
A Map of 3′ DNA Transduction Variants Mediated by Non-LTR Retroelements on 3202 Human Genomes



Abstract
Simple Summary
Abstract
1. Introduction
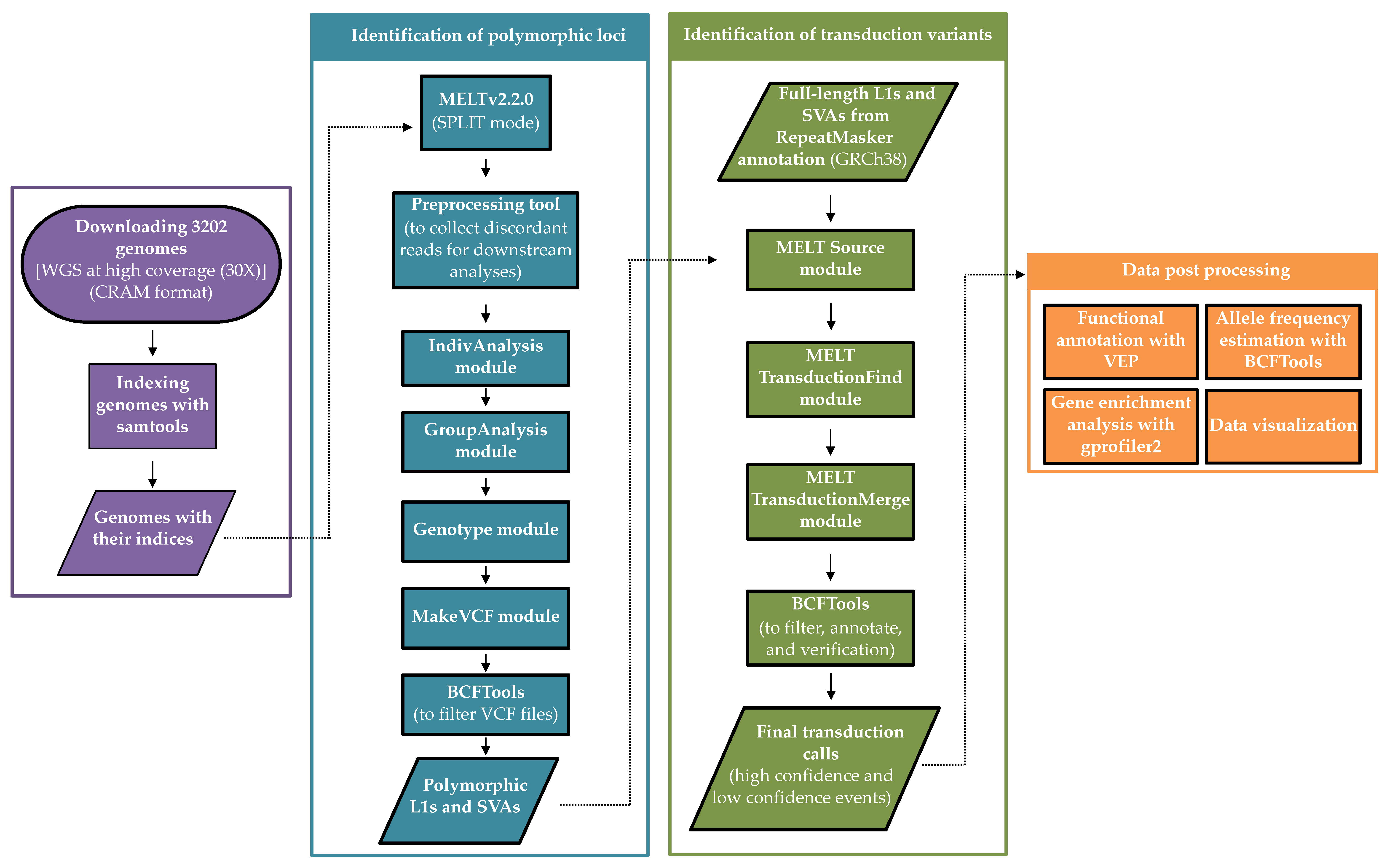
2. Materials and Methods
2.1. Data
2.2. Discovery of MEI Polymorphisms
- bcftools view -i ‘SR >= 3 && ASSESS >= 3′ <input.vcf> | bcftools view -i ‘FILTER=“PASS” || FILTER=“rSD”‘ | bcftools view -e ‘FILTER=“ac0” || FILTER=“hDP” || FILTER=“lc”‘ -o <output.filtered.vcf>

2.3. Identification of 3′ Transduction Events
2.4. Data Post-Processing (Validation, Classification, and Allele Frequency Estimation)
- bcftools plugin fill-tags <input.transductions.vcf> -o <output.transductions.AF.vcf > -- -S sample.txt -t AF,AC
2.5. Functional Annotation
- vep --cache --distance 2000,1000 --regulatory --numbers --variant_class --overlaps --gene_phenotype --canonical --symbol --tab --terms SO --pick -i <tab-delimited format> -o <output.tsv>
2.6. Functional Enrichment Analysis
- functional_analysis <- gost(<gene_list>, organism = “hsapiens”, ordered_query = FALSE,multi_query = FALSE, significant =TRUE, exclude_iea = TRUE, correction_method = “gSCS”, domain_scope = “annotated”, user_threshold = 0.01)
- gostplot(functional_analysis, capped = TRUE, interactive = FALSE)
- publish_gosttable(functional_analysis, highlight_terms = functional_analysis$result[c(1:3,7,9, 4:6, 8, 10),], use_colors = TRUE,show_columns = c("source", "term_name", "term_size", "intersection_size"),filename = NULL)
2.7. Data Visualization
3. Results and Discussion
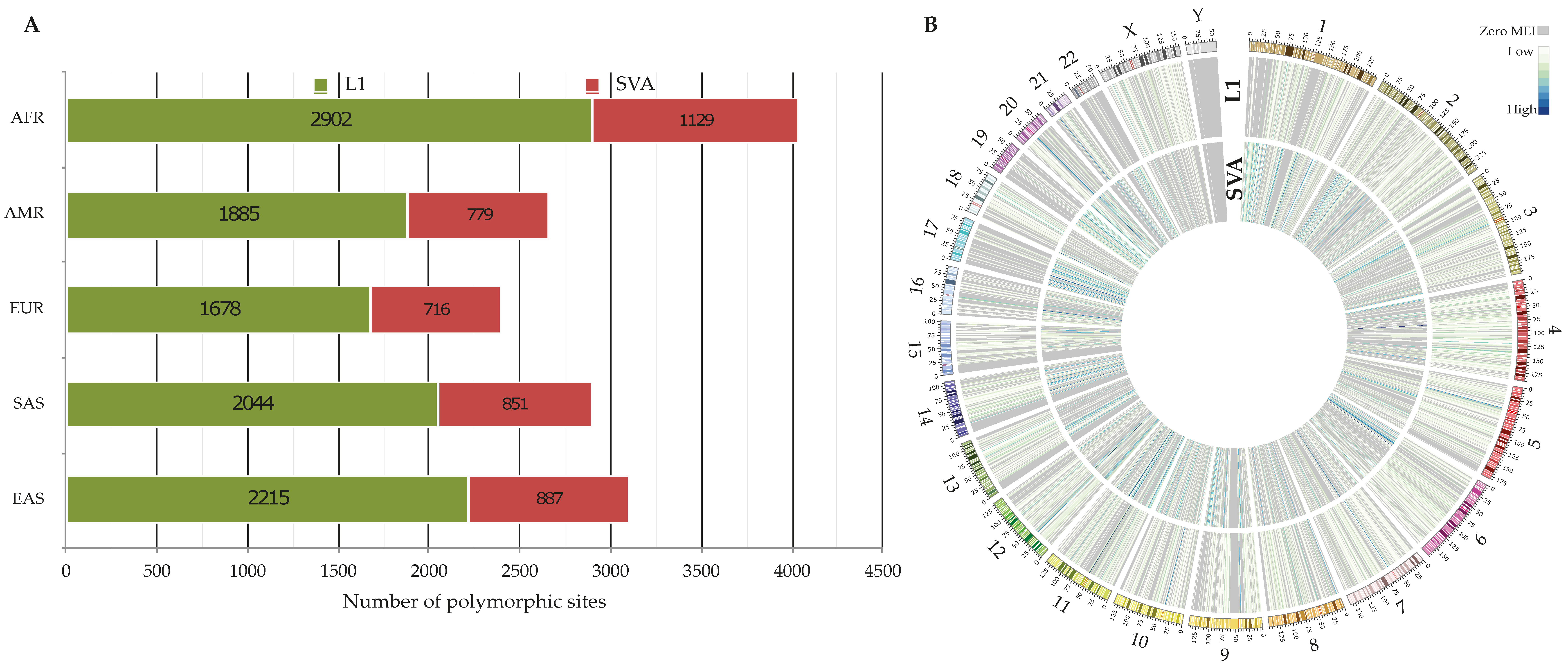
3.1. Polymorphic Non-Reference MEIs
3.2. Transduction Variants
3.2.1. Number of Identified 3′ Transductions
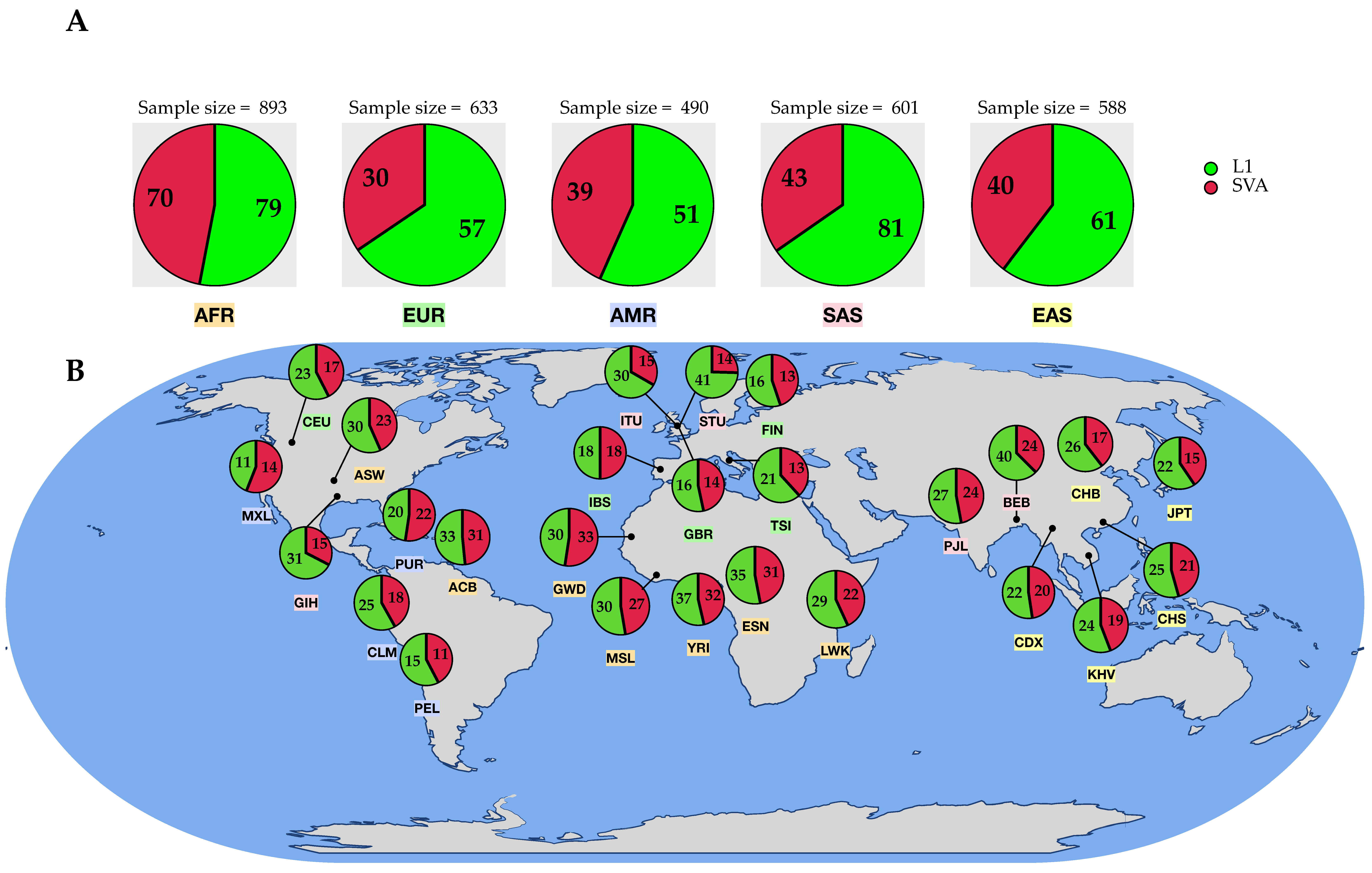
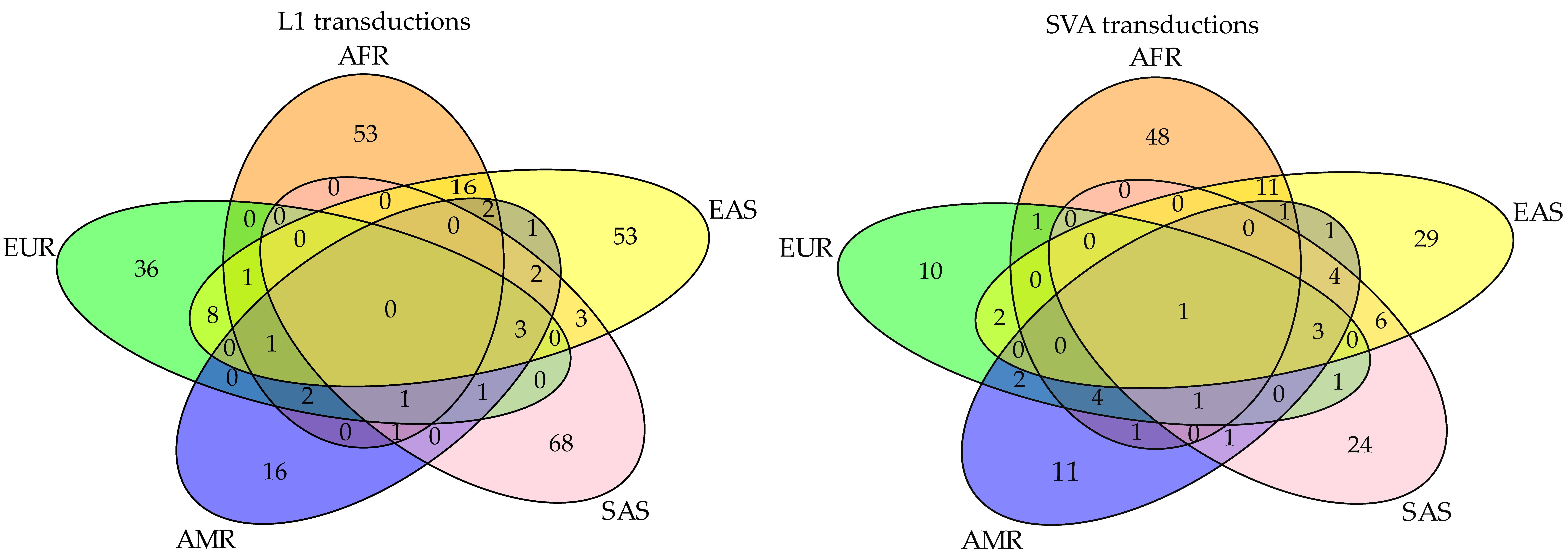
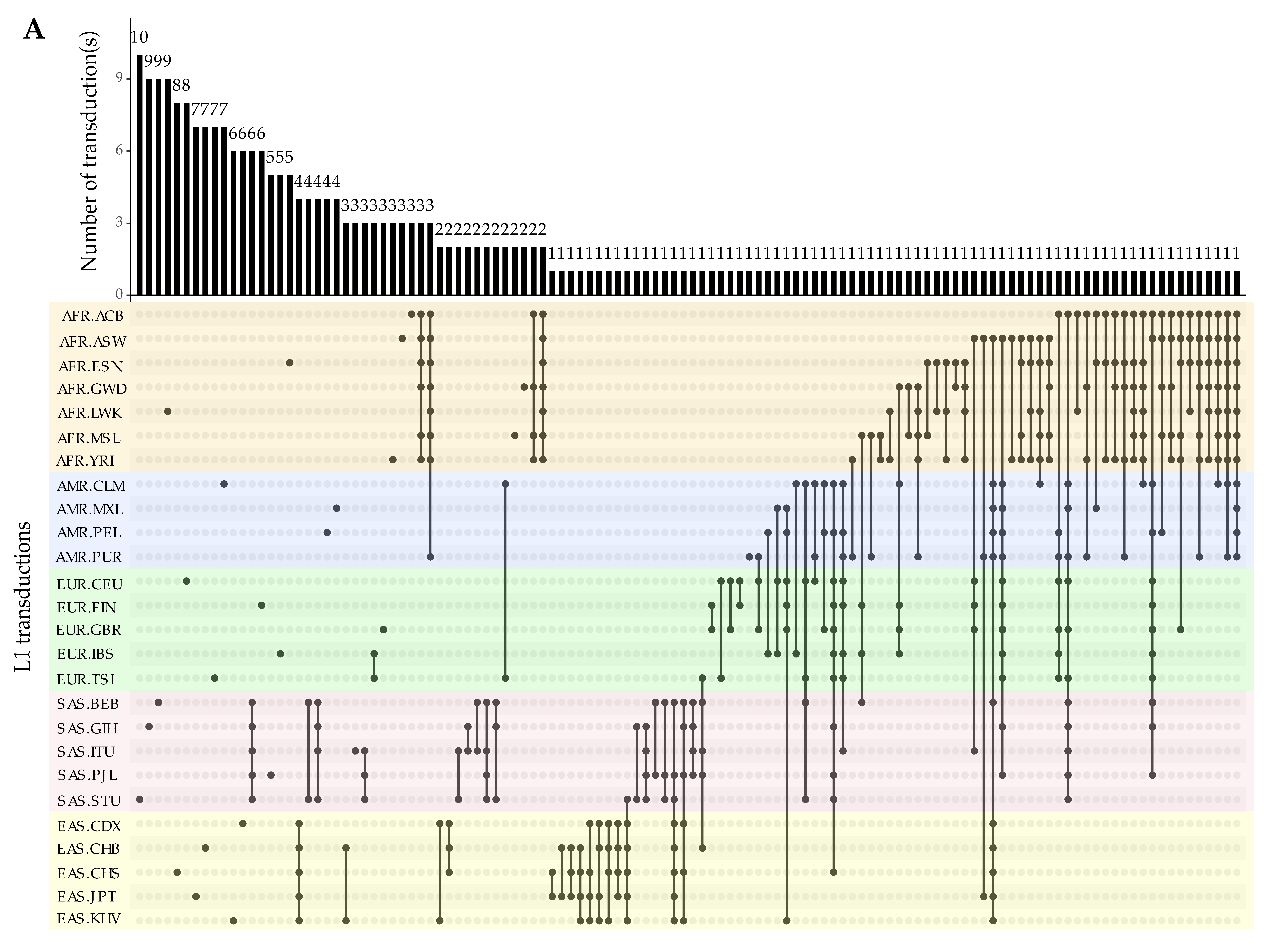
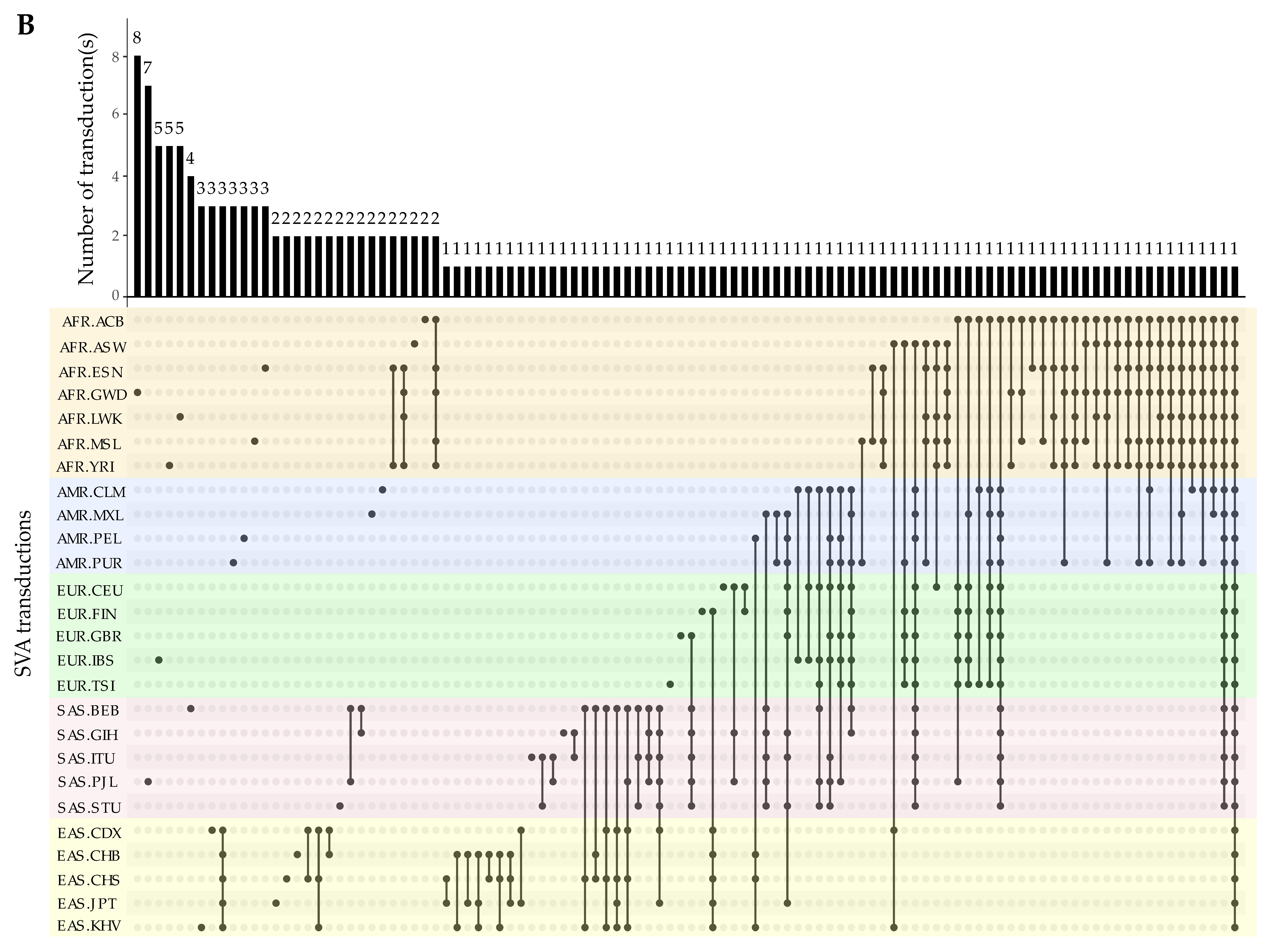
3.2.2. Distribution of 3′ Transductions across the Populations
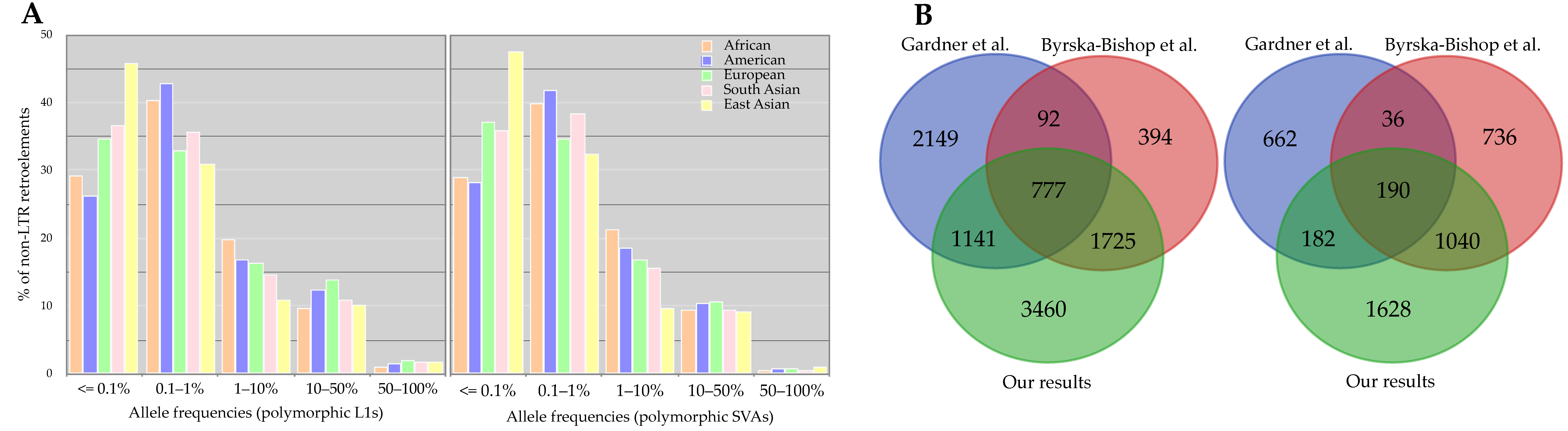
3.2.3. Length and Allele Frequency of 3′ Transductions
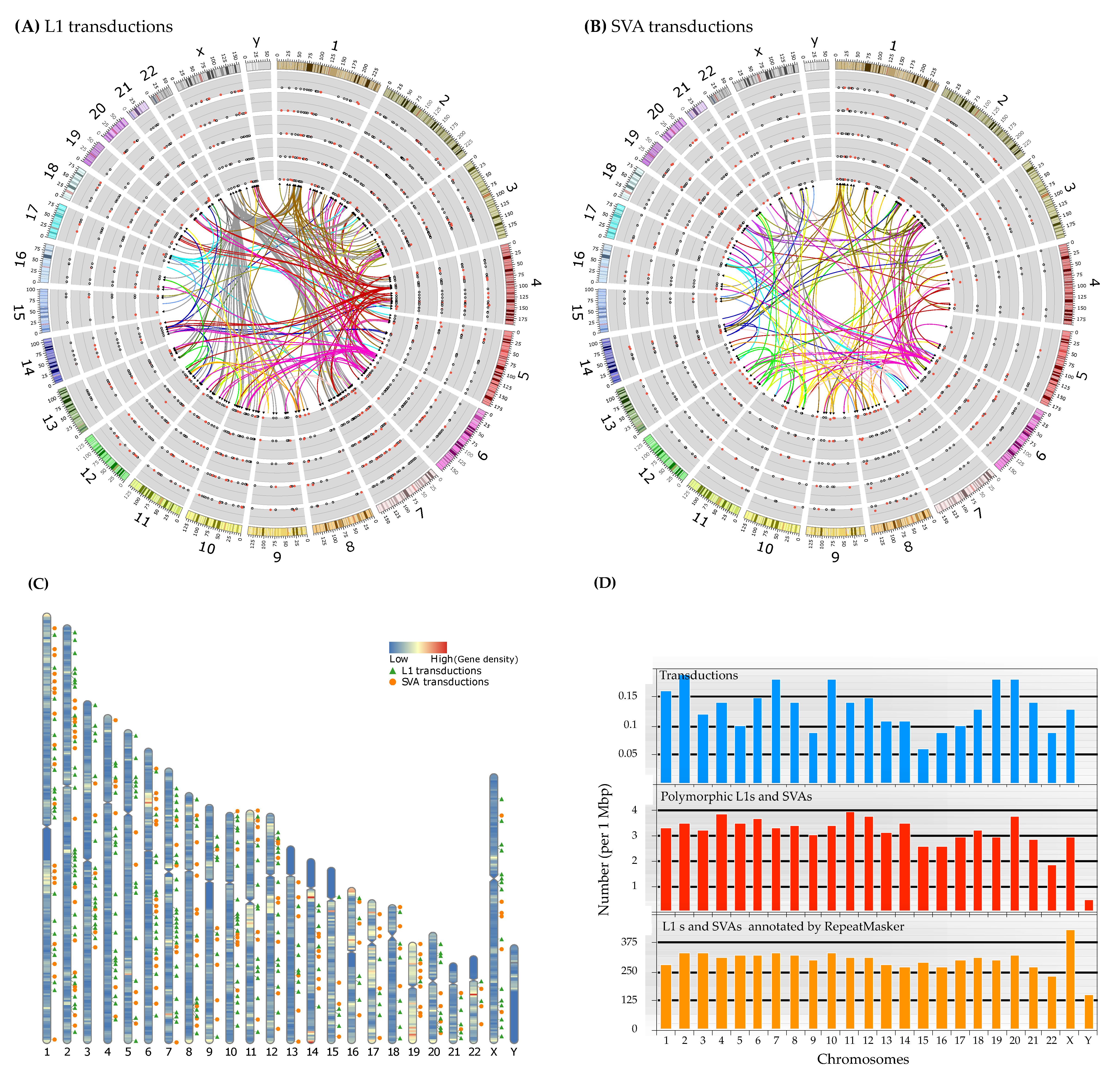
3.2.4. Progenitors and Offspring of 3′ Transductions
3.2.5. Insertion Location and Functional Impact
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoyt, S.J.; Storer, J.M.; Hartley, G.A.; Grady, P.G.S.; Gershman, A.; de Lima, L.G.; Limouse, C.; Halabian, R.; Wojenski, L.; Rodriguez, M.; et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 2022, 376, eabk3112. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Devine, S.E. Which transposable elements are active in the human genome? Trends Genet. 2007, 23, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Goodier, J.L.; Ostertag, E.M.; Kazazian, H.H. Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum. Mol. Genet. 2000, 9, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.V.; DeBerardinis, R.J.; Kazazian, H.H., Jr. Exon shuffling by L1 retrotransposition. Science 1999, 283, 1530–1534. [Google Scholar] [CrossRef]
- Pickeral, O.K.; Makalowski, W.; Boguski, M.S.; Boeke, J.D. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000, 10, 411–415. [Google Scholar] [CrossRef]
- Szak, S.T.; Pickeral, O.K.; Landsman, D.; Boeke, J.D. Identifying related L1 retrotransposons by analyzing 3′ transduced sequences. Genome Biol. 2003, 4, R30. [Google Scholar] [CrossRef]
- Xing, J.; Wang, H.; Belancio, V.P.; Cordaux, R.; Deininger, P.L.; Batzer, M.A. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 17608–17613. [Google Scholar] [CrossRef]
- Solyom, S.; Ewing, A.D.; Rahrmann, E.P.; Doucet, T.; Nelson, H.H.; Burns, M.B.; Harris, R.S.; Sigmon, D.F.; Casella, A.; Erlanger, B.; et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012, 22, 2328–2338. [Google Scholar] [CrossRef]
- Tubio, J.M.C.; Li, Y.; Ju, Y.S.; Martincorena, I.; Cooke, S.L.; Tojo, M.; Gundem, G.; Pipinikas, C.P.; Zamora, J.; Raine, K.; et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 2014, 345, 1251343. [Google Scholar] [CrossRef]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Serra Mari, R.; et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372, eabf7117. [Google Scholar] [CrossRef]
- Bae, J.; Lee, K.W.; Islam, M.N.; Yim, H.S.; Park, H.; Rho, M. iMGEins: Detecting novel mobile genetic elements inserted in individual genomes. BMC Genom. 2018, 19, 944. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H., Jr. SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol. 2010, 20, 234–245. [Google Scholar] [CrossRef]
- Pradhan, B.; Cajuso, T.; Katainen, R.; Sulo, P.; Tanskanen, T.; Kilpivaara, O.; Pitkanen, E.; Aaltonen, L.A.; Kauppi, L.; Palin, K. Detection of subclonal L1 transductions in colorectal cancer by long-distance inverse-PCR and Nanopore sequencing. Sci. Rep. 2017, 7, 14521. [Google Scholar] [CrossRef] [PubMed]
- Siva, N. 1000 Genomes project. Nat. Biotechnol. 2008, 26, 256. [Google Scholar] [CrossRef]
- Konkel, M.K.; Walker, J.A.; Hotard, A.B.; Ranck, M.C.; Fontenot, C.C.; Storer, J.; Stewart, C.; Marth, G.T.; Genomes, C.; Batzer, M.A. Sequence Analysis and Characterization of Active Human Alu Subfamilies Based on the 1000 Genomes Pilot Project. Genome Biol. Evol. 2015, 7, 2608–2622. [Google Scholar] [CrossRef]
- Zheng-Bradley, X.; Flicek, P. Applications of the 1000 Genomes Project resources. Brief. Funct. Genom. 2017, 16, 163–170. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- McVean, G.A.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Durbin, R.M.; Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De La Vega, F.M.; Donnelly, P.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Gardner, E.J.; Lam, V.K.; Harris, D.N.; Chuang, N.T.; Scott, E.C.; Pittard, W.S.; Mills, R.E.; Genomes Project, C.; Devine, S.E. The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 2017, 27, 1916–1929. [Google Scholar] [CrossRef]
- Huang, C.R.; Schneider, A.M.; Lu, Y.; Niranjan, T.; Shen, P.; Robinson, M.A.; Steranka, J.P.; Valle, D.; Civin, C.I.; Wang, T.; et al. Mobile interspersed repeats are major structural variants in the human genome. Cell 2010, 141, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Kural, D.; Stromberg, M.P.; Walker, J.A.; Konkel, M.K.; Stutz, A.M.; Urban, A.E.; Grubert, F.; Lam, H.Y.; Lee, W.P.; et al. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011, 7, e1002236. [Google Scholar] [CrossRef] [PubMed]
- Witherspoon, D.J.; Zhang, Y.; Xing, J.; Watkins, W.S.; Ha, H.; Batzer, M.A.; Jorde, L.B. Mobile element scanning (ME-Scan) identifies thousands of novel Alu insertions in diverse human populations. Genome Res. 2013, 23, 1170–1181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rishishwar, L.; Tellez Villa, C.E.; Jordan, I.K. Transposable element polymorphisms recapitulate human evolution. Mob. DNA 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, C.M.; Collier, P.; Rahbari, R.; Beck, C.R.; Wagstaff, J.F.; Igoe, S.; Moran, J.V.; Badge, R.M. Transduction-specific ATLAS reveals a cohort of highly active L1 retrotransposons in human populations. Hum. Mutat. 2013, 34, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Teng, X.; Zhou, H.; Shi, Y.; Li, Y.; Tang, Y.; Zhang, P.; Luo, H.; Kang, Q.; Xu, T.; et al. Characterizing mobile element insertions in 5675 genomes. Nucleic Acids Res. 2022, 50, 2493–2508. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D. Transposable element detection from whole genome sequence data. Mob. DNA 2015, 6, 24. [Google Scholar] [CrossRef]
- Feusier, J.; Watkins, W.S.; Thomas, J.; Farrell, A.; Witherspoon, D.J.; Baird, L.; Ha, H.; Xing, J.; Jorde, L.B. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019, 29, 1567–1577. [Google Scholar] [CrossRef]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 2021. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tasse, A.M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2019, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xing, J.; Grover, D.; Hedges, D.J.; Han, K.; Walker, J.A.; Batzer, M.A. SVA elements: A hominid-specific retroposon family. J. Mol. Biol. 2005, 354, 994–1007. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, 709. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Zhaodong Hao, D.L. Ying Ge, Jisen Shi, Dolf Weijers, Guangchuang Yu, Jinhui Chen, RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Tang, W.; Mun, S.; Joshi, A.; Han, K.; Liang, P. Mobile elements contribute to the uniqueness of human genome with 15,000 human-specific insertions and 14 Mbp sequence increase. DNA Res. 2018, 25, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Wigginton, J.E.; Cutler, D.J.; Abecasis, G.R. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 2005, 76, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Cole, J.W.; Grond-Ginsbach, C. Departure from Hardy Weinberg Equilibrium and Genotyping Error. Front. Genet. 2017, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Abramovs, N.; Brass, A.; Tassabehji, M. Hardy-Weinberg Equilibrium in the Large Scale Genomic Sequencing Era. Front. Genet. 2020, 11, 210. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Werling, D.M.; Brand, H.; An, J.-Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef]
- Campbell, M.C.; Tishkoff, S.A. African genetic diversity: Implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genom. Hum. Genet. 2008, 9, 403–433. [Google Scholar] [CrossRef]
- Jakobsson, M.; Scholz, S.W.; Scheet, P.; Gibbs, J.R.; VanLiere, J.M.; Fung, H.C.; Szpiech, Z.A.; Degnan, J.H.; Wang, K.; Guerreiro, R.; et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008, 451, 998–1003. [Google Scholar] [CrossRef]
- Lohmueller, K.E.; Indap, A.R.; Schmidt, S.; Boyko, A.R.; Hernandez, R.D.; Hubisz, M.J.; Sninsky, J.J.; White, T.J.; Sunyaev, S.R.; Nielsen, R.; et al. Proportionally more deleterious genetic variation in European than in African populations. Nature 2008, 451, 994–997. [Google Scholar] [CrossRef]
- Bailey, J.A.; Carrel, L.; Chakravarti, A.; Eichler, E.E. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: The Lyon repeat hypothesis. Proc. Natl. Acad. Sci. USA 2000, 97, 6634–6639. [Google Scholar] [CrossRef]
- Darmon, S.K.; Lutz, C.S. Novel upstream and downstream sequence elements contribute to polyadenylation efficiency. RNA Biol. 2012, 9, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Ustyantsev, I.G.; Golubchikova, J.S.; Borodulina, O.R.; Kramerov, D.A. Canonical and noncanonical RNA polyadenylation. Mol. Biol. 2017, 51, 226–236. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Webster, T.H.; Couse, M.; Grande, B.M.; Karlins, E.; Phung, T.N.; Richmond, P.A.; Whitford, W.; Wilson, M.A. Identifying, understanding, and correcting technical artifacts on the sex chromosomes in next-generation sequencing data. GigaScience 2019, 8, giz074. [Google Scholar] [CrossRef]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.N.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef]
- Erwin, J.A.; Marchetto, M.C.; Gage, F.H. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 2014, 15, 497–506. [Google Scholar] [CrossRef]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Abugessaisa, I., Kasukawa, T., Eds.; Springer Nature: Singapore, 2021; pp. 27–56. [Google Scholar]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
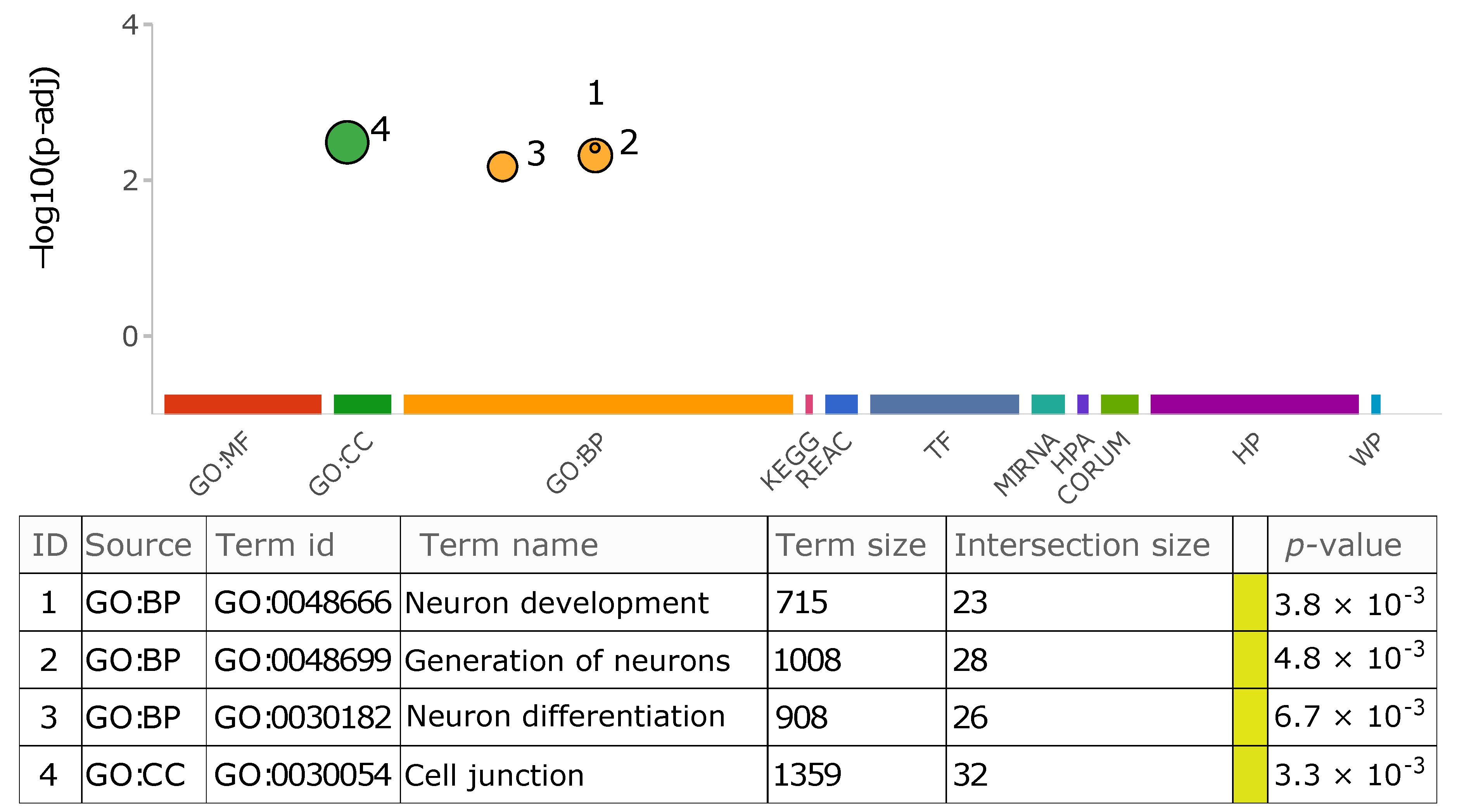{kind=link}
| Super Population Code (Name) | Population Code (Name) | Sample Size |
|---|---|---|
| AFR (African) | ASW (Americans of African Ancestry in SW, USA) | 74 |
| YRI (Yoruba in Ibadan, Nigeria) | 178 | |
| LWK (Luhya in Webuye, Kenya) | 99 | |
| GWD (Gambian in Western Divisions in the Gambia) | 178 | |
| MSL (Mende in Sierra Leone) | 99 | |
| ESN (Esan in Nigeria) | 149 | |
| ACB (African Caribbeans in Barbados) | 116 | |
| AMR (Ad Mixed American) | MXL (Mexican Ancestry from Los Angeles, USA) | 97 |
| PUR (Puerto Ricans from Puerto Rico) | 139 | |
| CLM (Colombians in Medellin, Colombia) | 132 | |
| PEL (Peruvians in Lima, Peru) | 122 | |
| EUR (European) | CEU (Utah Residents, CEPH, with Northern and Western European Ancestry) | 179 |
| TSI (Toscani in Italy) | 107 | |
| FIN (Finnish in Finland) | 99 | |
| GBR (British in England and Scotland) | 91 | |
| IBS (Iberian Population in Spain) | 157 | |
| SAS (South Asian) | GIH (Gujarati Indian in Houston, Texas) | 103 |
| PJL (Punjabi from Lahore, Pakistan) | 146 | |
| BEB (Bengali from Bangladesh) | 131 | |
| STU (Sri Lankan Tamil) | 114 | |
| ITU (Indian Telugu in the UK) | 107 | |
| EAS (East Asian) | CHB (Han Chinese in Beijing, China) | 103 |
| JPT (Japanese in Tokyo, Japan) | 104 | |
| CHS (Southern Han Chinese) | 163 | |
| CDX (Chinese Dai in Xishuangbanna, China) | 93 | |
| KHV (Kinh in Ho Chi Minh City, Vietnam) | 122 |
| L1 | SVA | |
|---|---|---|
| Number of polymorphic sites | 7103 * | 3040 * |
| Minimum length of MEI (bp) | 33 | 31 |
| Maximum length of MEI (bp) | 6019 | 1316 |
| Mean length of MEI (bp) | 2980 | 949 |
| Median length of MEI (bp) | 1975 | 1240 |
| L1 | SVA | |
|---|---|---|
| Total number of identified transductions (unfiltered) | 505 | 361 |
| Number of transductions after removing those overlapping with segmental duplicates | 466 | 342 |
| Number of low-confidence transductions | 198 | 180 |
| Number of high-confidence transductions | 268 | 162 |
| Number of progenitors | 58 | 63 |
| Minimum length of transduced sequences (bp) * | 8 | 7 |
| Maximum length of transduced sequences (bp) * | 997 | 995 |
| Mean length of transduced sequences (bp) * | 206.4 | 205.9 |
| Median length of transduced sequences (bp) * | 63 | 71 |
| Retroelement | Source Type | Sub-Family Type | Number |
|---|---|---|---|
| L1 | Present in GRCh38 | L1HS | 217 |
| Non-reference | Sub-family undetermined | 32 | |
| L1Ta | 3 | ||
| L1T1d | 16 | ||
| SVA | Present in GRCh38 | SVA_F | 83 |
| SVA_E | 71 | ||
| Non-reference | SVA 1 | 8 |
| Source Locus | Source Element (Source Type) | Polyadenylation Signal 2 | Upstream Sequence Element 3 | Downstream Sequence Element 4 |
|---|---|---|---|---|
| chrX:11707248-11713279 | L1HS (reference) | AAUAAA | UGUAN | No |
| chrX:11935296-11941314 | L1HS (reference) | AAUAAA | UGUAN | No |
| chr6:13190801 1 | L1ta1d (non-reference) | AAUAAA | - | GU reach |
| chr6_GL000253v2_alt :4065841-4067131 | SVA_F (reference) | AUUAAA | - | GU reach |
| chr7:20667753-20669743 | SVA_E (reference) | AAUAAA | - | GU reach |
| chr6:122847780-122849162 | SVA_F (reference) | - | - | GU reach |
| chr6:56893617-56896059 | SVA_E (reference) | AAUAAA | - | GU reach |
| L1 | SVA | |
|---|---|---|
| Intergenic insertion | 82 | 43 |
| Intronic insertion | 156 | 91 |
| Exonic insertion (coding genes) | 0 | 1 1 |
| Exonic insertion (non-coding genes) | 2 | 4 |
| Regulatory region insertion | 13 | 7 |
| TF binding site insertion | 1 | 0 |
| 5′ UTR insertion/3′ UTR insertion | 0/1 | 0/1 |
| Upstream gene/Downstream gene 2 | 7/6 | 10/5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halabian, R.; Makałowski, W. A Map of 3′ DNA Transduction Variants Mediated by Non-LTR Retroelements on 3202 Human Genomes. Biology 2022, 11, 1032. https://doi.org/10.3390/biology11071032
Halabian R, Makałowski W. A Map of 3′ DNA Transduction Variants Mediated by Non-LTR Retroelements on 3202 Human Genomes. Biology. 2022; 11(7):1032. https://doi.org/10.3390/biology11071032
Chicago/Turabian StyleHalabian, Reza, and Wojciech Makałowski. 2022. "A Map of 3′ DNA Transduction Variants Mediated by Non-LTR Retroelements on 3202 Human Genomes" Biology 11, no. 7: 1032. https://doi.org/10.3390/biology11071032
APA StyleHalabian, R., & Makałowski, W. (2022). A Map of 3′ DNA Transduction Variants Mediated by Non-LTR Retroelements on 3202 Human Genomes. Biology, 11(7), 1032. https://doi.org/10.3390/biology11071032