
Neuronal Depolarization Induced RNA m5C Methylation Changes in Mouse Cortical Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
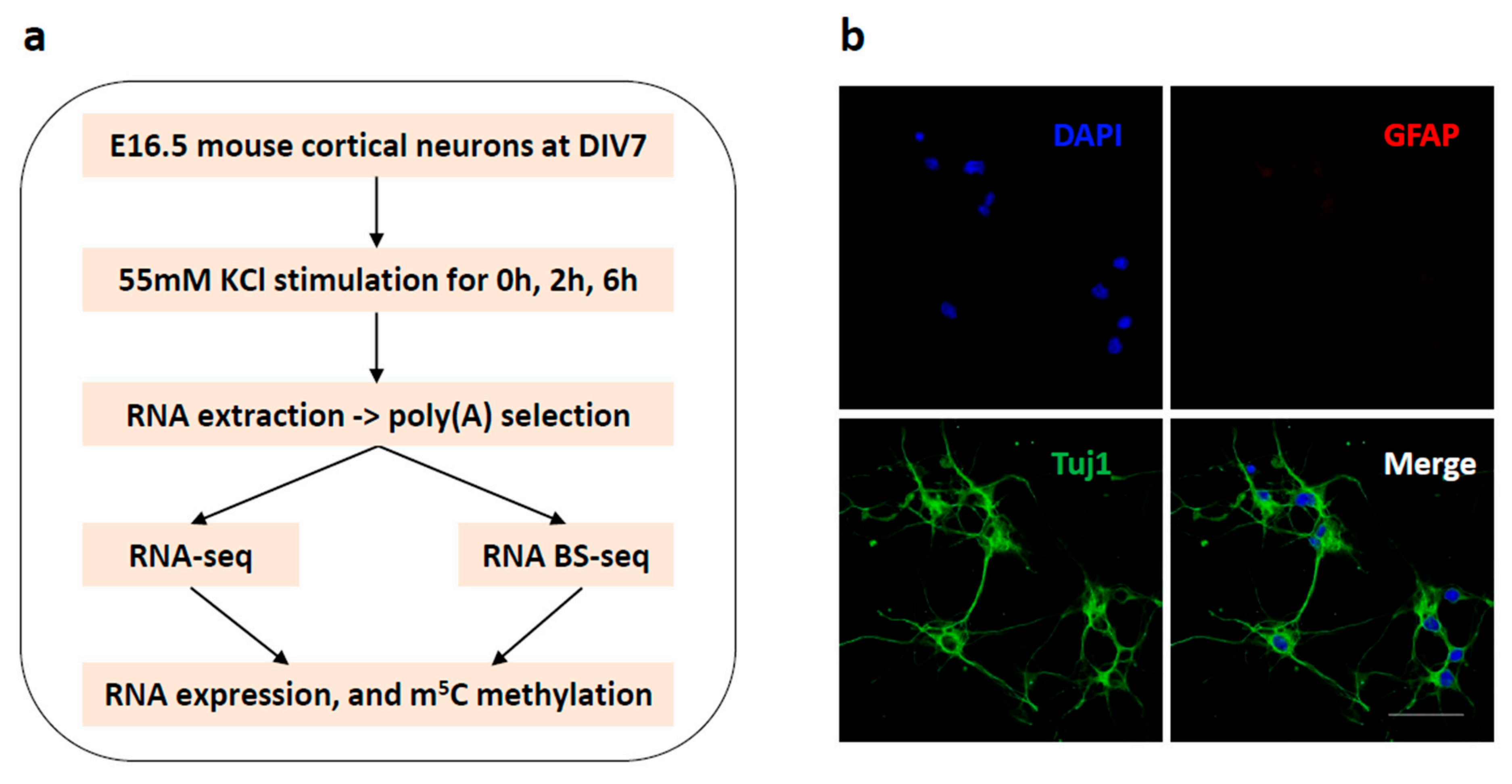
2.1. Animal
2.2. Primary Mouse Cortical Neuronal Culture
2.3. Membrane Depolarization and RNA Isolation
2.4. Immunostaining
2.5. RNA-seq Library Construction
2.6. RNA-seq Data Analysis
2.7. Generation of Spike-In Unmethylated mRNA Control
2.8. RNA BS-seq Library Construction
2.9. RNA BS-seq Data Processing
2.10. Determination of m5C Distribution, Differential Methylation and Correlation Analysis
3. Results
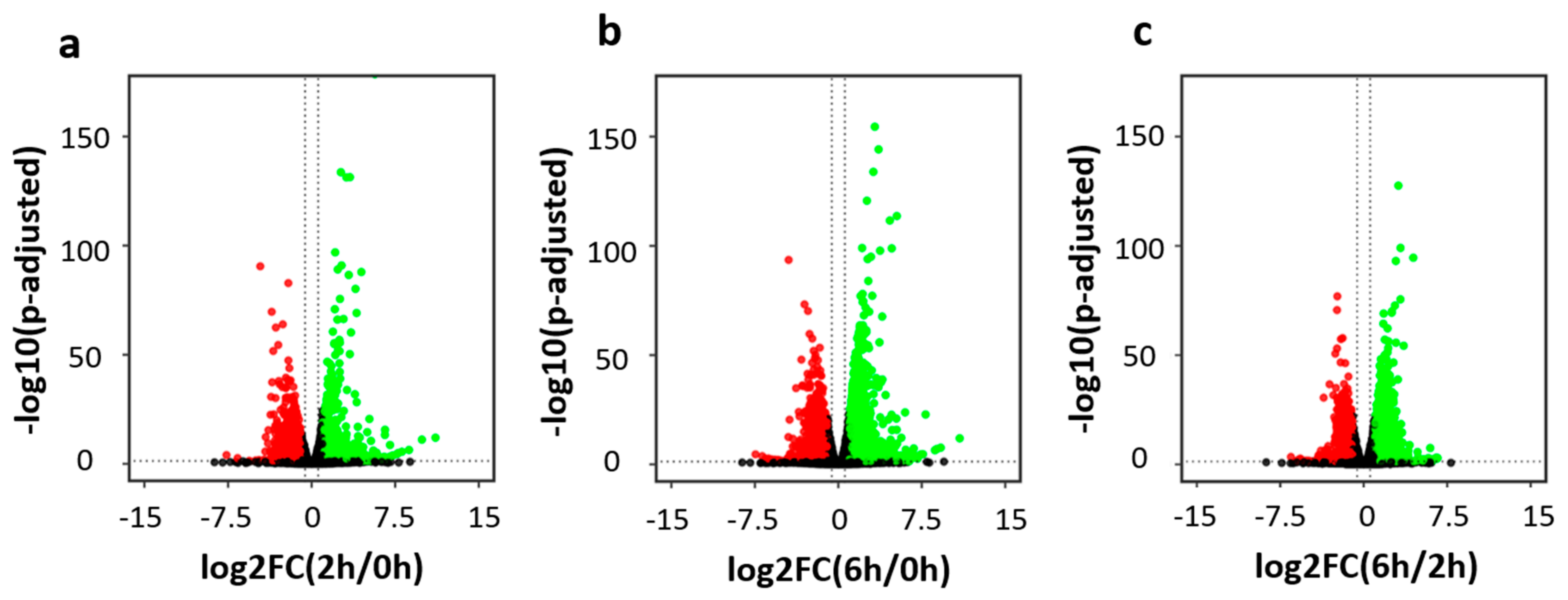
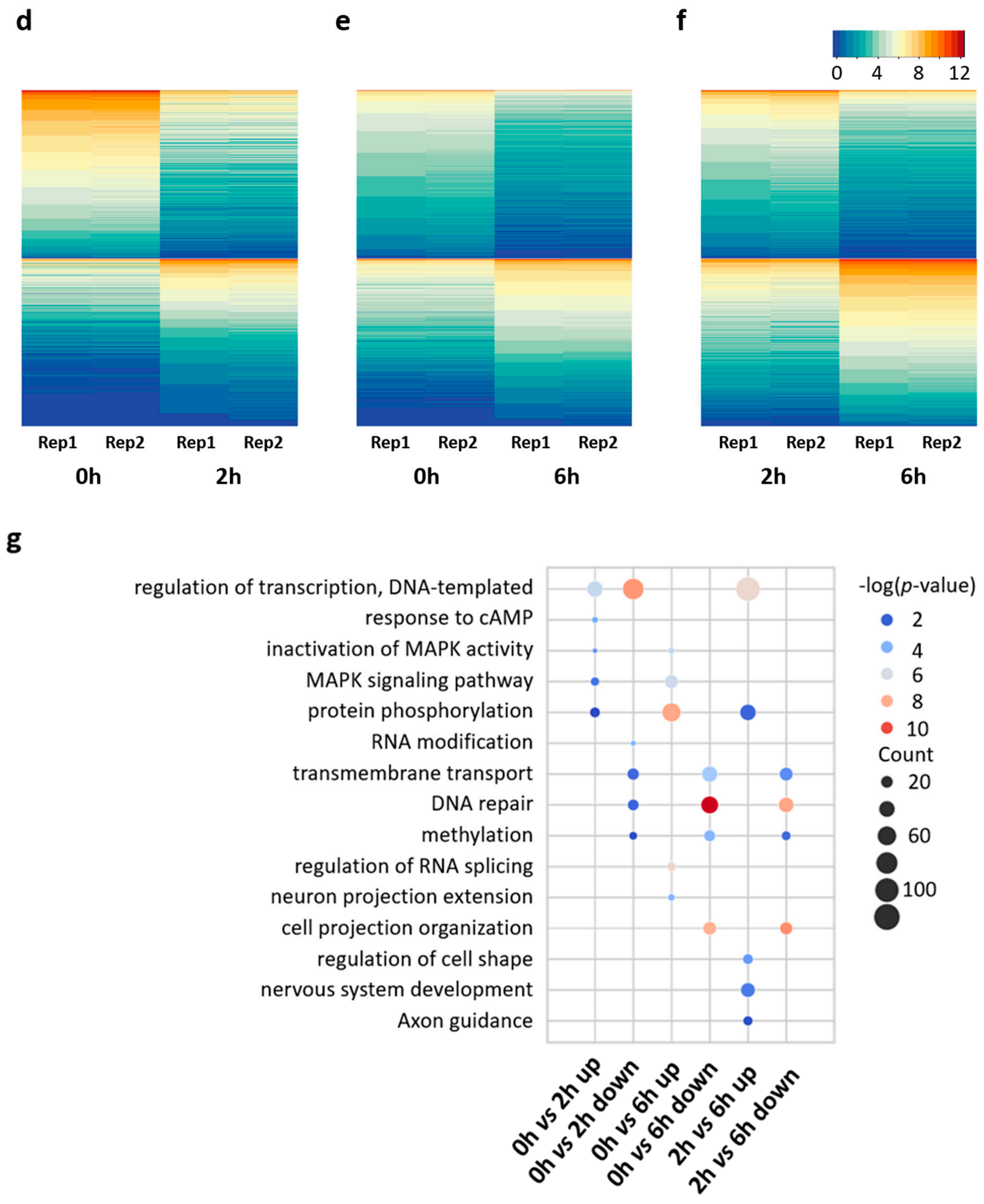
3.1. Robust Transcriptional Dynamics Induced by Neuronal Depolarization
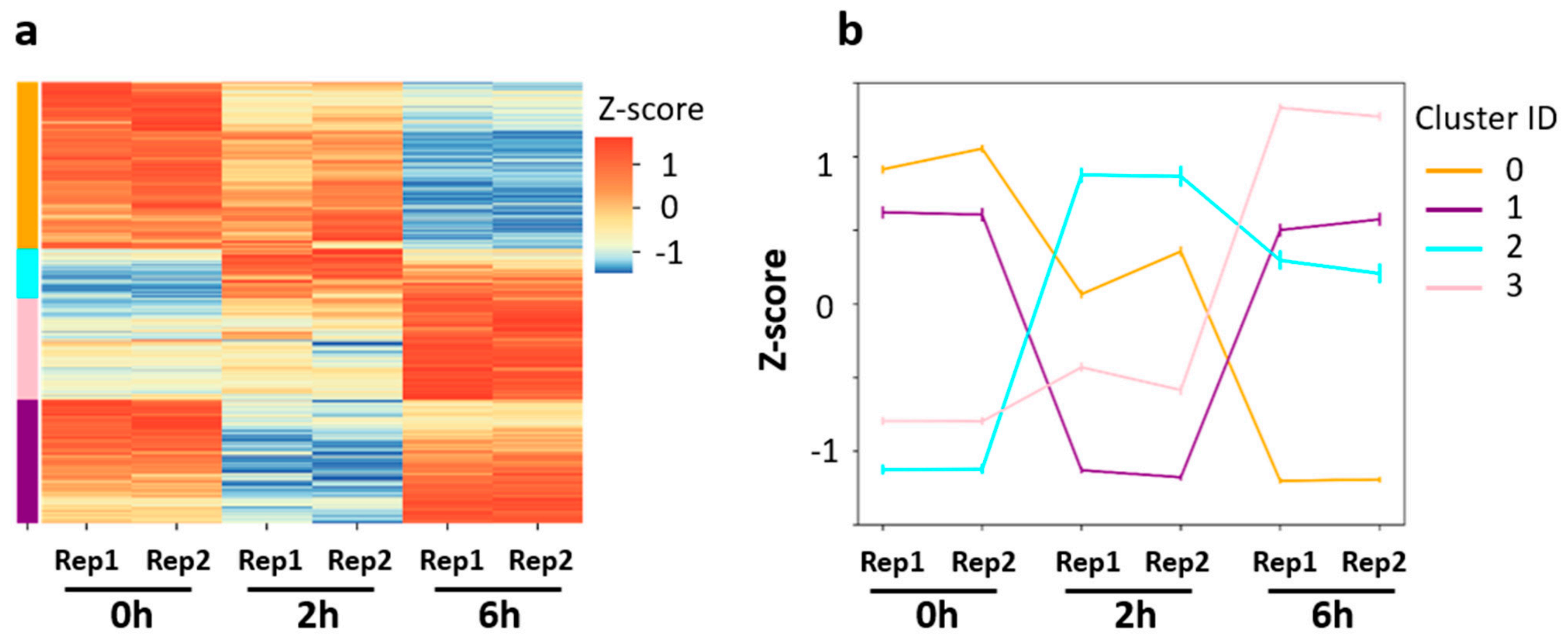
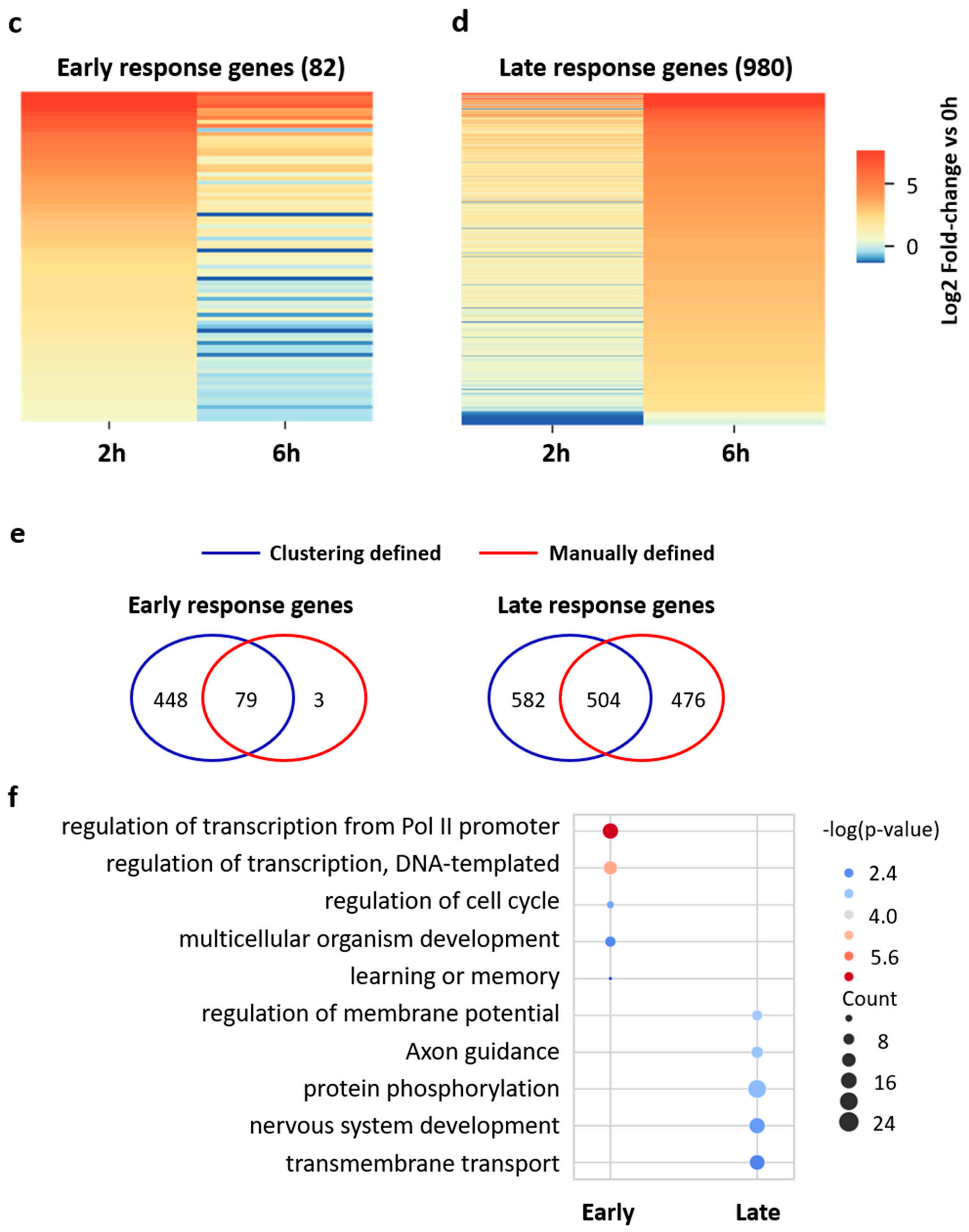
3.2. Distinct Gene Expression Patterns in the Early and Late Phases of Neuronal Activation
3.3. Transcriptome-wide mRNA m5C Modification in Mouse Cortical Neurons
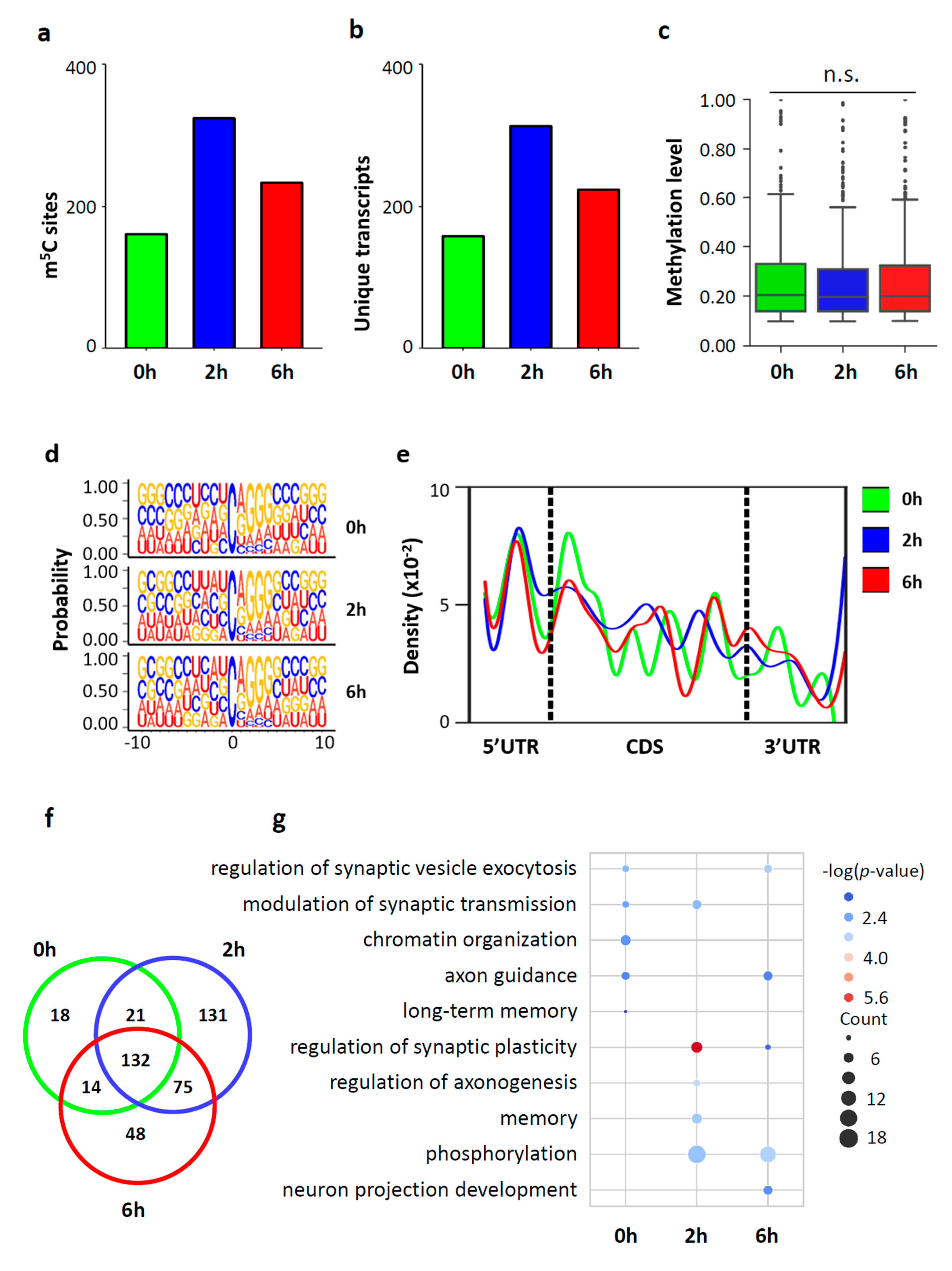
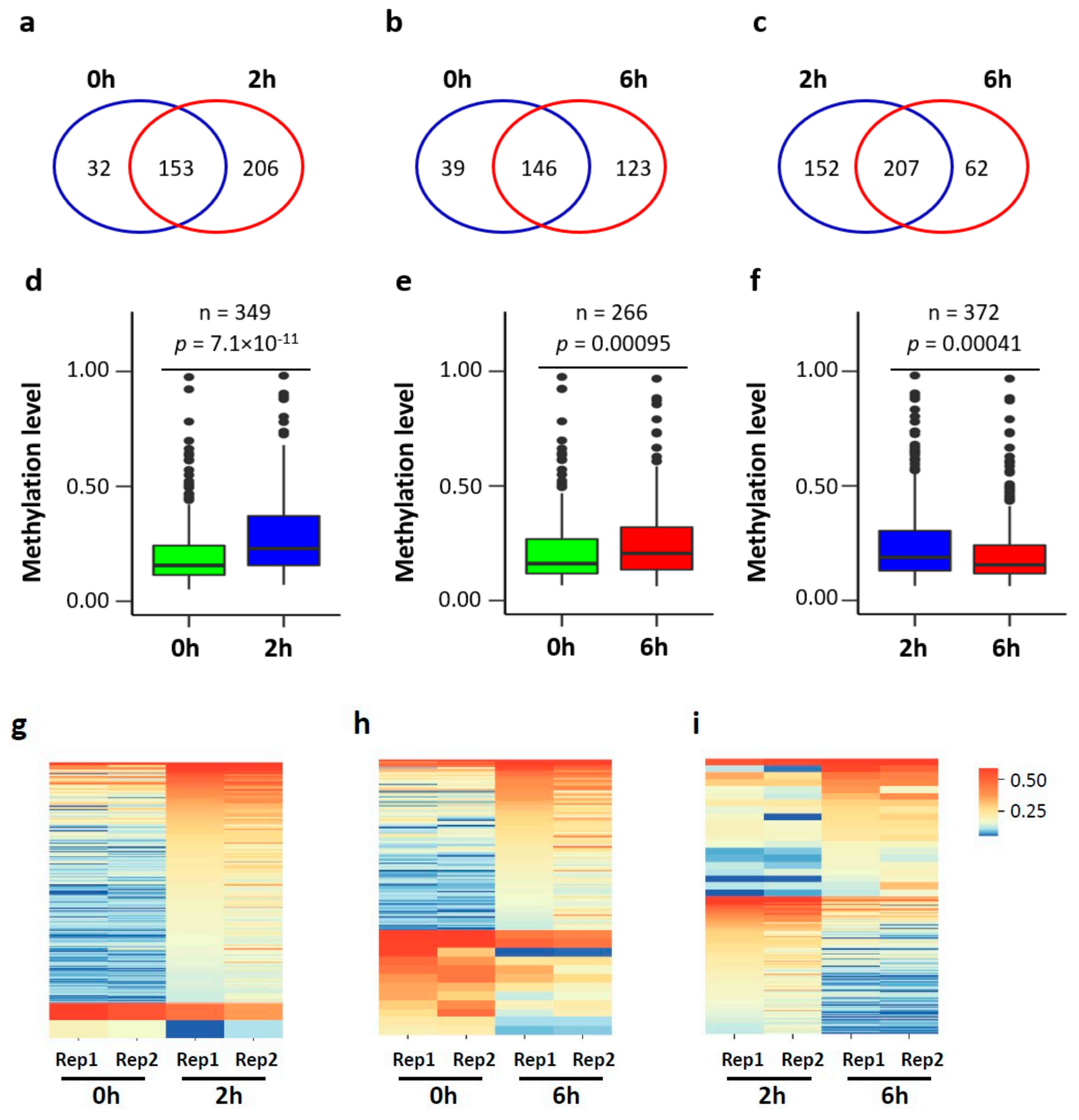
3.4. Dynamic m5C Landscape upon Neuronal Activation
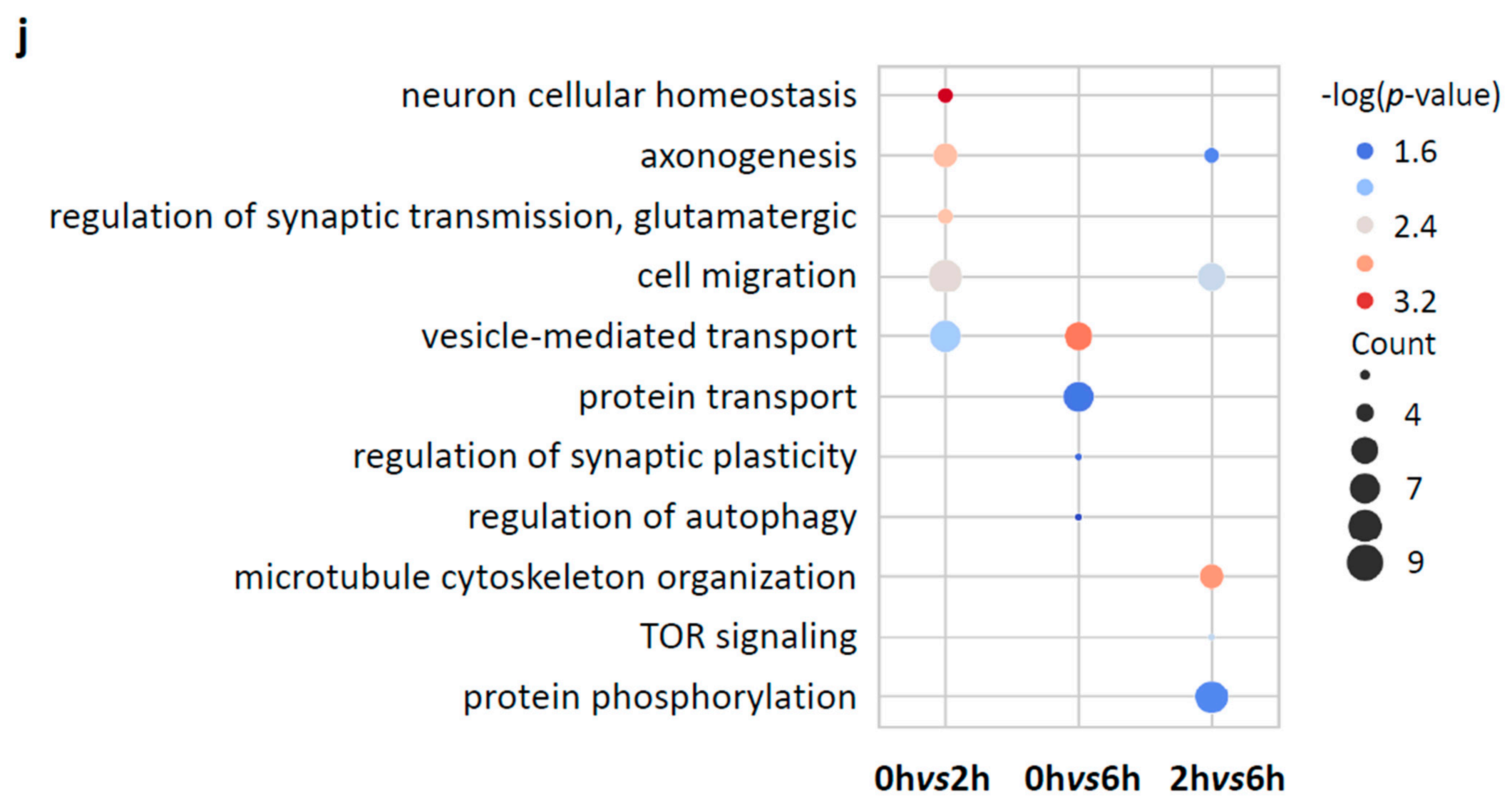
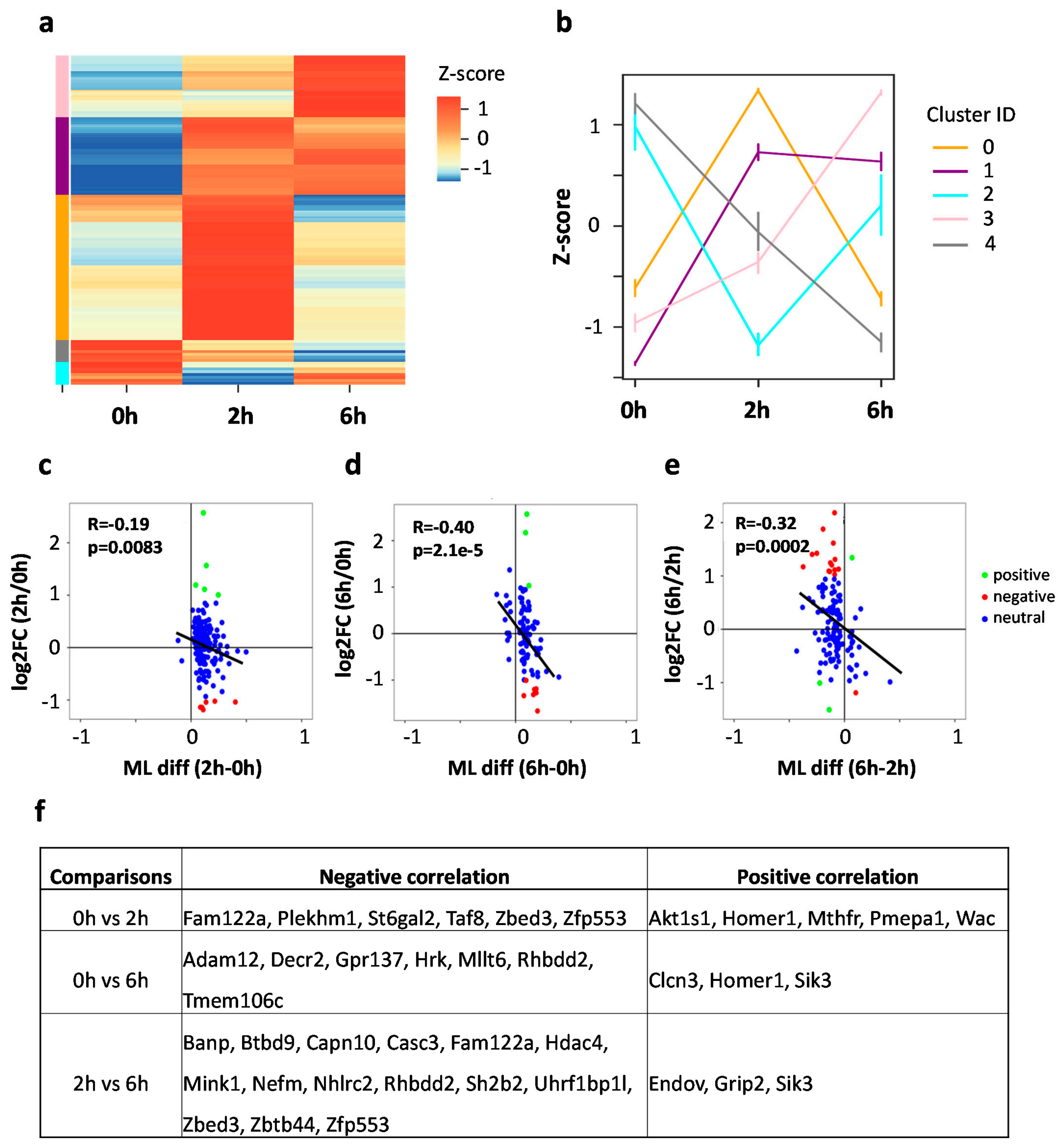
3.5. mRNA m5C Methylation Negatively Correlates with Expression in Neurons upon Neuronal Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, J.; Yi, C. Chemical Modifications to RNA: A New Layer of Gene Expression Regulation. ACS Chem. Biol. 2017, 12, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F. Bringing order to translation: The contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008, 9, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef]
- Fu, L.; Guerrero, C.R.; Zhong, N.; Amato, N.J.; Liu, Y.; Liu, S.; Cai, Q.; Ji, D.; Jin, S.G.; Niedernhofer, L.J.; et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 2014, 136, 11582–11585. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.M.; van Delft, P.; Mendil, L.; Bachman, M.; Smollett, K.; Werner, F.; Miska, E.A.; Balasubramanian, S. Formation and abundance of 5-hydroxymethylcytosine in RNA. ChemBioChem 2015, 16, 752–755. [Google Scholar] [CrossRef] [Green Version]
- Basanta-Sanchez, M.; Wang, R.; Liu, Z.; Ye, X.; Li, M.; Shi, X.; Agris, P.F.; Zhou, Y.; Huang, Y.; Sheng, J. TET1-Mediated Oxidation of 5-Formylcytosine (5fC) to 5-Carboxycytosine (5caC) in RNA. ChemBioChem 2017, 18, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.Y.; Micura, R.; Lusser, A. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, F.; Tu, R.; Duan, B.; Yang, Z.; Ping, Z.; Song, X.; Chen, S.; Price, A.; Li, H.; Scott, A.; et al. Drosophila YBX1 homolog YPS promotes ovarian germ line stem cell development by preferentially recognizing 5-methylcytosine RNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 3603–3609. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Han, X.; Yang, W.L.; Zhang, M.; Ma, H.L.; Sun, B.F.; Li, A.; Xia, J.; Chen, J.; et al. RNA 5-Methylcytosine Facilitates the Maternal-to-Zygotic Transition by Preventing Maternal mRNA Decay. Mol. Cell 2019, 75, 1188–1202. [Google Scholar] [CrossRef]
- Chen, X.; Li, A.; Sun, B.F.; Yang, Y.; Han, Y.N.; Yuan, X.; Chen, R.X.; Wei, W.S.; Liu, Y.; Gao, C.C.; et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 2019, 21, 978–990. [Google Scholar] [CrossRef]
- Abbasi-Moheb, L.; Mertel, S.; Gonsior, M.; Nouri-Vahid, L.; Kahrizi, K.; Cirak, S.; Wieczorek, D.; Motazacker, M.M.; Esmaeeli-Nieh, S.; Cremer, K.; et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 847–855. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.J.; Lee, J.H.; Lee, J.E.; Blanco, S.; Nickerson, E.; Gabriel, S.; Frye, M.; Al-Gazali, L.; Gleeson, J.G. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 2012, 49, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef]
- Flores, J.V.; Cordero-Espinoza, L.; Oeztuerk-Winder, F.; Andersson-Rolf, A.; Selmi, T.; Blanco, S.; Tailor, J.; Dietmann, S.; Frye, M. Cytosine-5 RNA Methylation Regulates Neural Stem Cell Differentiation and Motility. Stem Cell Rep. 2017, 8, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Blaze, J.; Navickas, A.; Phillips, H.L.; Heissel, S.; Plaza-Jennings, A.; Miglani, S.; Asgharian, H.; Foo, M.; Katanski, C.D.; Watkins, C.P.; et al. Neuronal Nsun2 deficiency produces tRNA epitranscriptomic alterations and proteomic shifts impacting synaptic signaling and behavior. Nat. Commun. 2021, 12, 4913. [Google Scholar] [CrossRef]
- Awah, C.U.; Winter, J.; Mazdoom, C.M.; Ogunwobi, O.O. NSUN6, an RNA methyltransferase of 5-mC controls glioblastoma response to temozolomide (TMZ) via NELFB and RPS6KB2 interaction. Cancer Biol. Ther. 2021, 22, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Zhang, C.; Qi, Z.; Li, X.; Lou, Y.; Kang, Y.; Deng, W.; Lv, Y.; Wang, C.; Wang, W.; et al. Alteration of mRNA 5-Methylcytosine Modification in Neurons After OGD/R and Potential Roles in Cell Stress Response and Apoptosis. Front. Genet. 2021, 12, 633681. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.H.; Nedivi, E. Activity-regulated genes as mediators of neural circuit plasticity. Prog. Neurobiol. 2011, 94, 223–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.E.; Greenberg, M.E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 2011, 3, a005744. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef]
- Su, Y.; Shin, J.; Zhong, C.; Wang, S.; Roychowdhury, P.; Lim, J.; Kim, D.; Ming, G.L.; Song, H. Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci. 2017, 20, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.N.; Vierbuchen, T.; Hemberg, M.; Rubin, A.A.; Ling, E.; Couch, C.H.; Stroud, H.; Spiegel, I.; Farh, K.K.; Harmin, D.A.; et al. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat. Neurosci. 2014, 17, 1330–1339. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Xu, X.; He, J.; Murray, A.; Sun, M.A.; Wei, X.; Wang, X.; McCoig, E.; Xie, E.; Jiang, X.; et al. EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity. Nat. Commun. 2019, 10, 3892. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, Z.; Xu, X.; Pacholec, C.; Xie, H. Systematic evaluation of parameters in RNA bisulfite sequencing data generation and analysis. NAR Genom. Bioinform. 2022, 4, lqac045. [Google Scholar] [CrossRef] [PubMed]
- Rieder, D.; Amort, T.; Kugler, E.; Lusser, A.; Trajanoski, Z. meRanTK: Methylated RNA analysis ToolKit. Bioinformatics 2016, 32, 782–785. [Google Scholar] [CrossRef] [Green Version]
- Schumann, U.; Zhang, H.N.; Sibbritt, T.; Pan, A.; Horvath, A.; Gross, S.; Clark, S.J.; Yang, L.; Preiss, T. Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol. 2020, 18, 40. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Wei, Z.; Panneerdoss, S.; Timilsina, S.; Zhu, J.; Mohammad, T.A.; Lu, Z.L.; de Magalhaes, J.P.; Chen, Y.; Rong, R.; Huang, Y.; et al. Topological Characterization of Human and Mouse m(5)C Epitranscriptome Revealed by Bisulfite Sequencing. Int. J. Genom. 2018, 2018, 1351964. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.Y.; Schaukowitch, K.; Farbiak, L.; Kilaru, G.; Kim, T.K. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat. Neurosci. 2016, 19, 75–83. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Johnson, Z.; Xie, H. Neuronal Depolarization Induced RNA m5C Methylation Changes in Mouse Cortical Neurons. Biology 2022, 11, 988. https://doi.org/10.3390/biology11070988
Xu X, Johnson Z, Xie H. Neuronal Depolarization Induced RNA m5C Methylation Changes in Mouse Cortical Neurons. Biology. 2022; 11(7):988. https://doi.org/10.3390/biology11070988
Chicago/Turabian StyleXu, Xiguang, Zachary Johnson, and Hehuang Xie. 2022. "Neuronal Depolarization Induced RNA m5C Methylation Changes in Mouse Cortical Neurons" Biology 11, no. 7: 988. https://doi.org/10.3390/biology11070988
APA StyleXu, X., Johnson, Z., & Xie, H. (2022). Neuronal Depolarization Induced RNA m5C Methylation Changes in Mouse Cortical Neurons. Biology, 11(7), 988. https://doi.org/10.3390/biology11070988