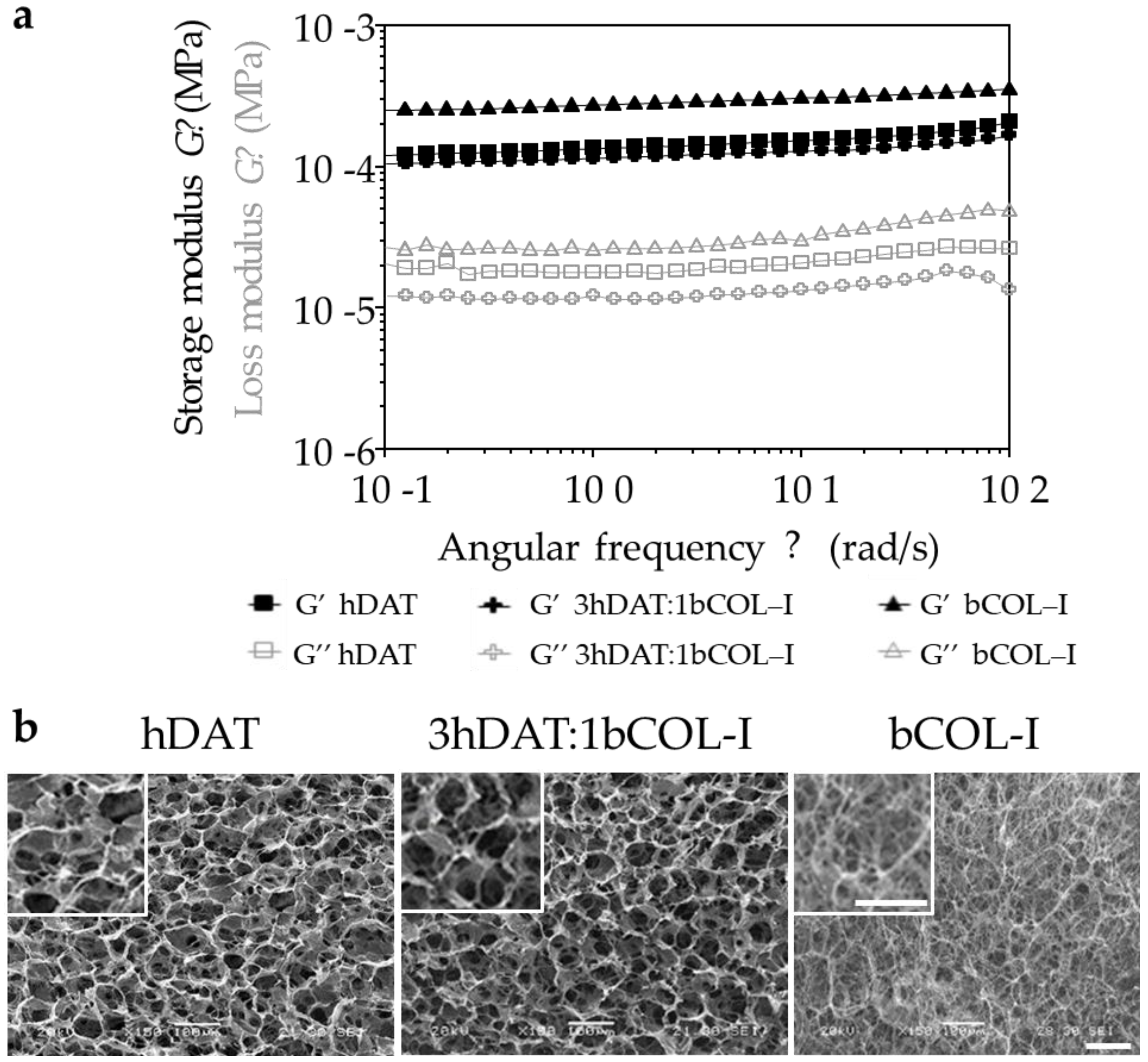
3.1. hDAT and 3hDAT:1bCOL-I Solid Foams Degradation Is Slower Than bCOL-I
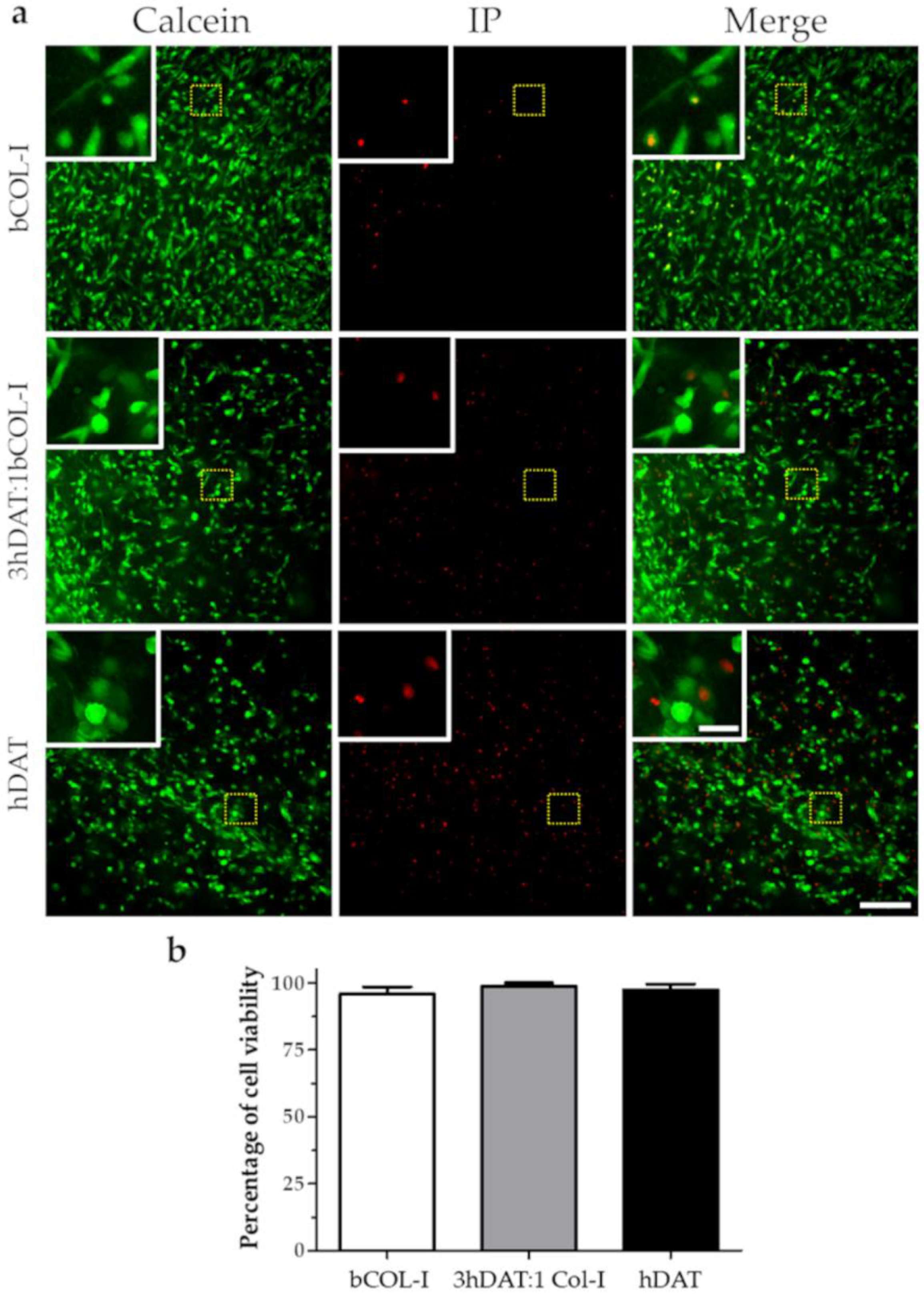
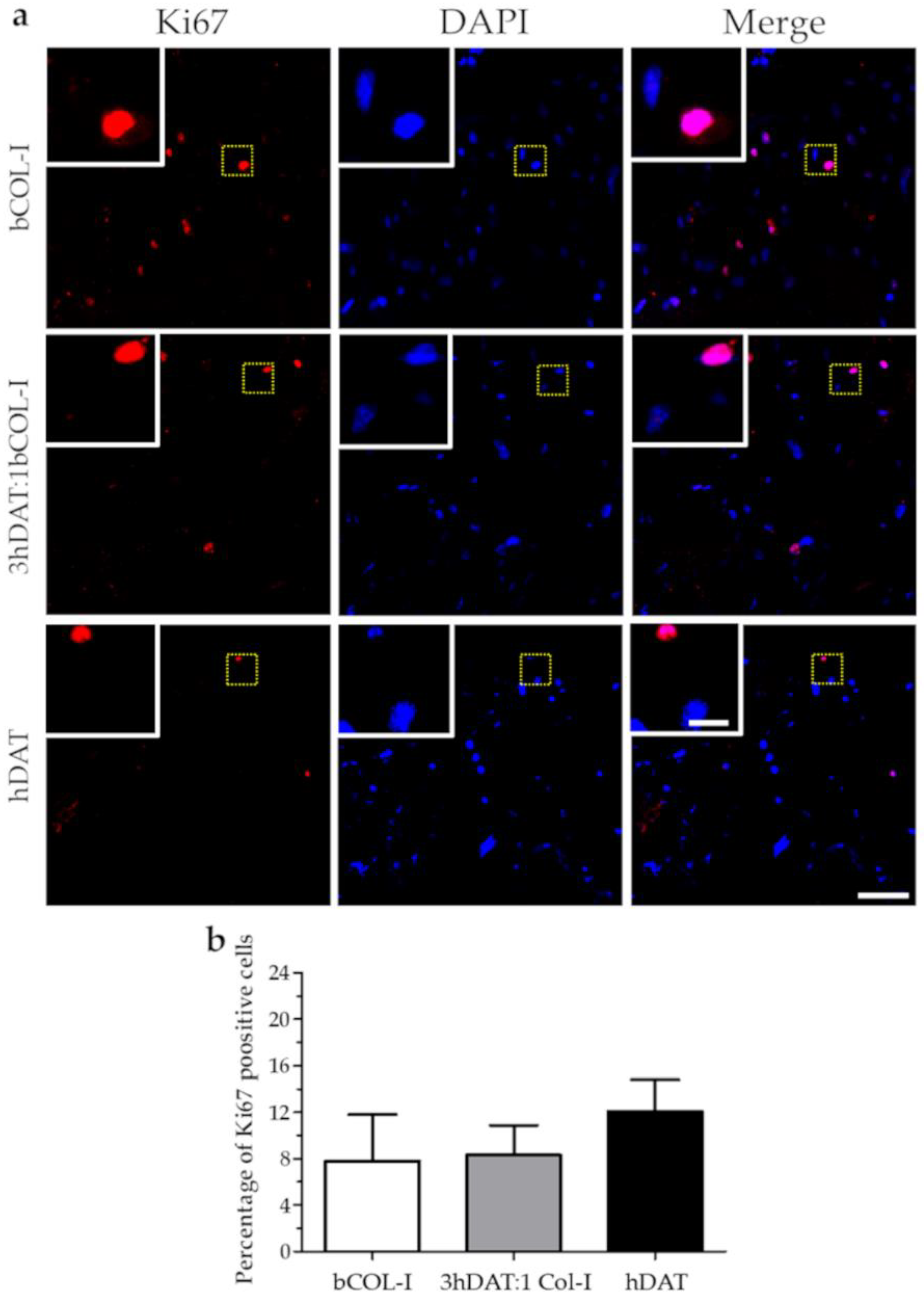
3.2. hDPSC Viability and Proliferation
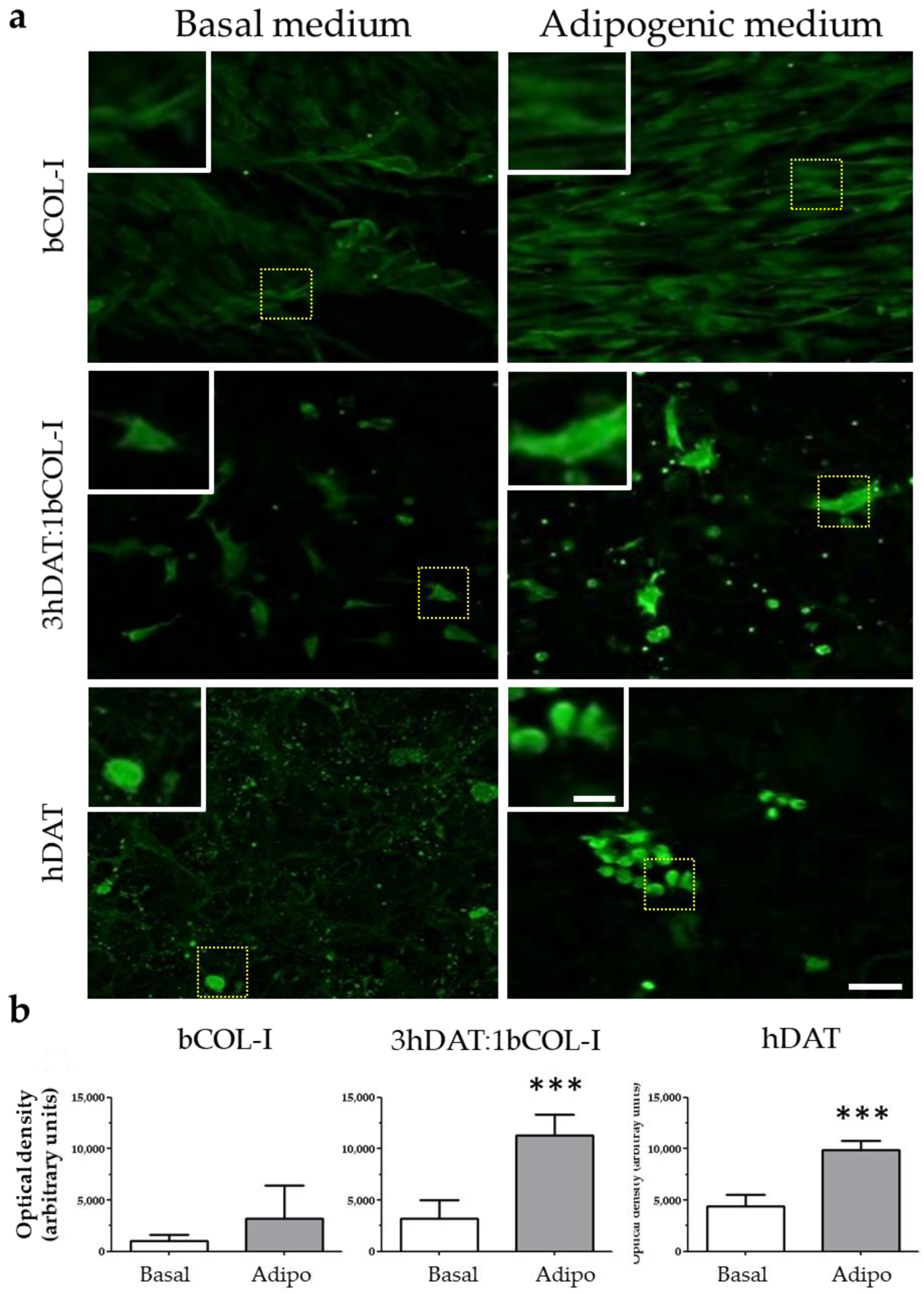
3.3. hDPSCs Cultured on Solid Foams Expressed Adipogenic Markers
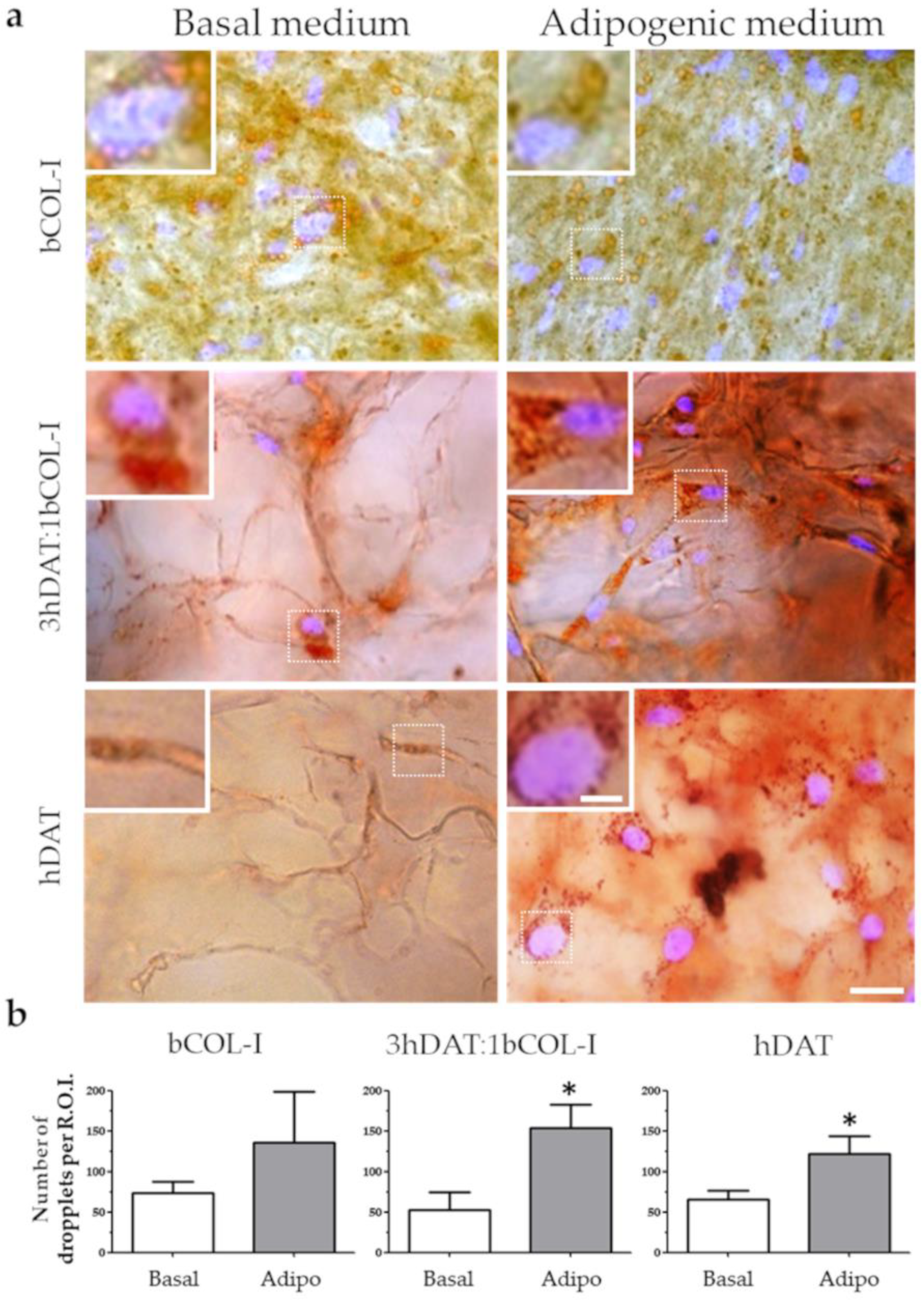
3.4. PPAR-γ Immunofluorescence and Oil Red Staining
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hajer, G.R.; van Haeften, T.W.; Visseren, F.L.J. Adipose Tissue Dysfunction in Obesity, Diabetes, and Vascular Diseases. Eur. Heart J. 2008, 29, 2959–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelin, G.; Silveira, A.L.M.; Martins, L.B.; Ferreira, A.V.; Clément, K. Deciphering the Cellular Interplays Underlying Obesity-Induced Adipose Tissue Fibrosis. J. Clin. Investig. 2019, 129, 4032–4040. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose Tissue Plasticity: How Fat Depots Respond Differently to Pathophysiological Cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [Green Version]
- Obesity. Available online: https://www.who.int/news-room/facts-in-pictures/detail/6-facts-on-obesity (accessed on 7 June 2022).
- Tremmel, M.; Gerdtham, U.-G.; Nilsson, P.M.; Saha, S. Economic Burden of Obesity: A Systematic Literature Review. Int. J. Environ. Res. Public Health 2017, 14, 435. [Google Scholar] [CrossRef]
- Finkelstein, E.A.; Khavjou, O.A.; Thompson, H.; Trogdon, J.G.; Pan, L.; Sherry, B.; Dietz, W. Obesity and Severe Obesity Forecasts through 2030. Am. J. Prev. Med. 2012, 42, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Bellas, E.; Vunjak-Novakovic, G.; Kaplan, D.L. Adipogenic Differentiation of Human Adipose-Derived Stem Cells on 3D Silk Scaffolds. Methods Mol. Biol. 2011, 702, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, J.C.; Lin, Y.-C.; Brayfield, C.A.; Minteer, D.M.; Li, H.; Rubin, J.P.; Marra, K.G. Adipogenesis of Human Adipose-Derived Stem Cells within Three-Dimensional Hollow Fiber-Based Bioreactors. Tissue Eng. Part. C Methods 2012, 18, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Carmona, A.; Stojkova, K.; Garcia Huitron, E.I.; Goddi, A.; Bhushan, A.; Cohen, R.N.; Brey, E.M. A 3D Human Adipose Tissue Model within a Microfluidic Device. Lab Chip 2021, 21, 435–446. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide Trends in Body-Mass Index, Underweight, Overweight, and Obesity from 1975 to 2016: A Pooled Analysis of 2416 Population-Based Measurement Studies in 128·9 Million Children, Adolescents, and Adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.; Brown, T.; Alarcon, A.; Williams, C.; Wu, X.; Abbott, R.D.; Gimble, J.; Frazier, T. Fat-On-A-Chip Models for Research and Discovery in Obesity and Its Metabolic Comorbidities. Tissue Eng. Part. B Rev. 2020, 26, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Abbott, R.D.; Zieba, A.; Borowsky, F.E.; Kaplan, D.L. Development of a Three-Dimensional Adipose Tissue Model for Studying Embryonic Exposures to Obesogenic Chemicals. Ann. Biomed. Eng. 2017, 45, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharm. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Gibler, P.; Gimble, J.; Hamel, K.; Rogers, E.; Henderson, M.; Wu, X.; Olesky, S.; Frazier, T. Human Adipose-Derived Stromal/Stem Cell Culture and Analysis Methods for Adipose Tissue Modeling In Vitro: A Systematic Review. Cells 2021, 10, 1378. [Google Scholar] [CrossRef]
- De Sousa Neto, I.V.; Durigan, J.L.Q.; da Silva, A.S.R.; de Cássia Marqueti, R. Adipose Tissue Extracellular Matrix Remodeling in Response to Dietary Patterns and Exercise: Molecular Landscape, Mechanistic Insights, and Therapeutic Approaches. Biology 2022, 11, 765. [Google Scholar] [CrossRef]
- Reing, J.E.; Zhang, L.; Myers-Irvin, J.; Cordero, K.E.; Freytes, D.O.; Heber-Katz, E.; Bedelbaeva, K.; McIntosh, D.; Dewilde, A.; Braunhut, S.J.; et al. Degradation Products of Extracellular Matrix Affect Cell Migration and Proliferation. Tissue Eng. Part. A 2009, 15, 605–614. [Google Scholar] [CrossRef]
- Mendibil, U.; Ruiz-Hernandez, R.; Retegi-Carrion, S.; Garcia-Urquia, N.; Olalde-Graells, B.; Abarrategi, A. Tissue-Specific Decellularization Methods: Rationale and Strategies to Achieve Regenerative Compounds. Int. J. Mol. Sci. 2020, 21, 5447. [Google Scholar] [CrossRef]
- Nelson, C.M.; Bissell, M.J. Of Extracellular Matrix, Scaffolds, and Signaling: Tissue Architecture Regulates Development, Homeostasis, and Cancer. Annu Rev. Cell Dev. Biol. 2006, 22, 287–309. [Google Scholar] [CrossRef] [Green Version]
- Keane, T.J.; Swinehart, I.T.; Badylak, S.F. Methods of Tissue Decellularization Used for Preparation of Biologic Scaffolds and in Vivo Relevance. Methods 2015, 84, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Parmaksiz, M.; Dogan, A.; Odabas, S.; Elçin, A.E.; Elçin, Y.M. Clinical Applications of Decellularized Extracellular Matrices for Tissue Engineering and Regenerative Medicine. Biomed. Mater. 2016, 11, 022003. [Google Scholar] [CrossRef]
- Badylak, S.F. The Extracellular Matrix as a Biologic Scaffold Material. Biomaterials 2007, 28, 3587–3593. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, T.W.; Sellaro, T.L.; Badylak, S.F. Decellularization of Tissues and Organs. Biomaterials 2006, 27, 3675–3683. [Google Scholar] [CrossRef] [PubMed]
- Crapo, P.M.; Gilbert, T.W.; Badylak, S.F. An Overview of Tissue and Whole Organ Decellularization Processes. Biomaterials 2011, 32, 3233–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, T.W. Strategies for Tissue and Organ Decellularization. J. Cell Biochem. 2012, 113, 2217–2222. [Google Scholar] [CrossRef] [PubMed]
- Cicuéndez, M.; Casarrubios, L.; Feito, M.J.; Madarieta, I.; Garcia-Urkia, N.; Murua, O.; Olalde, B.; Briz, N.; Diez-Orejas, R.; Portolés, M.T. Effects of Human and Porcine Adipose Extracellular Matrices Decellularized by Enzymatic or Chemical Methods on Macrophage Polarization and Immunocompetence. Int. J. Mol. Sci. 2021, 22, 3847. [Google Scholar] [CrossRef]
- Kornmuller, A.; Brown, C.F.C.; Yu, C.; Flynn, L.E. Fabrication of Extracellular Matrix-Derived Foams and Microcarriers as Tissue-Specific Cell Culture and Delivery Platforms. J. Vis. Exp. 2017, 122, e55436. [Google Scholar] [CrossRef] [Green Version]
- Madarieta, I.; García-Urquia, N.; Fernandez García, R. Method for Producing a Decellularized Tissue Matrix. Patent WO/2017/114902, 6 July 2017. [Google Scholar]
- Luzuriaga, J.; García-Gallastegui, P.; García-Urkia, N.; Pineda, J.R.; Irastorza, I.; Fernandez-San-Argimiro, F.-J.; Briz, N.; Olalde, B.; Unda, F.; Madarieta, I.; et al. Osteogenic Differentiation of Human Dental Pulp Stem Cells in Decellularised Adipose Tissue Solid Foams. Eur. Cell Mater. 2022, 43, 112–129. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- McKee, C.; Chaudhry, G.R. Advances and Challenges in Stem Cell Culture. Colloids Surf. B Biointerfaces 2017, 159, 62–77. [Google Scholar] [CrossRef]
- Rowe, R.G.; Daley, G.Q. Induced Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Diomede, F.; Fonticoli, L.; Marconi, G.D.; Della Rocca, Y.; Rajan, T.S.; Trubiani, O.; Murmura, G.; Pizzicannella, J. Decellularized Dental Pulp, Extracellular Vesicles, and 5-Azacytidine: A New Tool for Endodontic Regeneration. Biomedicines 2022, 10, 403. [Google Scholar] [CrossRef] [PubMed]
- Diomede, F.; Marconi, G.D.; Guarnieri, S.; D’Attilio, M.; Cavalcanti, M.F.X.B.; Mariggiò, M.A.; Pizzicannella, J.; Trubiani, O. A Novel Role of Ascorbic Acid in Anti-Inflammatory Pathway and ROS Generation in HEMA Treated Dental Pulp Stem Cells. Materials 2019, 13, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurrekoetxea, M.; Garcia-Gallastegui, P.; Irastorza, I.; Luzuriaga, J.; Uribe-Etxebarria, V.; Unda, F.; Ibarretxe, G. Dental Pulp Stem Cells as a Multifaceted Tool for Bioengineering and the Regeneration of Craniomaxillofacial Tissues. Front. Physiol. 2015, 6, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal Human Dental Pulp Stem Cells (DPSCs) in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzuriaga, J.; Pastor-Alonso, O.; Encinas, J.M.; Unda, F.; Ibarretxe, G.; Pineda, J.R. Human Dental Pulp Stem Cells Grown in Neurogenic Media Differentiate Into Endothelial Cells and Promote Neovasculogenesis in the Mouse Brain. Front. Physiol. 2019, 10, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Etxebarria, V.; Luzuriaga, J.; Garcia-Gallastegui, P.; Agliano, A.; Unda, F.; Ibarretxe, G. Notch/Wnt Cross-Signalling Regulates Stemness of Dental Pulp Stem Cells through Expression of Neural Crest and Core Pluripotency Factors. Eur. Cell Mater. 2017, 34, 249–270. [Google Scholar] [CrossRef]
- Monterubbianesi, R.; Bencun, M.; Pagella, P.; Woloszyk, A.; Orsini, G.; Mitsiadis, T.A. A Comparative in Vitro Study of the Osteogenic and Adipogenic Potential of Human Dental Pulp Stem Cells, Gingival Fibroblasts and Foreskin Fibroblasts. Sci. Rep. 2019, 9, 1761. [Google Scholar] [CrossRef]
- Redmond, J.; McCarthy, H.; Buchanan, P.; Levingstone, T.J.; Dunne, N.J. Advances in Biofabrication Techniques for Collagen-Based 3D in Vitro Culture Models for Breast Cancer Research. Mater. Sci Eng. C Mater. Biol. Appl 2021, 122, 111944. [Google Scholar] [CrossRef]
- Irastorza, I.; Luzuriaga, J.; Martinez-Conde, R.; Ibarretxe, G.; Unda, F. Adhesion, Integration and Osteogenesis of Human Dental Pulp Stem Cells on Biomimetic Implant Surfaces Combined with Plasma Derived Products. Eur. Cell Mater. 2019, 38, 201–214. [Google Scholar] [CrossRef]
- Uribe-Etxebarria, V.; García-Gallastegui, P.; Pérez-Garrastachu, M.; Casado-Andrés, M.; Irastorza, I.; Unda, F.; Ibarretxe, G.; Subirán, N. Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells. Cells 2020, 9, 652. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kinkel, A.D.; Fernyhough, M.E.; Helterline, D.L.; Vierck, J.L.; Oberg, K.S.; Vance, T.J.; Hausman, G.J.; Hill, R.A.; Dodson, M.V. Oil Red-O Stains Non-Adipogenic Cells: A Precautionary Note. Cytotechnology 2004, 46, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ASTM International. ASTM F3354-19 Standard Guide for Evaluating Extracellular Matrix Decellularization Processes; ASTM International: West Conshohocken, PA, USA, 2019. [Google Scholar]
- Gonzales, A.M.; Orlando, R.A. Role of Adipocyte-Derived Lipoprotein Lipase in Adipocyte Hypertrophy. Nutr. Metab. 2007, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Darlington, G.J.; Ross, S.E.; MacDougald, O.A. The Role of C/EBP Genes in Adipocyte Differentiation. J. Biol. Chem. 1998, 273, 30057–30060. [Google Scholar] [CrossRef] [Green Version]
- Gervois, P.; Fruchart, J.-C. PPAR gamma: A major nuclear receptor in adipogenesis. Med. Sci. 2003, 19, 20–22. [Google Scholar] [CrossRef] [Green Version]
- Boutari, C.; Mantzoros, C.S. A 2022 Update on the Epidemiology of Obesity and a Call to Action: As Its Twin COVID-19 Pandemic Appears to Be Receding, the Obesity and Dysmetabolism Pandemic Continues to Rage On. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef]
- Stranahan, A.M. Visceral Adiposity, Inflammation, and Hippocampal Function in Obesity. Neuropharmacology 2022, 205, 108920. [Google Scholar] [CrossRef]
- Olshansky, S.J.; Passaro, D.J.; Hershow, R.C.; Layden, J.; Carnes, B.A.; Brody, J.; Hayflick, L.; Butler, R.N.; Allison, D.B.; Ludwig, D.S. A Potential Decline in Life Expectancy in the United States in the 21st Century. N. Engl. J. Med. 2005, 352, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Ebbeling, C.B.; Pawlak, D.B.; Ludwig, D.S. Childhood Obesity: Public-Health Crisis, Common Sense Cure. Lancet 2002, 360, 473–482. [Google Scholar] [CrossRef]
- Must, A.; Spadano, J.; Coakley, E.H.; Field, A.E.; Colditz, G.; Dietz, W.H. The Disease Burden Associated with Overweight and Obesity. JAMA 1999, 282, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Thomson, M.; Martin, A.; Long, E.; Logue, J.; Simpson, S.A. A Qualitative Exploration of Weight Management during COVID-19. Clin. Obes. 2022, 12, e12512. [Google Scholar] [CrossRef] [PubMed]
- Hussey, G.S.; Dziki, J.L.; Badylak, S.F. Extracellular Matrix-Based Materials for Regenerative Medicine. Nat. Rev. Mater. 2018, 3, 159–173. [Google Scholar] [CrossRef]
- Vasanthan, K.S.; Srinivasan, V.; Pandita, D. Extracellular Matrix Extraction Techniques and Applications in Biomedical Engineering. Regen. Med. 2021, 16, 775–802. [Google Scholar] [CrossRef] [PubMed]
- Flynn, L.E. The Use of Decellularized Adipose Tissue to Provide an Inductive Microenvironment for the Adipogenic Differentiation of Human Adipose-Derived Stem Cells. Biomaterials 2010, 31, 4715–4724. [Google Scholar] [CrossRef]
- Del Amo, C.; Fernández-San Argimiro, X.; Cascajo-Castresana, M.; Perez-Valle, A.; Madarieta, I.; Olalde, B.; Andia, I. Wound-Microenvironment Engineering through Advanced-Dressing Bioprinting. Int. J. Mol. Sci. 2022, 23, 2836. [Google Scholar] [CrossRef]
- Kaukua, N.; Shahidi, M.K.; Konstantinidou, C.; Dyachuk, V.; Kaucka, M.; Furlan, A.; An, Z.; Wang, L.; Hultman, I.; Ahrlund-Richter, L.; et al. Glial Origin of Mesenchymal Stem Cells in a Tooth Model System. Nature 2014, 513, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Luzuriaga, J.; Polo, Y.; Pastor-Alonso, O.; Pardo-Rodríguez, B.; Larrañaga, A.; Unda, F.; Sarasua, J.-R.; Pineda, J.R.; Ibarretxe, G. Advances and Perspectives in Dental Pulp Stem Cell Based Neuroregeneration Therapies. Int. J. Mol. Sci. 2021, 22, 3546. [Google Scholar] [CrossRef]
- Luzuriaga, J.; Pineda, J.R.; Irastorza, I.; Uribe-Etxebarria, V.; García-Gallastegui, P.; Encinas, J.M.; Chamero, P.; Unda, F.; Ibarretxe, G. BDNF and NT3 Reprogram Human Ectomesenchymal Dental Pulp Stem Cells to Neurogenic and Gliogenic Neural Crest Progenitors Cultured in Serum-Free Medium. Cell. Physiol. Biochem. 2019, 52, 1361–1380. [Google Scholar] [CrossRef]
- Lee, J.-H.; Woo, K.-J.; Kim, M.-A.; Hong, J.; Kim, J.; Kim, S.-H.; Han, K.-I.; Iwasa, M.; Kim, T.-J. Heat-Killed Enterococcus Faecalis Prevents Adipogenesis and High Fat Diet-Induced Obesity by Inhibition of Lipid Accumulation through Inhibiting C/EBP-α and PPAR-γ in the Insulin Signaling Pathway. Nutrients 2022, 14, 1308. [Google Scholar] [CrossRef]
- Farmer, S.R. Regulation of PPARgamma Activity during Adipogenesis. Int. J. Obes. 2005, 29 (Suppl. 1), S13–S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Formulation (wt%) | Water Absorption | Average Storage Modulus | Degradation Weight Loss (%) | ||
|---|---|---|---|---|---|---|
| (EWC, %) | S Ratio | G′ (Pa) | G″ (Pa) | |||
| hDAT | 100% hDAT | 92.4 ± 0.9 | 12.2 ± 1.6 | 152.8 ± 15.6 | 21.1 ± 3.2 | 22.9 ± 11.9 |
| 3hDAT:1 bCOL-I | 75% hDAT: 25% bCOL-I | 92.1 ± 0.7 | 11.7 ± 1.1 | 128.0 ± 11.1 | 13.6 ± 2.1 | 26.10 ± 11.5 |
| bCOL-I | 100% bCOL-I | 90.3 ± 0.1 | 9.3 ± 0.1 | 302.9 ± 22.2 | 32.7 ± 7.0 | 61.95 ± 15.99 |
| Primers | Sequence 5′-3′ | Annealing | Amplicon (bp) |
|---|---|---|---|
| β-ACTIN (upstream) | GTTGTCGACGACGAGCG | 58.5 | 93 |
| β-ACTIN (downstream) | GCACAGAGCCTCGCCTT | 59.7 | 93 |
| GAPDH (upstream) | CTTTTGCGTCGCCAG | 60.3 | 131 |
| GAPDH (downstream) | TTGATGGCAACAATATCCAC | 60.8 | 131 |
| CEBP (upstream) | AGCCTTGTTTGTACTGTATG | 54.3 | 199 |
| CEBP (downstream) | AAAATGGTGGTTTAGCAGAG | 58.3 | 199 |
| LPL (upstream) | ACACAGAGGTAGATATTGGAG | 53.8 | 143 |
| LPL (downstream) | CTTTTTCTGAGTCTCTCCTG | 52.9 | 143 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Urkia, N.; Luzuriaga, J.; Uribe-Etxebarria, V.; Irastorza, I.; Fernandez-San-Argimiro, F.J.; Olalde, B.; Briz, N.; Unda, F.; Ibarretxe, G.; Madarieta, I.; et al. Enhanced Adipogenic Differentiation of Human Dental Pulp Stem Cells in Enzymatically Decellularized Adipose Tissue Solid Foams. Biology 2022, 11, 1099. https://doi.org/10.3390/biology11081099
Garcia-Urkia N, Luzuriaga J, Uribe-Etxebarria V, Irastorza I, Fernandez-San-Argimiro FJ, Olalde B, Briz N, Unda F, Ibarretxe G, Madarieta I, et al. Enhanced Adipogenic Differentiation of Human Dental Pulp Stem Cells in Enzymatically Decellularized Adipose Tissue Solid Foams. Biology. 2022; 11(8):1099. https://doi.org/10.3390/biology11081099
Chicago/Turabian StyleGarcia-Urkia, Nerea, Jon Luzuriaga, Veronica Uribe-Etxebarria, Igor Irastorza, Francisco Javier Fernandez-San-Argimiro, Beatriz Olalde, Nerea Briz, Fernando Unda, Gaskon Ibarretxe, Iratxe Madarieta, and et al. 2022. "Enhanced Adipogenic Differentiation of Human Dental Pulp Stem Cells in Enzymatically Decellularized Adipose Tissue Solid Foams" Biology 11, no. 8: 1099. https://doi.org/10.3390/biology11081099
APA StyleGarcia-Urkia, N., Luzuriaga, J., Uribe-Etxebarria, V., Irastorza, I., Fernandez-San-Argimiro, F. J., Olalde, B., Briz, N., Unda, F., Ibarretxe, G., Madarieta, I., & Pineda, J. R. (2022). Enhanced Adipogenic Differentiation of Human Dental Pulp Stem Cells in Enzymatically Decellularized Adipose Tissue Solid Foams. Biology, 11(8), 1099. https://doi.org/10.3390/biology11081099