
The Effect of Traumatic Brain Injury on Sleep Architecture and Circadian Rhythms in Mice—A Comparison of High-Frequency Head Impact and Controlled Cortical Injury


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
- Animals
- High-frequency Head Impact (HF-HI) injury model
- Controlled Cortical Impact (CCI) injury model
- EEG/EMG Implantation
- Circadian Wheel-Running Behavior
- Sleep Architecture
- Circadian Gene Analysis
- Statistics
3. Results
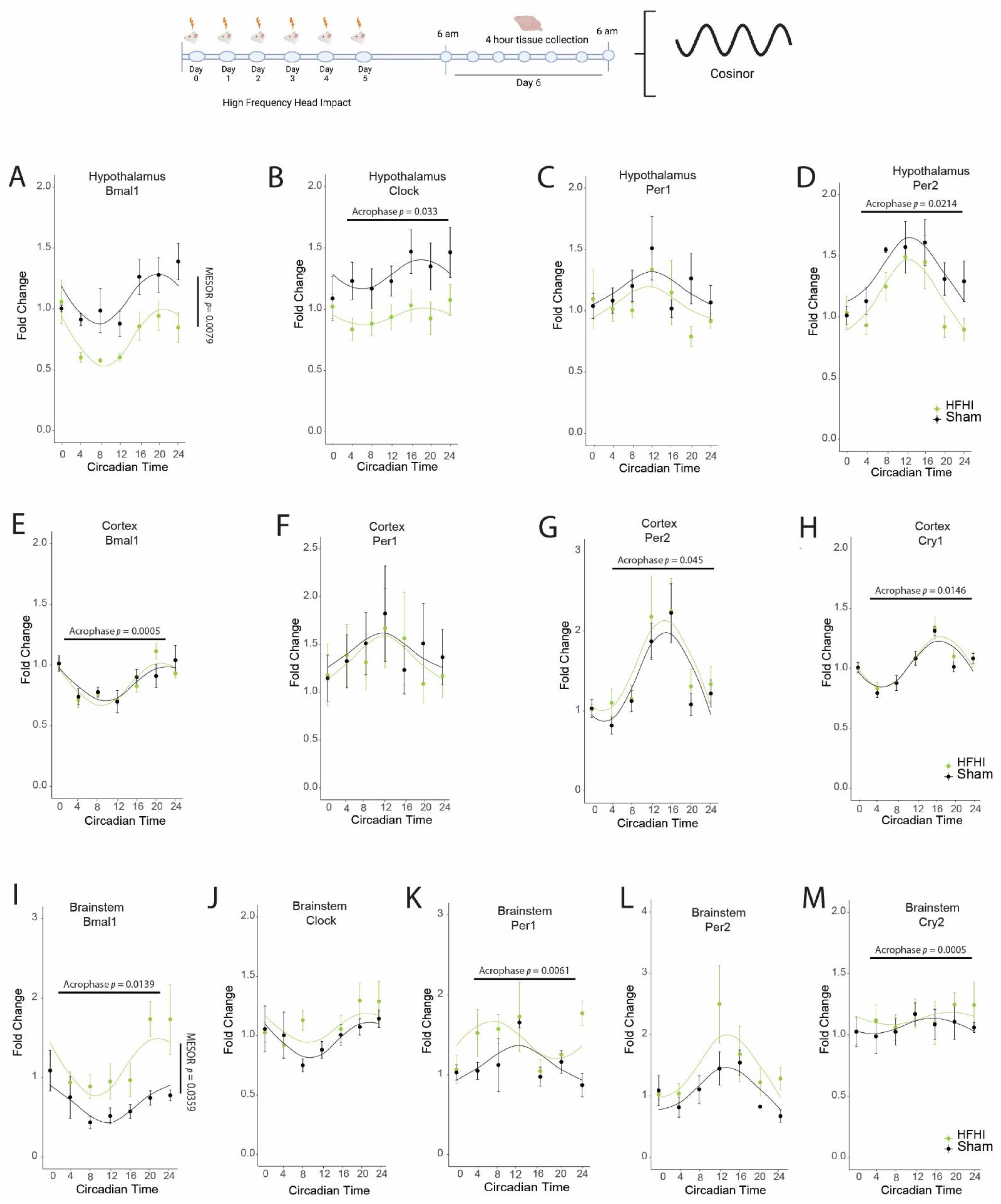
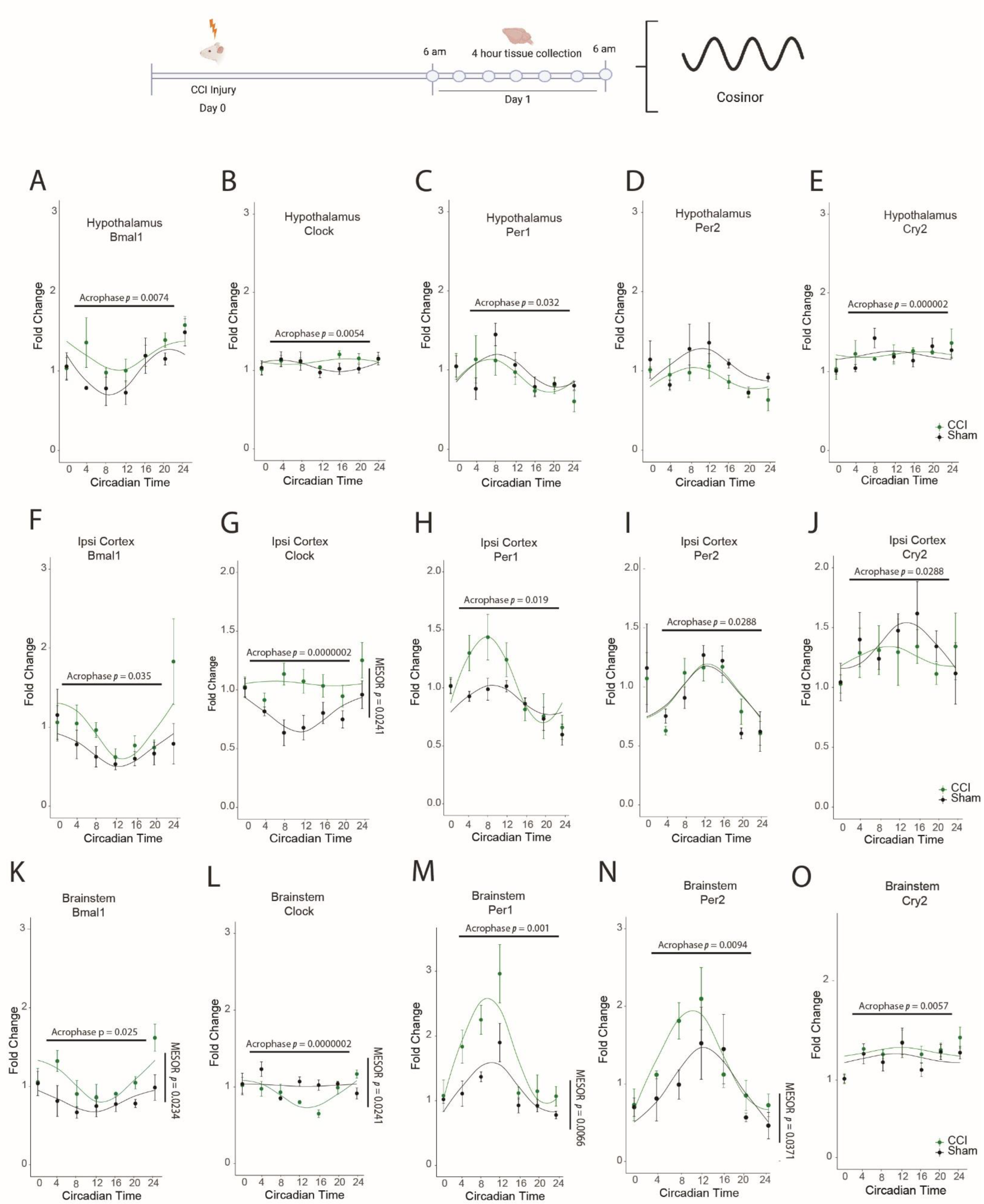
3.1. HFHI and CCI Cause Dysregulation in the Diurnal Expression of Brain Core Circadian Clock Genes
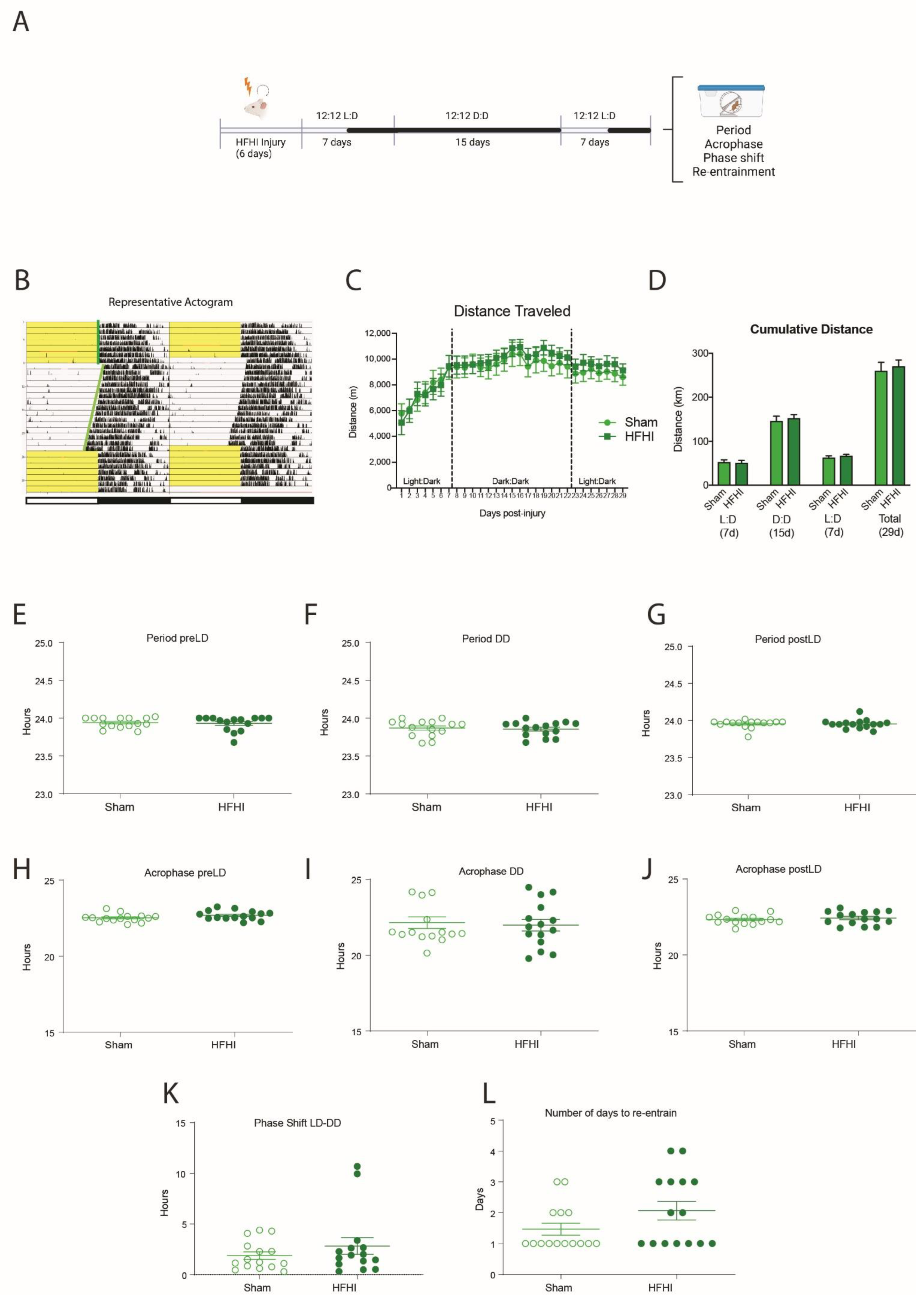
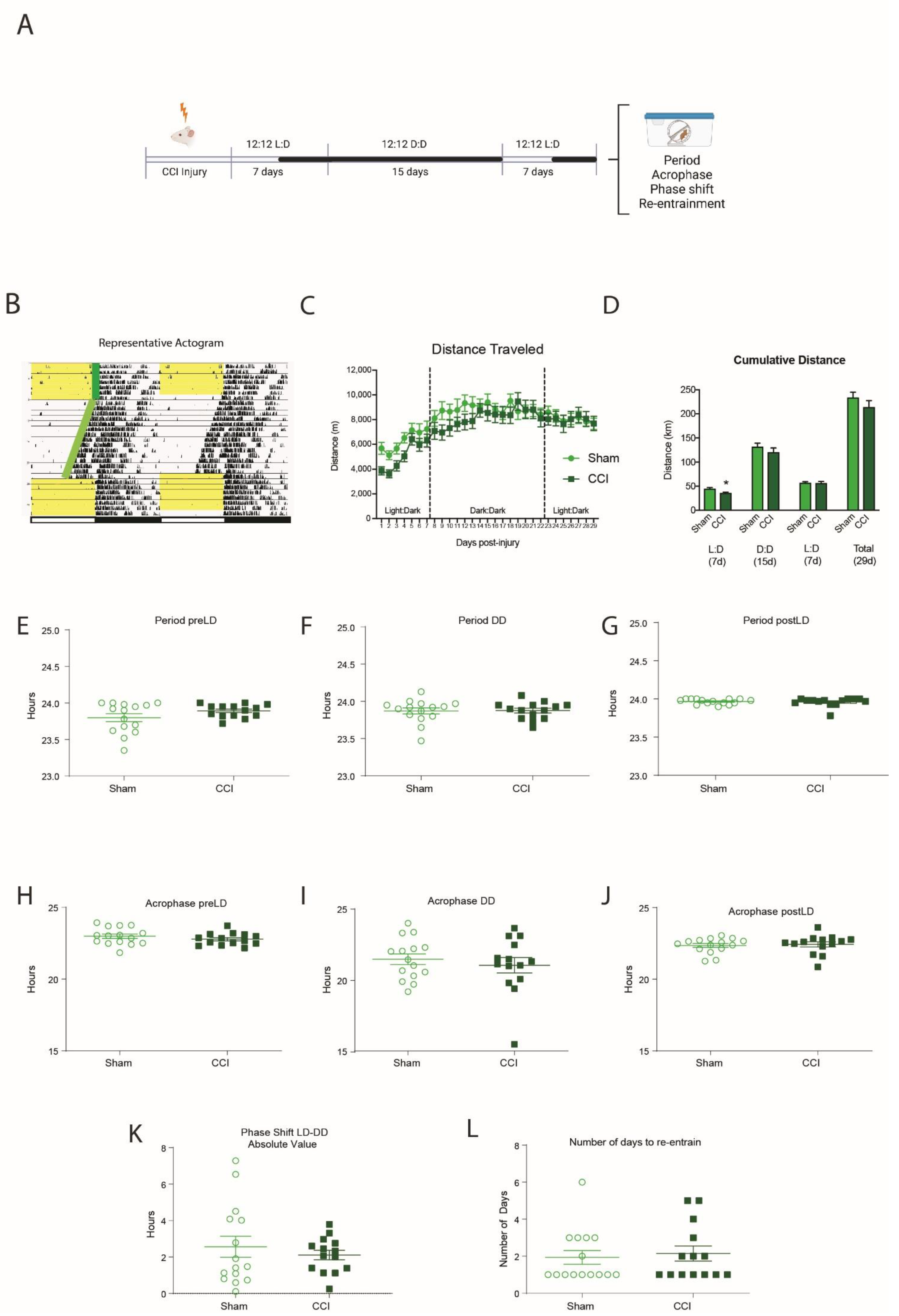
3.2. HFHI and CCI Do Not Change Circadian Wheel-Running Behavior
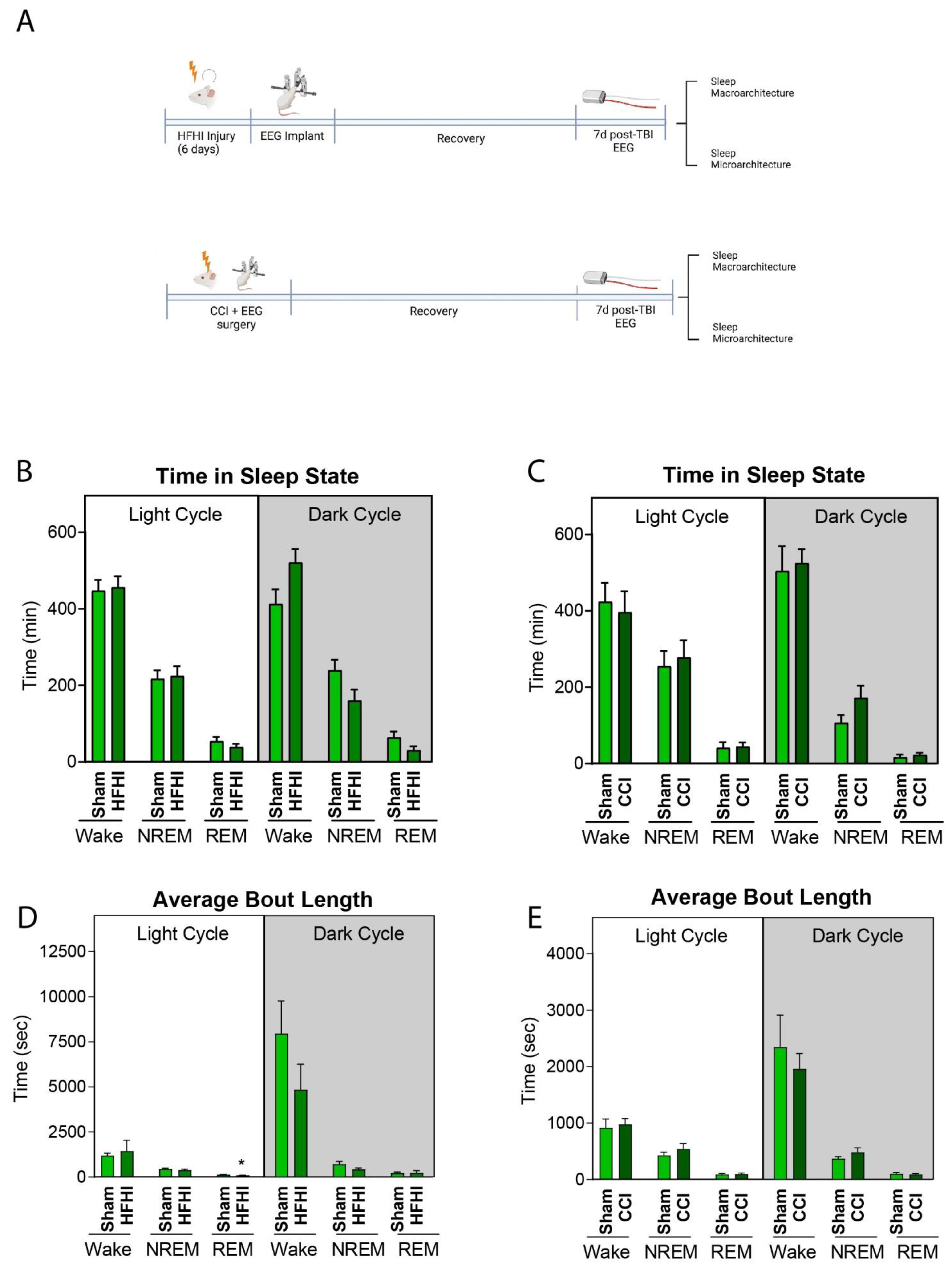
3.3. HFHI and CCI Do Not Alter Sleep Macro-Architecture One-Week Post-Injury
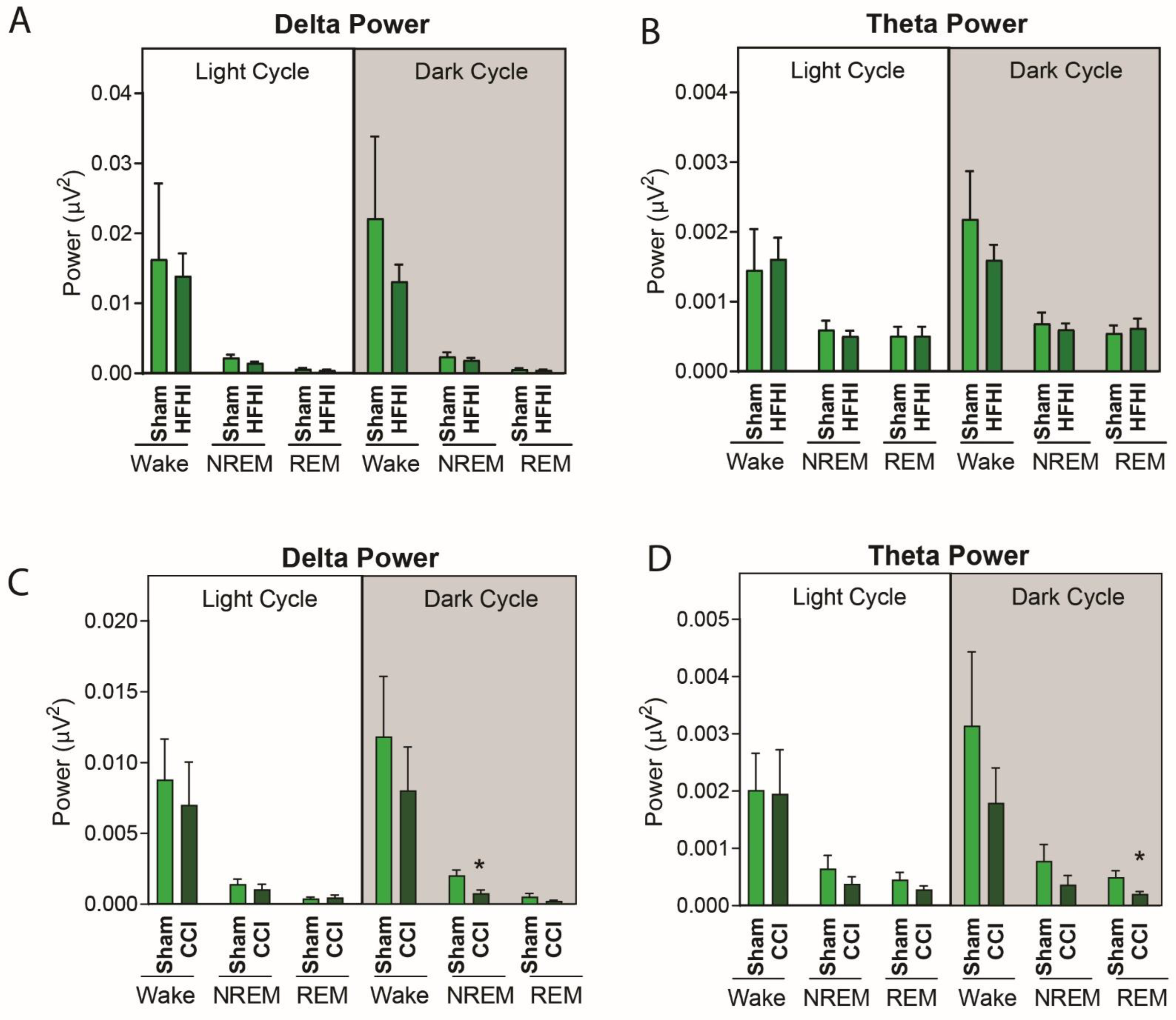
3.4. Sleep Micro-Architecture Is Changed in CCI, but Not Hfhi Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR Surveill Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mathias, J.L.; Alvaro, P.K. Prevalence of sleep disturbances, disorders, and problems following traumatic brain injury: A meta-analysis. Sleep Med. 2012, 13, 898–905. [Google Scholar] [CrossRef]
- Grima, N.; Ponsford, J.; Rajaratnam, S.M.; Mansfield, D.; Pase, M.P. Sleep Disturbances in Traumatic Brain Injury: A Meta-Analysis. J. Clin. Sleep Med. 2016, 12, 419–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.; Bergey, A.; Hill, H.; Efron, D.; McCann, U. Sleep disturbance after mild traumatic brain injury: Indicator of injury? J. Neuropsychiatry Clin. Neurosci. 2011, 23, 201–205. [Google Scholar] [CrossRef]
- Chan, L.G.; Feinstein, A. Persistent Sleep Disturbances Independently Predict Poorer Functional and Social Outcomes 1 Year After Mild Traumatic Brain Injury. J. Head Trauma Rehabil. 2015, 30, E67–E75. [Google Scholar] [CrossRef]
- Wickwire, E.M.; Williams, S.G.; Roth, T.; Capaldi, V.F.; Jaffe, M.; Moline, M.; Motamedi, G.K.; Morgan, G.W.; Mysliwiec, V.; Germain, A. Sleep, sleep disorders, and mild traumatic brain injury. What we know and what we need to know: Findings from a national working group. Neurotherapeutics 2016, 13, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Zuzuárregui, J.R.P.; Bickart, K.; Kutscher, S.J. A review of sleep disturbances following traumatic brain injury. Sleep Sci. Pract. 2018, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Castriotta, R.J.; Wilde, M.C.; Lai, J.M.; Atanasov, S.; Masel, B.E.; Kuna, S.T. Prevalence and consequences of sleep disorders in traumatic brain injury. J. Clin. Sleep Med. 2007, 3, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Baumann, C.R.; Werth, E.; Stocker, R.; Ludwig, S.; Bassetti, C.L. Sleep-wake disturbances 6 months after traumatic brain injury: A prospective study. Brain 2007, 130, 1873–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, J.; Werth, E.; Kaiser, P.R.; Bassetti, C.L.; Baumann, C.R. Sleep-wake disturbances 3 years after traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1402–1405. [Google Scholar] [CrossRef] [Green Version]
- Ouellet, M.C.; Beaulieu-Bonneau, S.; Morin, C.M. Insomnia in patients with traumatic brain injury: Frequency, characteristics, and risk factors. J. Head Trauma Rehabil. 2006, 21, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Beetar, J.T.; Guilmette, T.J.; Sparadeo, F.R. Sleep and pain complaints in symptomatic traumatic brain injury and neurologic populations. Arch. Phys. Med. Rehabil. 1996, 77, 1298–1302. [Google Scholar] [CrossRef]
- Bryan, C.J. Repetitive Traumatic Brain Injury (or Concussion) Increases Severity of Sleep Disturbance among Deployed Military Personnel. Sleep 2013, 36, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, G.R.; Brady, R.D.; Sun, M.; McDonald, S.J.; Shultz, S.R.; Mychasiuk, R. The interaction of the circadian and immune system: Desynchrony as a pathological outcome to traumatic brain injury. Neurobiol. Sleep Circadian Rhythm. 2020, 9, 100058. [Google Scholar] [CrossRef]
- Nagtegaal, J.E.; Kerkhof, G.A.; Smits, M.G.; Swart, A.C.; van der Meer, Y.G. Traumatic brain injury-associated delayed sleep phase syndrome. Funct. Neurol. 1997, 12, 345–348. [Google Scholar]
- Smits, M.G.; Nagtegaal, J.E. Post-traumatic delayed sleep phase syndrome. Neurology 2000, 55, 902–903. [Google Scholar] [CrossRef]
- Quinto, C.; Gellido, C.; Chokroverty, S.; Masdeu, J. Posttraumatic delayed sleep phase syndrome. Neurology 2000, 54, 250–252. [Google Scholar] [CrossRef]
- Nakase-Richardson, R.; Sherer, M.; Barnett, S.D.; Yablon, S.A.; Evans, C.C.; Kretzmer, T.; Schwartz, D.J.; Modarres, M. Prospective evaluation of the nature, course, and impact of acute sleep abnormality after traumatic brain injury. Arch. Phys. Med. Rehabil 2013, 94, 875–882. [Google Scholar] [CrossRef]
- Ayalon, L.; Borodkin, K.; Dishon, L.; Kanety, H.; Dagan, Y. Circadian rhythm sleep disorders following mild traumatic brain injury. Neurology 2007, 68, 1136–1140. [Google Scholar] [CrossRef]
- Iber, C.; Ancoli-Israel, S.; Chesson, A.; Quan, S. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology, and Technical Specifications. Am. Acad. Sleep Med. 2007, 176, 2012. [Google Scholar]
- Carskadon, M.A.R.A. Monitoring and staging human sleep. In Principles and Practice of Sleep Medicine, 4th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2005. [Google Scholar]
- Carskadon, M.A.; Dement, W.C. Monitoring and staging human sleep. In Principles and Practice of Sleep Medicine, 5th ed.; Keenan, S., Hirshkowitz, M., Eds.; Elsevier Saunders: St. Louis, MI, USA, 2011; pp. 16–26. [Google Scholar]
- Girardeau, G.; Lopes-Dos-Santos, V. Brain neural patterns and the memory function of sleep. Science 2021, 374, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Sandsmark, D.K.; Elliott, J.E.; Lim, M.M. Sleep-Wake Disturbances After Traumatic Brain Injury: Synthesis of Human and Animal Studies. Sleep 2017, 40, zsx044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willie, J.T.; Lim, M.M.; Bennett, R.E.; Azarion, A.A.; Schwetye, K.E.; Brody, D.L. Controlled cortical impact traumatic brain injury acutely disrupts wakefulness and extracellular orexin dynamics as determined by intracerebral microdialysis in mice. J. Neurotrauma 2012, 29, 1908–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazra, A.; Macolino, C.; Elliott, M.B.; Chin, J. Delayed thalamic astrocytosis and disrupted sleep-wake patterns in a preclinical model of traumatic brain injury. J. Neurosci. Res. 2014, 92, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.M.; Elkind, J.; Xiong, G.; Galante, R.; Zhu, J.; Zhang, L.; Lian, J.; Rodin, J.; Kuzma, N.N.; Pack, A.I.; et al. Dietary therapy mitigates persistent wake deficits caused by mild traumatic brain injury. Sci. Transl. Med. 2013, 5, 215ra173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, R.K.; Striz, M.; Bachstetter, A.D.; Van Eldik, L.J.; Donohue, K.D.; O’Hara, B.F.; Lifshitz, J. Diffuse brain injury induces acute post-traumatic sleep. PLoS ONE 2014, 9, e82507. [Google Scholar] [CrossRef] [Green Version]
- Rowe, R.K.; Harrison, J.L.; O’Hara, B.F.; Lifshitz, J. Recovery of neurological function despite immediate sleep disruption following diffuse brain injury in the mouse: Clinical relevance to medically untreated concussion. Sleep 2014, 37, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Rowe, R.K.; Harrison, J.L.; O’Hara, B.F.; Lifshitz, J. Diffuse brain injury does not affect chronic sleep patterns in the mouse. Brain Inj. 2014, 28, 504–510. [Google Scholar] [CrossRef]
- Petraglia, A.L.; Plog, B.A.; Dayawansa, S.; Chen, M.; Dashnaw, M.L.; Czerniecka, K.; Walker, C.T.; Viterise, T.; Hyrien, O.; Iliff, J.J.; et al. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: A novel mouse model of chronic traumatic encephalopathy. J. Neurotrauma 2014, 31, 1211–1224. [Google Scholar] [CrossRef] [Green Version]
- Sabir, M.; Gaudreault, P.O.; Freyburger, M.; Massart, R.; Blanchet-Cohen, A.; Jaber, M.; Gosselin, N.; Mongrain, V. Impact of traumatic brain injury on sleep structure, electrocorticographic activity and transcriptome in mice. Brain Behav. Immun. 2015, 47, 118–130. [Google Scholar] [CrossRef]
- Buchele, F.; Morawska, M.M.; Schreglmann, S.R.; Penner, M.; Muser, M.; Baumann, C.R.; Noain, D. Novel Rat Model of Weight Drop-Induced Closed Diffuse Traumatic Brain Injury Compatible with Electrophysiological Recordings of Vigilance States. J. Neurotrauma 2016, 33, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Forcelli, P.A.; Wilkins, T.; Zapple, D.N.; Parsadanian, M.; Burns, M.P. The effect of injury severity on behavior: A phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J. Neurotrauma 2012, 29, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Sloley, S.S.; Main, B.S.; Winston, C.N.; Harvey, A.C.; Kaganovich, A.; Korthas, H.T.; Caccavano, A.P.; Zapple, D.N.; Wu, J.Y.; Partridge, J.G.; et al. High-frequency head impact causes chronic synaptic adaptation and long-term cognitive impairment in mice. Nat. Commun. 2021, 12, 2613. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.M.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 2013, 8, e53376. [Google Scholar] [CrossRef]
- Main, B.S.; Sloley, S.S.; Villapol, S.; Zapple, D.N.; Burns, M.P. A Mouse Model of Single and Repetitive Mild Traumatic Brain Injury. J. Vis. Exp. 2017, 124, e55713. [Google Scholar] [CrossRef]
- Ziogas, N.K.; Koliatsos, V.E. Primary Traumatic Axonopathy in Mice Subjected to Impact Acceleration: A Reappraisal of Pathology and Mechanisms with High-Resolution Anatomical Methods. J. Neurosci. 2018, 38, 4031–4047. [Google Scholar] [CrossRef]
- Ledger, C.; Karameh, W.K.; Munoz, D.G.; Fischer, C.E.; Schweizer, T.A. Gender role in sleep disturbances among older adults with traumatic brain injury. Int. Rev. Psychiatry 2020, 32, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Pocivavsek, A.; Moussa, C.E.; Thompson, R.; Matsuoka, Y.; Faden, A.I.; Rebeck, G.W.; Burns, M.P. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 2009, 15, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Main, B.S.; Villapol, S.; Sloley, S.S.; Barton, D.J.; Parsadanian, M.; Agbaegbu, C.; Stefos, K.; McCann, M.S.; Washington, P.M.; Rodriguez, O.C.; et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol. Neurodegener. 2018, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Neckel, N.D.; Dai, H.; Burns, M.P. A Novel Multi-Dimensional Analysis of Rodent Gait Reveals the Compensation Strategies Used during Spontaneous Recovery from Spinal Cord and Traumatic Brain Injury. J. Neurotrauma 2020, 37, 517–527. [Google Scholar] [CrossRef]
- Cornelissen, G. Cosinor-based rhythmometry. Theor Biol Med. Model. 2014, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, D.R.; Sell, S.L.; Micci, M.-A.; Crookshanks, J.M.; Parsley, M.; Uchida, T.; Prough, D.S.; DeWitt, D.S.; Hellmich, H.L. Traumatic Brain Injury-Induced Dysregulation of the Circadian Clock. PLoS ONE 2012, 7, e46204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Hsieh, W.; Chung, J.R.; Lan, T.H.; Wang, Y. Repetitive mild traumatic brain injury alters diurnal locomotor activity and response to the light change in mice. Sci Rep. 2019, 9, 14067. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Boucher, M.; Conley, G.; Li, Y.; Zhang, J.; Morriss, N.; Meehan Iii, W.P.; Mannix, R. Traumatic Brain Injury-Related Optic Nerve Damage. J. Neuropathol. Exp. Neurol. 2022, 81, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, M.; Patton, A.P.; Chesham, J.E.; Maywood, E.S.; Hastings, M.H. Astrocytes Control Circadian Timekeeping in the Suprachiasmatic Nucleus via Glutamatergic Signaling. Neuron 2017, 93, 1420–1435.e1425. [Google Scholar] [CrossRef] [Green Version]
- Ebling, F.J. The role of glutamate in the photic regulation of the suprachiasmatic nucleus. Prog. Neurobiol. 1996, 50, 109–132. [Google Scholar] [CrossRef]
- Almeida-Suhett, C.P.; Prager, E.M.; Pidoplichko, V.; Figueiredo, T.H.; Marini, A.M.; Li, Z.; Eiden, L.E.; Braga, M.F. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp. Neurol. 2015, 273, 11–23. [Google Scholar] [CrossRef]
- Winston, C.N.; Chellappa, D.; Wilkins, T.; Barton, D.J.; Washington, P.M.; Loane, D.J.; Zapple, D.N.; Burns, M.P. Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J. Neurotrauma 2013, 30, 1966–1972. [Google Scholar] [CrossRef]
- Gao, X.; Deng, P.; Xu, Z.C.; Chen, J. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS ONE 2011, 6, e24566. [Google Scholar] [CrossRef] [Green Version]
- Villapol, S.; Loane, D.J.; Burns, M.P. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 2017, 65, 1423–1438. [Google Scholar] [CrossRef]
- Rao, V.L.; Baskaya, M.K.; Dogan, A.; Rothstein, J.D.; Dempsey, R.J. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J. Neurochem. 1998, 70, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Debruyne, J.P.; Noton, E.; Lambert, C.M.; Maywood, E.S.; Weaver, D.R.; Reppert, S.M. A clock shock: Mouse CLOCK is not required for circadian oscillator function. Neuron 2006, 50, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karelina, K.; Schneiderman, K.; Shah, S.; Fitzgerald, J.; Cruz, R.V.; Oliverio, R.; Whitehead, B.; Yang, J.; Weil, Z.M. Moderate Intensity Treadmill Exercise Increases Survival of Newborn Hippocampal Neurons and Improves Neurobehavioral Outcomes after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sabirzhanov, B.; Wu, J.; Faden, A.I.; Stoica, B.A. Voluntary Exercise Preconditioning Activates Multiple Antiapoptotic Mechanisms and Improves Neurological Recovery after Experimental Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Skopin, M.D.; Kabadi, S.V.; Viechweg, S.S.; Mong, J.A.; Faden, A.I. Chronic decrease in wakefulness and disruption of sleep-wake behavior after experimental traumatic brain injury. J. Neurotrauma 2015, 32, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Modarres, M.H.; Kuzma, N.N.; Kretzmer, T.; Pack, A.I.; Lim, M.M. EEG slow waves in traumatic brain injury: Convergent findings in mouse and man. Neurobiol. Sleep Circadian Rhythm. 2017, 2, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.R.; Lazic, S.E.; Ogilvie, R.D. Polysomnographic and quantitative EEG analysis of subjects with long-term insomnia complaints associated with mild traumatic brain injury. Clin. Neurophysiol. 2008, 119, 429–438. [Google Scholar] [CrossRef]
- Gosselin, N.; Lassonde, M.; Petit, D.; Leclerc, S.; Mongrain, V.; Collie, A.; Montplaisir, J. Sleep following sport-related concussions. Sleep Med. 2009, 10, 35–46. [Google Scholar] [CrossRef]
- Arbour, C.; Khoury, S.; Lavigne, G.J.; Gagnon, K.; Poirier, G.; Montplaisir, J.Y.; Carrier, J.; Gosselin, N. Are NREM sleep characteristics associated to subjective sleep complaints after mild traumatic brain injury? Sleep Med. 2015, 16, 534–539. [Google Scholar] [CrossRef]
- Khoury, S.; Chouchou, F.; Amzica, F.; Giguere, J.F.; Denis, R.; Rouleau, G.A.; Lavigne, G.J. Rapid EEG activity during sleep dominates in mild traumatic brain injury patients with acute pain. J. Neurotrauma 2013, 30, 633–641. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korthas, H.T.; Main, B.S.; Harvey, A.C.; Buenaventura, R.G.; Wicker, E.; Forcelli, P.A.; Burns, M.P. The Effect of Traumatic Brain Injury on Sleep Architecture and Circadian Rhythms in Mice—A Comparison of High-Frequency Head Impact and Controlled Cortical Injury. Biology 2022, 11, 1031. https://doi.org/10.3390/biology11071031
Korthas HT, Main BS, Harvey AC, Buenaventura RG, Wicker E, Forcelli PA, Burns MP. The Effect of Traumatic Brain Injury on Sleep Architecture and Circadian Rhythms in Mice—A Comparison of High-Frequency Head Impact and Controlled Cortical Injury. Biology. 2022; 11(7):1031. https://doi.org/10.3390/biology11071031
Chicago/Turabian StyleKorthas, Holly T., Bevan S. Main, Alex C. Harvey, Ruchelle G. Buenaventura, Evan Wicker, Patrick A. Forcelli, and Mark P. Burns. 2022. "The Effect of Traumatic Brain Injury on Sleep Architecture and Circadian Rhythms in Mice—A Comparison of High-Frequency Head Impact and Controlled Cortical Injury" Biology 11, no. 7: 1031. https://doi.org/10.3390/biology11071031
APA StyleKorthas, H. T., Main, B. S., Harvey, A. C., Buenaventura, R. G., Wicker, E., Forcelli, P. A., & Burns, M. P. (2022). The Effect of Traumatic Brain Injury on Sleep Architecture and Circadian Rhythms in Mice—A Comparison of High-Frequency Head Impact and Controlled Cortical Injury. Biology, 11(7), 1031. https://doi.org/10.3390/biology11071031