
The Complete Genome of the “Flavescence Dorée” Phytoplasma Reveals Characteristics of Low Genome Plasticity

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Material Source
2.2. DNA Extraction and Enrichment
2.3. MinION and Illumina Sequencing
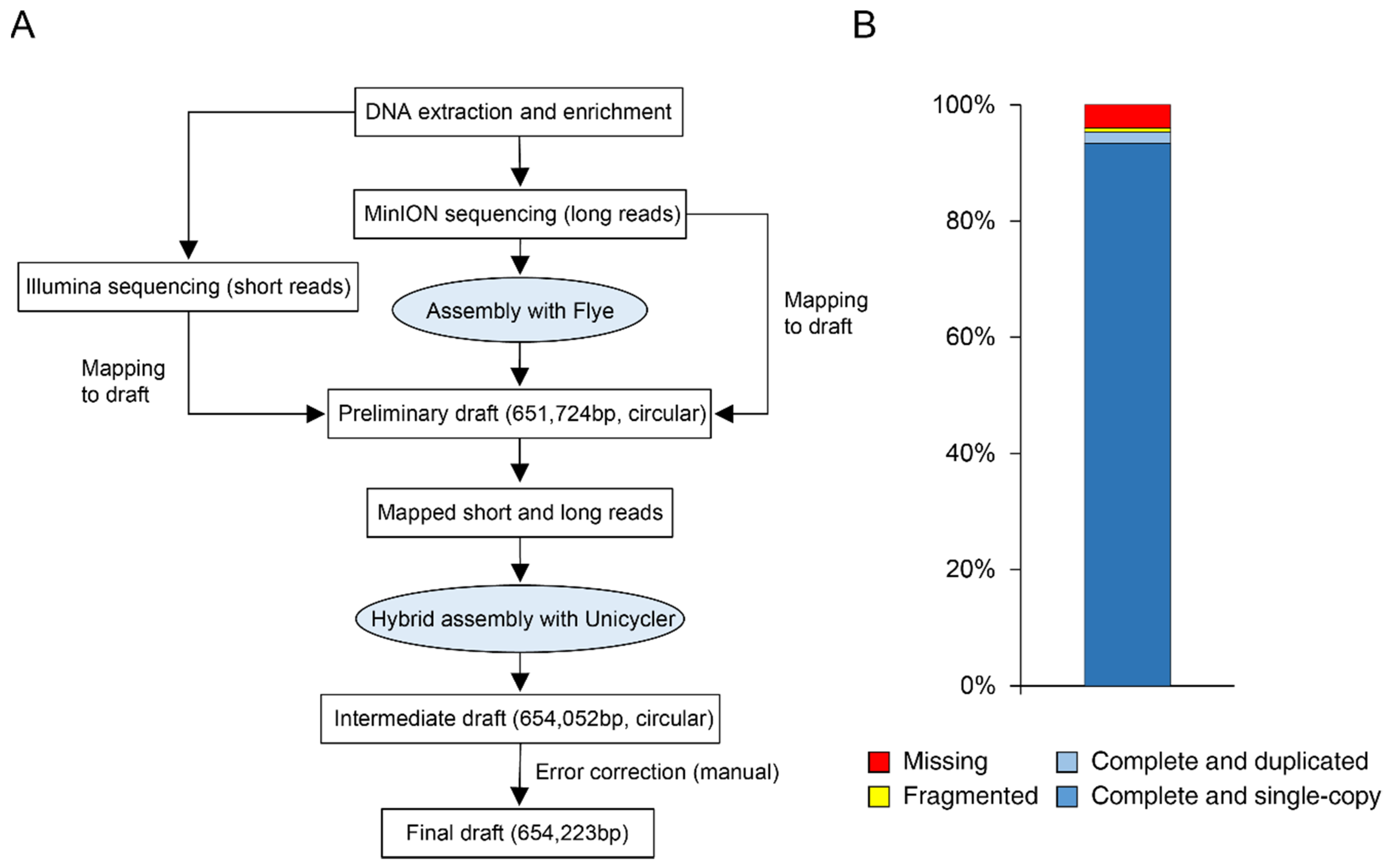
2.4. De Novo Genome Assembly and Quality Assessment
2.5. Functional Annotation
2.6. Genome Comparison and Phylogenetic Analyses
3. Results
3.1. Genome Assembly and Quality Assessment
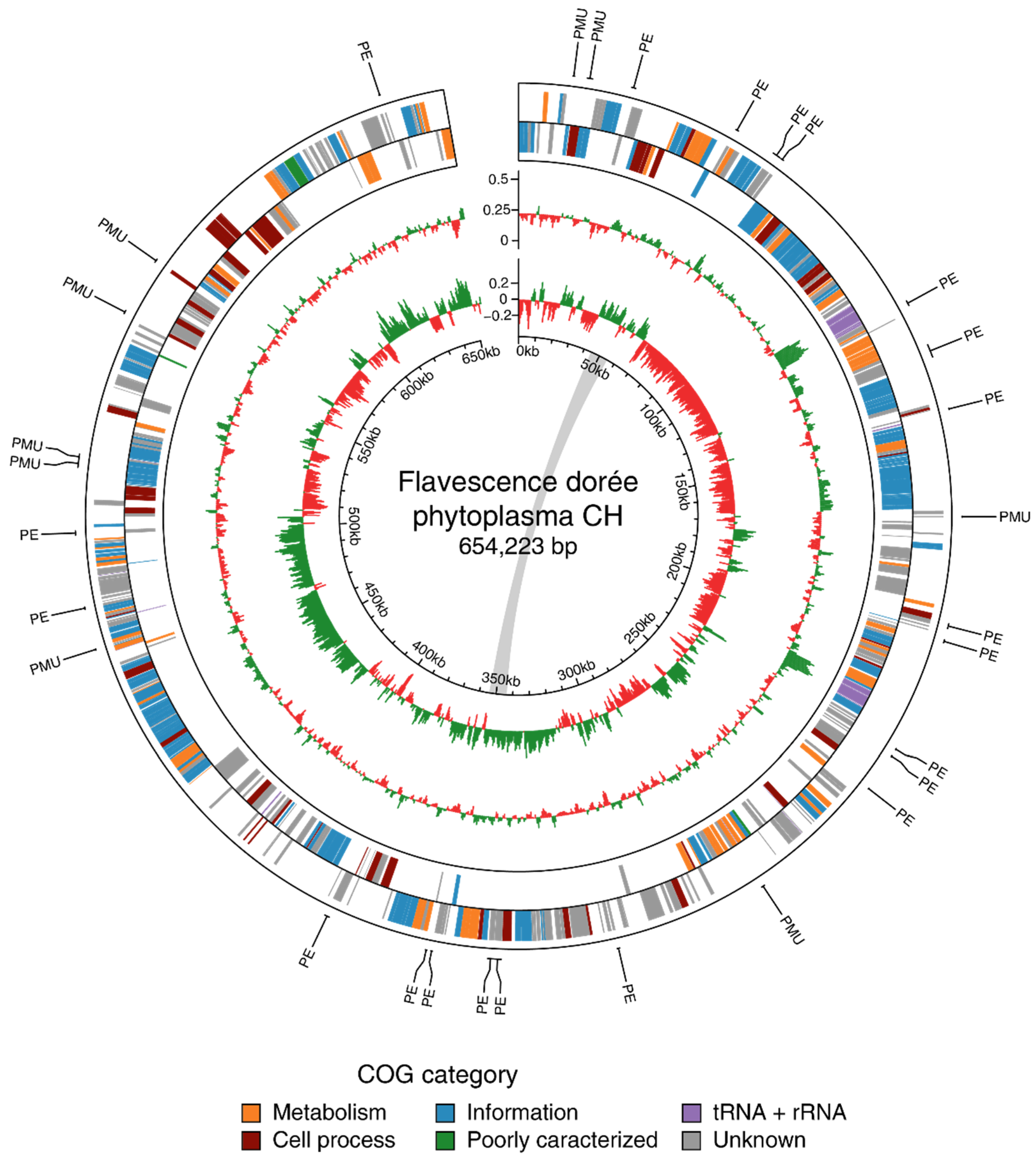
3.2. Genome Characteristics and Annotation
3.2.1. General Features
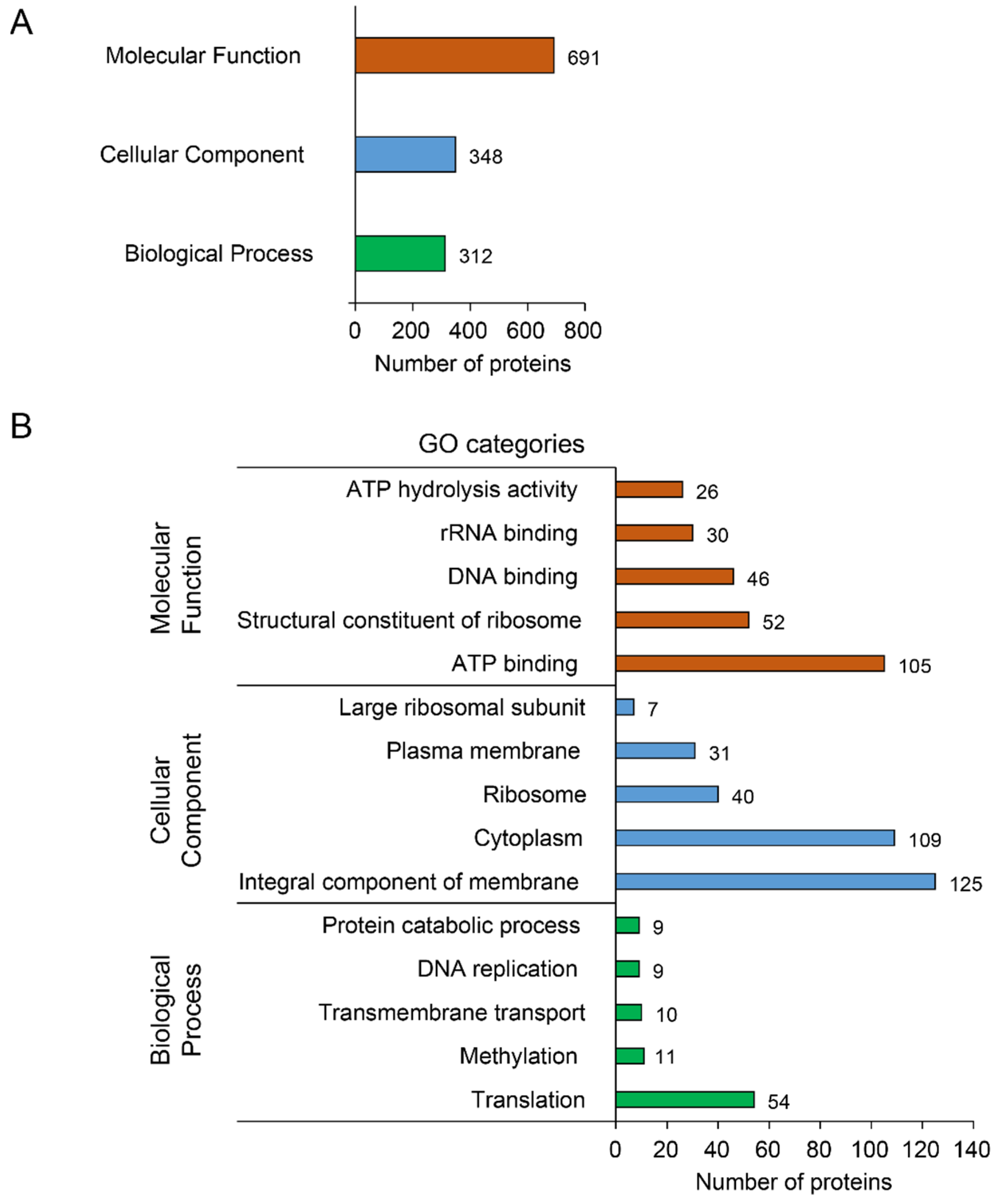
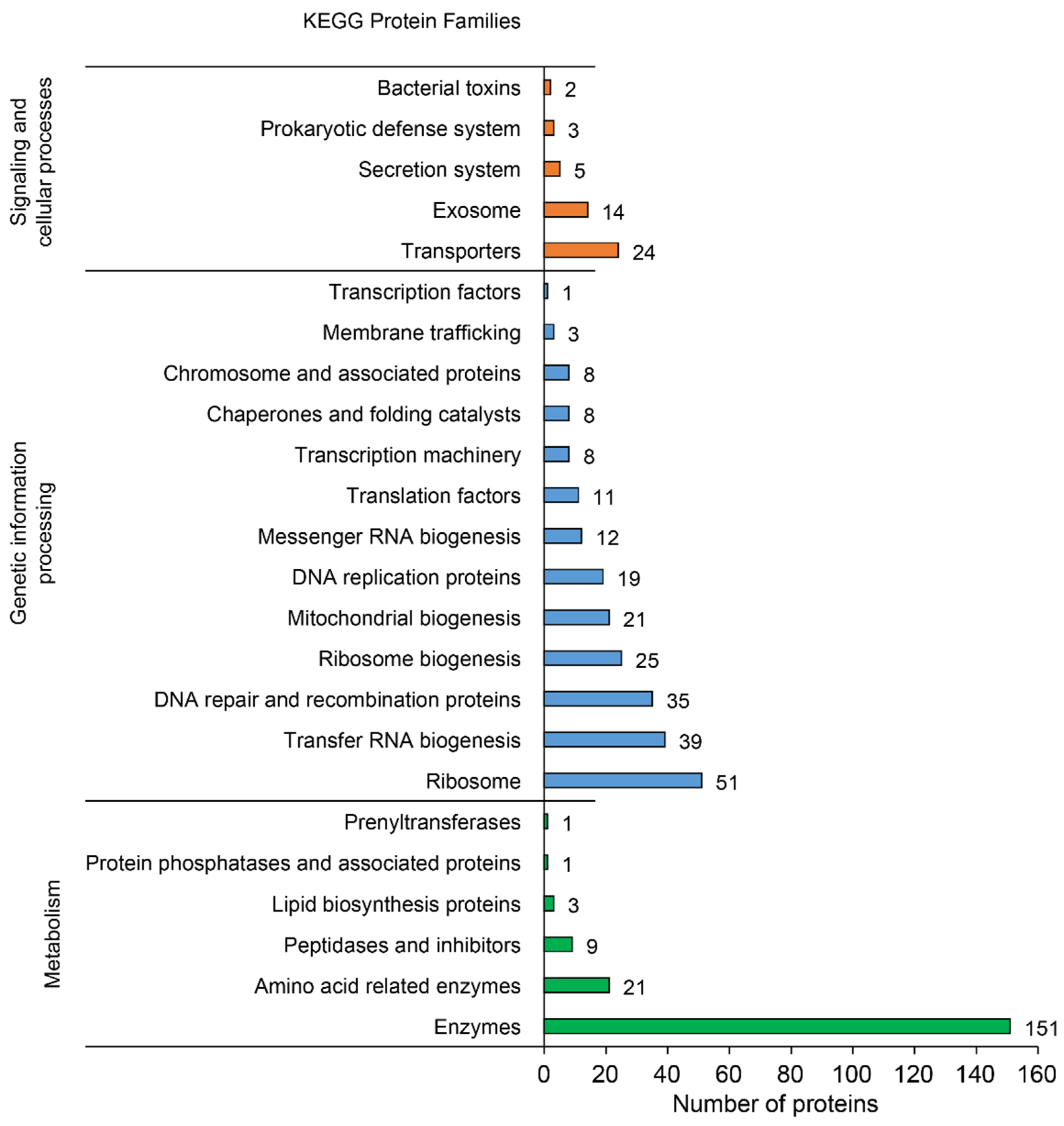
3.2.2. Functional Annotation
3.3. Key Features
3.3.1. Transporters and Metabolic Genes
3.3.2. Effector Genes and Potential Mobile Units
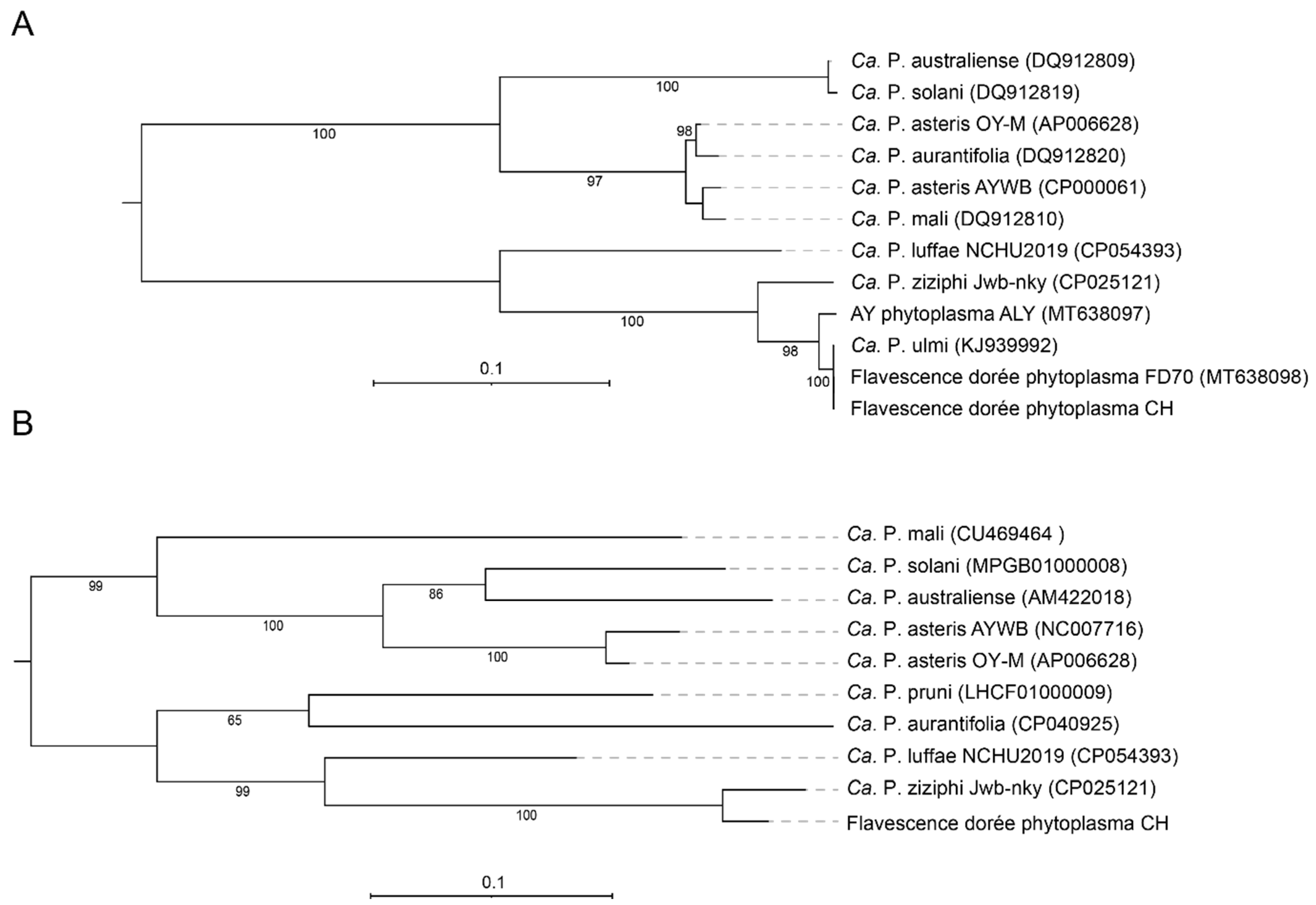
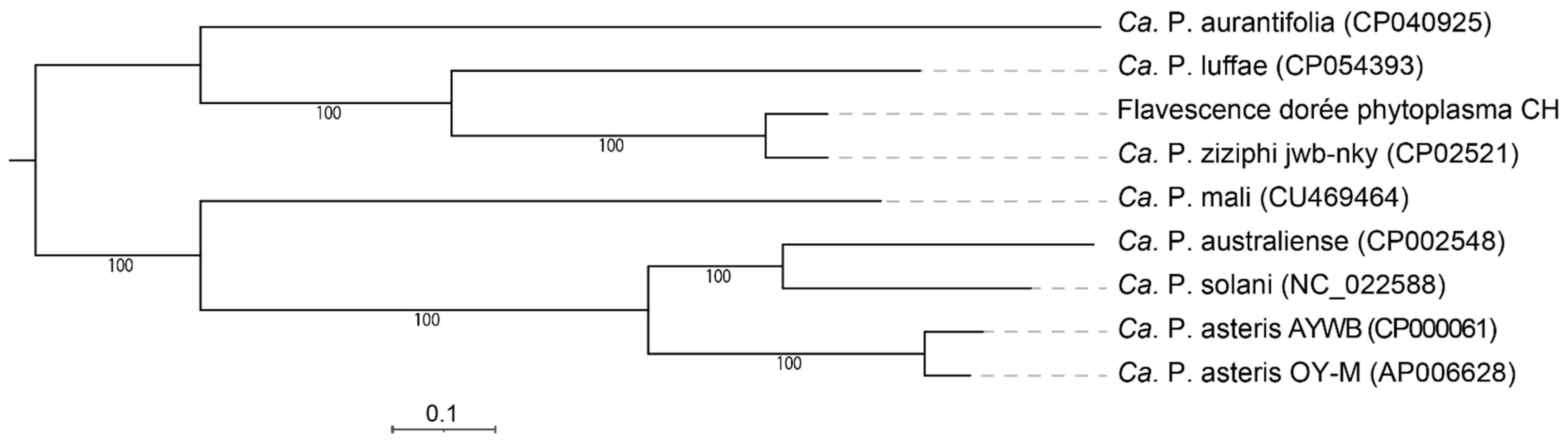
3.4. Phylogeny and Taxonomy
3.5. Full Genome Comparison
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hogenhout, S.A.; Oshima, K.; Ammarel, D.; Kakizawa, S.; Kingdom, H.N.; Namba, S. Phytoplasmas: Bacteria that manipulate plants and insects. Mol. Plant Pathol. 2008, 9, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Namba, S. Molecular and biological properties of phytoplasmas. Proc. Jpn. Acad. Ser. B 2019, 95, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Bertaccini, A.; Duduk, B.; Paltrinieri, S.; Contaldo, N. Phytoplasmas and Phytoplasma Diseases: A Severe Threat to Agriculture. Am. J. Plant Sci. 2014, 5, 1763–1788. [Google Scholar] [CrossRef]
- Lee, I.M.; Davis, R.E.; Gundersen-Rindal, D.E. Phytoplasma: Phytopathogenic mollicutes. Annu. Rev. Microbiol. 2000, 54, 221–255. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, H.; Maejima, K.; Tokuda, R.; Kitazawa, Y.; Nijo, T.; Wei, W.; Kumita, K.; Miyazaki, A.; Namba, S.; Yamaji, Y. Spatiotemporal dynamics and quantitative analysis of phytoplasmas in insect vectors. Sci. Rep. 2020, 10, 4291. [Google Scholar] [CrossRef] [PubMed]
- Trivellone, V.; Dietrich, C.H. Evolutionary Diversification in Insect Vector–Phytoplasma–Plant Associations. Ann. Entomol. Soc. Am. 2021, 114, 137–150. [Google Scholar] [CrossRef]
- Thébaud, G.; Yvon, M.; Alary, R.; Sauvion, N.; Labonne, G. Efficient transmission of ‘Candidatus Phytoplasma prunorum’ is delayed by eight months due to a long latency in its host-alternating vector. Phytopathology 2009, 99, 265–273. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, J.; Ewing, A.; Miller, S.A.; Jancso Radek, A.; Shevchenko, D.V.; Tsukerman, K.; Walunas, T.; Lapidus, A.; Campbell, J.W.; et al. Living with genome instability: The adaptation of phytoplasmas to diverse environments of their insect and plant hosts. J. Bacteriol. 2006, 188, 3682–3696. [Google Scholar] [CrossRef]
- Oshima, K.; Ishii, Y.; Kakizawa, S.; Sugawara, K.; Neriya, Y.; Himeno, M.; Minato, N.; Miura, C.; Shiraishi, T.; Yamaji, Y.; et al. Dramatic Transcriptional Changes in an Intracellular Parasite Enable Host Switching between Plant and Insect. PLoS ONE 2011, 6, e23242. [Google Scholar] [CrossRef]
- Jollard, C.; Foissac, X.; Desqué, D.; Razan, F.; Garcion, C.; Beven, L.; Eveillard, S. Flavescence Dorée Phytoplasma Has Multiple ftsH Genes that Are Differentially Expressed in Plants and Insects. Int. J. Mol. Sci. 2019, 21, 150. [Google Scholar] [CrossRef]
- Doi, Y.; Teranaka, M.; Yora, K.; Asuyama, H. Mycoplasma- or PLT Group-like Microorganisms Found in the Phloem Elements of Plants Infected with Mulberry Dwarf, Potato Witches’ Broom, Aster Yellows, or Paulownia Witches’ Broom. Ann. Phytopath. Soc. Jpn. 1967, 33, 259–266. [Google Scholar] [CrossRef]
- Wei, W.; Lee, I.-M.; Davis, R.E.; Suo, X.; Zhao, Y. Automated RFLP pattern comparison and similarity coefficient calculation for rapid delineation of new and distinct phytoplasma 16Sr subgroup lineages. Int. J. Syst. Evol. Microbiol. 2008, 58, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Davis, R.E. Criteria for phytoplasma 16Sr group/subgroup delineation and the need of a platform for proper registration of new groups and subgroups. Int. J. Syst. Evol. Microbiol. 2016, 66, 2121–2123. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, W.; Lee, I.-M.; Shao, J.; Suo, X.; Davis, R.E. Construction of an interactive online phytoplasma classification tool, iPhyClassifier, and its application in analysis of the peach X-disease phytoplasma group (16SrIII). Int. J. Syst. Evol. Microbiol. 2009, 59, 2582–2593. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-C.; Chen, L.-L.; Lo, W.-S.; Lin, C.-P.; Kuo, C.-H. Comparative analysis of the peanut witches’-broom phytoplasma genome reveals horizontal transfer of potential mobile units and effectors. PLoS ONE 2013, 8, e62770. [Google Scholar] [CrossRef]
- Huang, C.T.; Cho, S.T.; Lin, Y.C.; Tan, C.M.; Chiu, Y.C.; Yang, J.Y.; Kuo, C.H. Comparative Genome Analysis of ‘Candidatus Phytoplasma luffae’ Reveals the Influential Roles of Potential Mobile Units in Phytoplasma Evolution. Front. Microbiol. 2022, 13, 773608. [Google Scholar] [CrossRef]
- Arashida, R.; Kakizawa, S.; Hoshi, A.; Ishii, Y.; Jung, H.Y.; Kagiwada, S.; Yamaji, Y.; Oshima, K.; Namba, S. Heterogeneic dynamics of the structures of multiple gene clusters in two pathogenetically different lines originating from the same phytoplasma. DNA Cell Biol. 2008, 27, 209–217. [Google Scholar] [CrossRef]
- Bai, X.; Correa, V.R.; Toruño, T.Y.; Ammar, E.D.; Kamoun, S.; Hogenhout, S.A. AY-WB phytoplasma secretes a protein that targets plant cell nuclei. Mol. Plant-Microbe Interact. 2009, 22, 18–30. [Google Scholar] [CrossRef]
- Anabestani, A.; Izadpanah, K.; Abbà, S.; Galetto, L.; Ghorbani, A.; Palmano, S.; Siampour, M.; Veratti, F.; Marzachì, C. Identification of putative effector genes and their transcripts in three strains related to ‘Candidatus Phytoplasma aurantifolia’. Microbiol. Res. 2017, 199, 57–66. [Google Scholar] [CrossRef]
- Deng, M.; Ma, F.; Zhang, X.; Huang, J.; Yang, J.; Chen, M.; Zhou, J.; Sun, Q.; Sun, J. Genome-wide identification of jujube witches’ broom phytoplasma effectors revealed the role of SJP3 in inducing phyllody. Sci. Hortic. 2021, 290, 110548. [Google Scholar] [CrossRef]
- Sugio, A.; Kingdom Heather, N.; MacLean Allyson, M.; Grieve Victoria, M.; Hogenhout Saskia, A. Phytoplasma protein effector SAP11 enhances insect vector reproduction by manipulating plant development and defense hormone biosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, E1254–E1263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ma, F.; Yao, Y.; Deng, M.; Chen, M.; Zhang, S.; Li, Y.; Yang, J.; Zhang, N.; Huang, J.; et al. Jujube witches’ broom phytoplasma effectors SJP1 and SJP2 induce lateral bud outgrowth by repressing the ZjBRC1-controlled auxin efflux channel. Plant Cell Environ. 2021, 44, 3257–3272. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.M.; Li, C.H.; Tsao, N.W.; Su, L.W.; Lu, Y.T.; Chang, S.H.; Lin, Y.Y.; Liou, J.C.; Hsieh, L.C.; Yu, J.Z.; et al. Phytoplasma SAP11 alters 3-isobutyl-2-methoxypyrazine biosynthesis in Nicotiana benthamiana by suppressing NbOMT1. J. Exp. Bot. 2016, 67, 4415–4425. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Tan, C.M.; Wu, C.T.; Lin, T.H.; Jiang, S.Y.; Liu, R.C.; Tsai, M.C.; Su, L.W.; Yang, J.Y. Alterations of plant architecture and phase transition by the phytoplasma virulence factor SAP11. J. Exp. Bot. 2018, 69, 5389–5401. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; MacLean, A.M.; Sugio, A.; Maqbool, A.; Busscher, M.; Cho, S.T.; Kamoun, S.; Kuo, C.H.; Immink, R.G.H.; Hogenhout, S.A. Parasitic modulation of host development by ubiquitin-independent protein degradation. Cell 2021, 184, 5201–5214.e12. [Google Scholar] [CrossRef]
- Orlovskis, Z.; Hogenhout, S.A. A Bacterial Parasite Effector Mediates Insect Vector Attraction in Host Plants Independently of Developmental Changes. Front. Plant Sci. 2016, 7, 885. [Google Scholar] [CrossRef]
- Hoshi, A.; Oshima, K.; Kakizawa, S.; Ishii, Y.; Ozeki, J.; Hashimoto, M.; Komatsu, K.; Kagiwada, S.; Yamaji, Y.; Namba, S. A unique virulence factor for proliferation and dwarfism in plants identified from a phytopathogenic bacterium. Proc. Natl. Acad. Sci. USA 2009, 106, 6416–6421. [Google Scholar] [CrossRef]
- Tomkins, M.; Kliot, A.; Marée, A.F.M.; Hogenhout, S.A. A multi-layered mechanistic modelling approach to understand how effector genes extend beyond phytoplasma to modulate plant hosts, insect vectors and the environment. Curr. Opin. Plant Biol. 2018, 44, 39–48. [Google Scholar] [CrossRef]
- Garcion, C.; Béven, L.; Foissac, X. Comparison of Current Methods for Signal Peptide Prediction in Phytoplasmas. Front. Microbiol. 2021, 12, 661524. [Google Scholar] [CrossRef]
- Arricau-Bouvery, N.; Duret, S.; Dubrana, M.P.; Batailler, B.; Desque, D.; Beven, L.; Danet, J.L.; Monticone, M.; Bosco, D.; Malembic-Maher, S.; et al. Variable Membrane Protein A of Flavescence Doree Phytoplasma Binds the Midgut Perimicrovillar Membrane of Euscelidius variegatus and Promotes Adhesion to Its Epithelial Cells. Appl. Environ. Microbiol. 2018, 84, e02487-17. [Google Scholar] [CrossRef]
- Boonrod, K.; Munteanu, B.; Jarausch, B.; Jarausch, W.; Krczal, G. An immunodominant membrane protein (Imp) of ‘Candidatus Phytoplasma mali’ binds to plant actin. Mol. Plant Microbe Interact. 2012, 25, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Galetto, L.; Bosco, D.; Balestrini, R.; Genre, A.; Fletcher, J.; Marzachi, C. The major antigenic membrane protein of “Candidatus Phytoplasma asteris” selectively interacts with ATP synthase and actin of leafhopper vectors. PLoS ONE 2011, 6, e22571. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, M.; Galetto, L.; Bosco, D.; Bulgarelli, A.; Vallino, M.; Veratti, F.; Marzachi, C. Role of the major antigenic membrane protein in phytoplasma transmission by two insect vector species. BMC Microbiol. 2015, 15, 193. [Google Scholar] [CrossRef] [PubMed]
- Chuche, J.; Thiéry, D. Biology and ecology of the Flavescence dorée vector Scaphoideus titanus: A review. Agron. Sustain. Dev. 2014, 34, 381–403. [Google Scholar] [CrossRef]
- EU. COUNCIL DIRECTIVE 2000/29/EC of 8 May 2000 on Protective Measures against the Introduction into the Community of Organisms Harmful to Plants or Plant Products and against Their Spread within the Community. OJI 2000, 169, 2000. [Google Scholar]
- Nijo, T.; Iwabuchi, N.; Tokuda, R.; Suzuki, T.; Matsumoto, O.; Miyazaki, A.; Maejima, K.; Oshima, K.; Namba, S.; Yamaji, Y. Enrichment of phytoplasma genome DNA through a methyl-CpG binding domain-mediated method for efficient genome sequencing. J. Gen. Plant Pathol. 2021, 87, 154–163. [Google Scholar] [CrossRef]
- Pelletier, C.; Salar, P.; Gillet, J.; Cloquemin, G.; Very, P.; Foissac, X.; Malembic-Maher, S. Triplex real-time PCR assay for sensitive and simultaneous detection of grapevine phytoplasmas of the 16SrV and 16SrXII-A groups with an endogenous analytical control. Vitis 2009, 48, 87–95. [Google Scholar] [CrossRef]
- Ripamonti, M.; Maron, F.; Cornara, D.; Marzachi, C.; Fereres, A.; Bosco, D. Leafhopper feeding behaviour on three grapevine cultivars with different susceptibilities to Flavescence doree. J. Insect. Physiol. 2022, 137, 104366. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Rodriguez, R.L.; Gunturu, S.; Harvey, W.T.; Rossello-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Araujo, F.A.; Barh, D.; Silva, A.; Guimaraes, L.; Ramos, R.T.J. GO FEAT: A rapid web-based functional annotation tool for genomic and transcriptomic data. Sci. Rep. 2018, 8, 1794. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Wang, J.; Song, L.; Jiao, Q.; Yang, S.; Gao, R.; Lu, X.; Zhou, G. Comparative genome analysis of jujube witches’-broom Phytoplasma, an obligate pathogen that causes jujube witches’-broom disease. BMC Genom. 2018, 19, 689. [Google Scholar] [CrossRef]
- Razin, S.; Yogev, D.; Naot, Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 1998, 62, 1094–1156. [Google Scholar] [CrossRef] [PubMed]
- Kube, M.; Schneider, B.; Kuhl, H.; Dandekar, T.; Heitmann, K.; Migdoll, A.M.; Reinhardt, R.; Seemuller, E. The linear chromosome of the plant-pathogenic mycoplasma ‘Candidatus Phytoplasma mali’. BMC Genom. 2008, 9, 306. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.M.; Randall, L.L. The Sec System: Protein Export in Escherichia coli. EcoSal Plus 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Seemuller, E.; Sule, S.; Kube, M.; Jelkmann, W.; Schneider, B. The AAA+ ATPases and HflB/FtsH proteases of ‘Candidatus Phytoplasma mali’: Phylogenetic diversity, membrane topology, and relationship to strain virulence. Mol. Plant Microbe Interact. 2013, 26, 367–376. [Google Scholar] [CrossRef]
- Jomantiene, R.; Zhao, Y.; Davis, R.E. Sequence-variable mosaics: Composites of recurrent transposition characterizing the genomes of phylogenetically diverse phytoplasmas. DNA Cell Biol. 2007, 26, 557–564. [Google Scholar] [CrossRef]
- Wei, W.; Davis, R.E.; Jomantiene, R.; Zhao, Y. Ancient, recurrent phage attacks and recombination shaped dynamic sequence-variable mosaics at the root of phytoplasma genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 11827–11832. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Lee, I.-M.; Gundersen-Rindal, D.; Davis, R.E.; Bartoszyk, I. Revised Classification Scheme of Phytoplasmas based on RFLP Analyses of 16S rRNA and Ribosomal Protein Gene Sequences. Int. J. Syst. Evol. Microbiol. 1998, 48, 1153–1169. [Google Scholar] [CrossRef]
- Lee, I.-M.; Hammond, R.E.; Davis, R.E.; Gundersen, D.E. Universal Amplification and Analysis of Pathogen 16S rDNA for Classification and Identification of Mycoplasmalike Organisms. Phytopathology 1993, 83, 834–842. [Google Scholar] [CrossRef]
- Malembic-Maher, S.; Desque, D.; Khalil, D.; Salar, P.; Bergey, B.; Danet, J.L.; Duret, S.; Dubrana-Ourabah, M.P.; Beven, L.; Ember, I.; et al. When a Palearctic bacterium meets a Nearctic insect vector: Genetic and ecological insights into the emergence of the grapevine Flavescence doree epidemics in Europe. PLoS Pathog. 2020, 16, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Zubert, C.; Ilic, A.M.; Duduk, B.; Kube, M. The Genome Reduction Excludes the Ribosomal Rescue System in Acholeplasmataceae. Microb. Physiol. 2022, 32, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bertaccini, A.; Arocha-Rosete, Y.; Contaldo, N.; Duduk, B.; Fiore, N.; Montano, H.G.; Kube, M.; Kuo, C.H.; Martini, M.; Oshima, K.; et al. Revision of the ‘Candidatus Phytoplasma’ species description guidelines. Int. J. Syst. Evol. Microbiol. 2022, 72, 005353. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.M.; Lin, Y.C.; Li, J.R.; Chien, Y.Y.; Wang, C.J.; Chou, L.; Wang, C.W.; Chiu, Y.C.; Kuo, C.H.; Yang, J.Y. Accelerating Complete Phytoplasma Genome Assembly by Immunoprecipitation-Based Enrichment and MinION-Based DNA Sequencing for Comparative Analyses. Front. Microbiol. 2021, 12, 766221. [Google Scholar] [CrossRef] [PubMed]
- Carle, P.; Malembic-Maher, S.; Arricau-Bouvery, N.; Desque, D.; Eveillard, S.; Carrere, S.; Foissac, X. ‘Flavescence dorée’ phytoplasma genome: A metabolism oriented towards glycolysis and protein degradation. Bull. Insectol. 2011, 64, S13–S14. [Google Scholar]
- Malembic-Maher, S.; Constable, F.; Cimerman, A.; Arnaud, G.; Carle, P.; Foissac, X.; Boudon-Padieu, E. A chromosome map of the Flavescence doree phytoplasma. Microbiology 2008, 154, 1454–1463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Music, M.S.; Samarzija, I.; Hogenhout, S.A.; Haryono, M.; Cho, S.T.; Kuo, C.H. The genome of ‘Candidatus Phytoplasma solani’ strain SA-1 is highly dynamic and prone to adopting foreign sequences. Syst. Appl. Microbiol. 2019, 42, 117–127. [Google Scholar] [CrossRef]
- Kube, M.; Mitrovic, J.; Duduk, B.; Rabus, R.; Seemuller, E. Current view on phytoplasma genomes and encoded metabolism. Sci. World J. 2012, 2012, 185942. [Google Scholar] [CrossRef]
- Oshima, K.; Kakizawa, S.; Nishigawa, H.; Jung, H.Y.; Wei, W.; Suzuki, S.; Arashida, R.; Nakata, D.; Miyata, S.; Ugaki, M.; et al. Reductive evolution suggested from the complete genome sequence of a plant-pathogenic phytoplasma. Nat. Genet. 2004, 36, 27–29. [Google Scholar] [CrossRef]
- Becker, A.; Schloder, P.; Steele, J.E.; Wegener, G. The regulation of trehalose metabolism in insects. Experientia 1996, 52, 433–439. [Google Scholar] [CrossRef]
- Lemoine, R.; La Camera, S.; Atanassova, R.; Dedaldechamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.L.; Laloi, M.; Coutos-Thevenot, P.; Maurousset, L.; et al. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Ziegler, H. Encyclopedia of Plant Physiology, Transport in Plants 1, Phloem Transport; Springer: Berlin/Heidelberg, Germany, 1975; Volume 1. [Google Scholar]
- Hekstra, D.; Tommassen, J. Functional exchangeability of the ABC proteins of the periplasmic binding protein-dependent transport systems Ugp and Mal of Escherichia coli. J. Bacteriol. 1993, 175, 6546–6552. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, L.I.; Vergara Meza, J.G.; Cabarca, S.; Costa-Martins, A.G.; Balan, A. Comparison of carbohydrate ABC importers from Mycobacterium tuberculosis. BMC Genom. 2021, 22, 841. [Google Scholar] [CrossRef] [PubMed]
- Economou, A. Following the leader: Bacterial protein export through the Sec pathway. Trends Microbiol. 1999, 7, 315–320. [Google Scholar] [CrossRef]
- MacLean, A.M.; Orlovskis, Z.; Kowitwanich, K.; Zdziarska, A.M.; Angenent, G.C.; Immink, R.G.; Hogenhout, S.A. Phytoplasma effector SAP54 hijacks plant reproduction by degrading MADS-box proteins and promotes insect colonization in a RAD23-dependent manner. PLoS Biol. 2014, 12, e1001835. [Google Scholar] [CrossRef]
- MacLean, A.M.; Sugio, A.; Makarova, O.V.; Findlay, K.C.; Grieve, V.M.; Toth, R.; Nicolaisen, M.; Hogenhout, S.A. Phytoplasma effector SAP54 induces indeterminate leaf-like flower development in Arabidopsis plants. Plant Physiol. 2011, 157, 831–841. [Google Scholar] [CrossRef]
- Pecher, P.; Moro, G.; Canale, M.C.; Capdevielle, S.; Singh, A.; MacLean, A.; Sugio, A.; Kuo, C.H.; Lopes, J.R.S.; Hogenhout, S.A. Phytoplasma SAP11 effector destabilization of TCP transcription factors differentially impact development and defence of Arabidopsis versus maize. PLoS Pathog. 2019, 15, e1008035. [Google Scholar] [CrossRef]
- Sugawara, K.; Honma, Y.; Komatsu, K.; Himeno, M.; Oshima, K.; Namba, S. The alteration of plant morphology by small peptides released from the proteolytic processing of the bacterial peptide TENGU. Plant Physiol. 2013, 162, 2005–2014. [Google Scholar] [CrossRef]
- Arricau-Bouvery, N.; Duret, S.; Dubrana, M.P.; Desque, D.; Eveillard, S.; Brocard, L.; Malembic-Maher, S.; Foissac, X. Interactions between the flavescence doree phytoplasma and its insect vector indicate lectin-type adhesion mediated by the adhesin VmpA. Sci. Rep. 2021, 11, 11222. [Google Scholar] [CrossRef]
- Trivellone, V.; Ripamonti, M.; Angelini, E.; Filippin, L.; Rossi, M.; Marzachi, C.; Galetto, L. Evidence suggesting interactions between immunodominant membrane protein Imp of Flavescence doree phytoplasma and protein extracts from distantly related insect species. J. Appl. Microbiol. 2019, 127, 1801–1813. [Google Scholar] [CrossRef]
- Toruno, T.Y.; Music, M.S.; Simi, S.; Nicolaisen, M.; Hogenhout, S.A. Phytoplasma PMU1 exists as linear chromosomal and circular extrachromosomal elements and has enhanced expression in insect vectors compared with plant hosts. Mol. Microbiol. 2010, 77, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.T.; Liefting, L.W.; Havukkala, I.; Beever, R.E. Comparison of the complete genome sequence of two closely related isolates of ‘Candidatus Phytoplasma australiense’ reveals genome plasticity. BMC Genom. 2013, 14, 529. [Google Scholar] [CrossRef] [PubMed]
- Shotland, Y.; Shifrin, A.; Ziv, T.; Teff, D.; Koby, S.; Kobiler, O.; Oppenheim, A.B. Proteolysis of bacteriophage lambda CII by Escherichia coli FtsH (HflB). J. Bacteriol. 2000, 182, 3111–3116. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain (Accession) | 16S rRNA Group | Genome Size (bp) | G/C Content (%) | No. of CDS | Protein-Coding Regions (%) | No. of tRNA Genes | No. of rRNA Operons |
|---|---|---|---|---|---|---|---|
| FDp strain CH (CP097583) | V | 654,223 | 21.7 | 506 | 77 | 32 | 2 |
| ‘Ca. P. ziziphi’ jwb-nky (CP02521) | V | 750,803 | 23.2 | 671 | 78 | 32 | 2 |
| ‘Ca. P. asteris’ AYWB (CP000061) | I | 706,569 | 26.9 | 636 | 73.9 | 32 | 2 |
| ‘Ca. P. asteris’ OY-M (AP006628) | I | 853,092 | 27.8 | 752 | 72.8 | 32 | 2 |
| ‘Ca. P. luffae’ (CP054393) | VIII | 769,143 | 23.3 | 725 | 80.1 | 31 | 2 |
| ‘Ca. P. australiense’ (CP002548) | XII | 959,779 | 27.2 | 928 | 75.5 | 35 | 2 |
| ‘Ca. P. mali’ (CU469464) | X | 601,943 | 21.4 | 500 | 78.2 | 32 | 2 |
| ‘Ca. P. aurantifolia’ (CP040925) | II | 635,584 | 24.5 | 471 | 66.1 | 24 | 2 |
| Protein ID | Function | SignalP5.0 | Phobius | Accession # of Orthologues in Other Phytoplasmas (Max. 2) |
|---|---|---|---|---|
| FlDop_00023 | Variable membrane protein B | SP | SP and 1 TM | VIO49504 (Alder yellows P.) |
| FlDop_00041 | Hypothetical protein (htmp2) | no prediction | TM | WP_225696264 (Ca. P. sp. AldY-WA1) WP_121463947 (Ca. P. ziziphi) |
| FlDop_00048 * | Hypothetical protein | SP | SP and 1 TM | none |
| FlDop_00049 * | Hypothetical protein | SP | SP and 1 TM | none |
| FlDop_00090 | Hypothetical protein | SP | SP | WP_225696128 (Ca. P. sp. AldY-WA1) WP_121464024 (Ca. P. ziziphi) |
| FlDop_00101 | Hypothetical protein | SP | TM | WP_238055118 (Ca. P. ziziphi) WP_121464035 (Ca. P. ziziphi) |
| FlDop_00112 | Hypothetical protein | SP | SP | AYJ01330 (Ca. P. ziziphi) |
| FlDop_00153 | Hypothetical protein (htmp5) | no prediction | TM | WP_121464226 (Ca. P. ziziphi) |
| FlDop_00158 * | Hypothetical protein | SP | SP | none |
| FlDop_00183 | Hypothetical protein | SP | SP | WP_026072021 (Poinsettia branch-inducing P.) WP_152411650 (Milkweed yellows P.) |
| FlDop_00185 | Hypothetical protein (SVM family) | SP | SP | WP_034172411 (Chrysanthemum yellows P.) WP_024563506 (Ca. P. tritici) |
| FlDop_00190 | Hypothetical protein | SP | SP | PQP79517 (Ca. P. phoenicium) WP_078123062 (Ca. P. aurantifolia) |
| FlDop_00246 * | Hypothetical protein | no prediction | SP | none |
| FlDop_00265 | Hypothetical protein | SP | TM | WP_121464113 (Ca. P. ziziphi) |
| FlDop_00266 | Hypothetical protein | no prediction | SP | WP_121464114 (Ca. P. ziziphi) |
| FlDop_00278 * | Hypothetical protein | SP | SP | none |
| FlDop_00279 * | Hypothetical protein (htmp1) | no prediction | TM | none |
| FlDop_00298 | Hypothetical protein | SP | TM | WP_012504569 (Ca. P. mali) WP_227807101 (Mulberry dwarf P.) |
| FlDop_00404 | Hypothetical protein (htmp3) | no prediction | TM | WP_225696004 (Ca. P. sp. AldY-WA1) WP_121463722 (Ca. P. ziziphi) |
| FlDop_00420 * | Hypothetical protein (htmp4) | SP | SP | none |
| FlDop_00531 | SAP21-like protein | SP | SP | QKX95099 (Rapeseed phyllody P.) WP_122225587 (Ca. P. solani) |
| FDp Strain CH | ‘Ca. P. solani’ | ‘Ca. P. asteris’ OY-M | ‘Ca. P. asteris’ AYWB | ‘Ca. P. ziziphi’ | ‘Ca. P. mali’ | ‘Ca. P. luffae’ | ‘Ca. P. aurantifolia’ | |
|---|---|---|---|---|---|---|---|---|
| ‘Ca. P. australiense’ (NC_021236) | 306 | 335 | 332 | 337 | 302 | 301 | 305 | 290 |
| ‘Ca. P. aurantifolia’ (CP040925) | 295 | 275 | 282 | 291 | 293 | 295 | 309 | |
| ‘Ca. P. luffae’ (CP054393) | 321 | 300 | 301 | 301 | 322 | 307 | ||
| ‘Ca. P. mali’ (NC_011047) | 300 | 299 | 303 | 301 | 302 | |||
| ‘Ca. P. ziziphi’ jwb-nky (CP02521) | 359 | 296 | 307 | 303 | ||||
| ‘Ca. P. asteris’ AYWB (NC_007716) | 299 | 329 | 384 | |||||
| ‘Ca. P. asteris’ OY-M (AP006628) | 302 | 343 | ||||||
| ‘Ca. P. solani’ (GCA_000970375) | 304 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debonneville, C.; Mandelli, L.; Brodard, J.; Groux, R.; Roquis, D.; Schumpp, O. The Complete Genome of the “Flavescence Dorée” Phytoplasma Reveals Characteristics of Low Genome Plasticity. Biology 2022, 11, 953. https://doi.org/10.3390/biology11070953
Debonneville C, Mandelli L, Brodard J, Groux R, Roquis D, Schumpp O. The Complete Genome of the “Flavescence Dorée” Phytoplasma Reveals Characteristics of Low Genome Plasticity. Biology. 2022; 11(7):953. https://doi.org/10.3390/biology11070953
Chicago/Turabian StyleDebonneville, Christophe, Léa Mandelli, Justine Brodard, Raphaël Groux, David Roquis, and Olivier Schumpp. 2022. "The Complete Genome of the “Flavescence Dorée” Phytoplasma Reveals Characteristics of Low Genome Plasticity" Biology 11, no. 7: 953. https://doi.org/10.3390/biology11070953
APA StyleDebonneville, C., Mandelli, L., Brodard, J., Groux, R., Roquis, D., & Schumpp, O. (2022). The Complete Genome of the “Flavescence Dorée” Phytoplasma Reveals Characteristics of Low Genome Plasticity. Biology, 11(7), 953. https://doi.org/10.3390/biology11070953