
The C-Terminal Repeat Units of SpaA Mediate Adhesion of Erysipelothrix rhusiopathiae to Host Cells and Regulate Its Virulence

 , ,
, ,  and
and 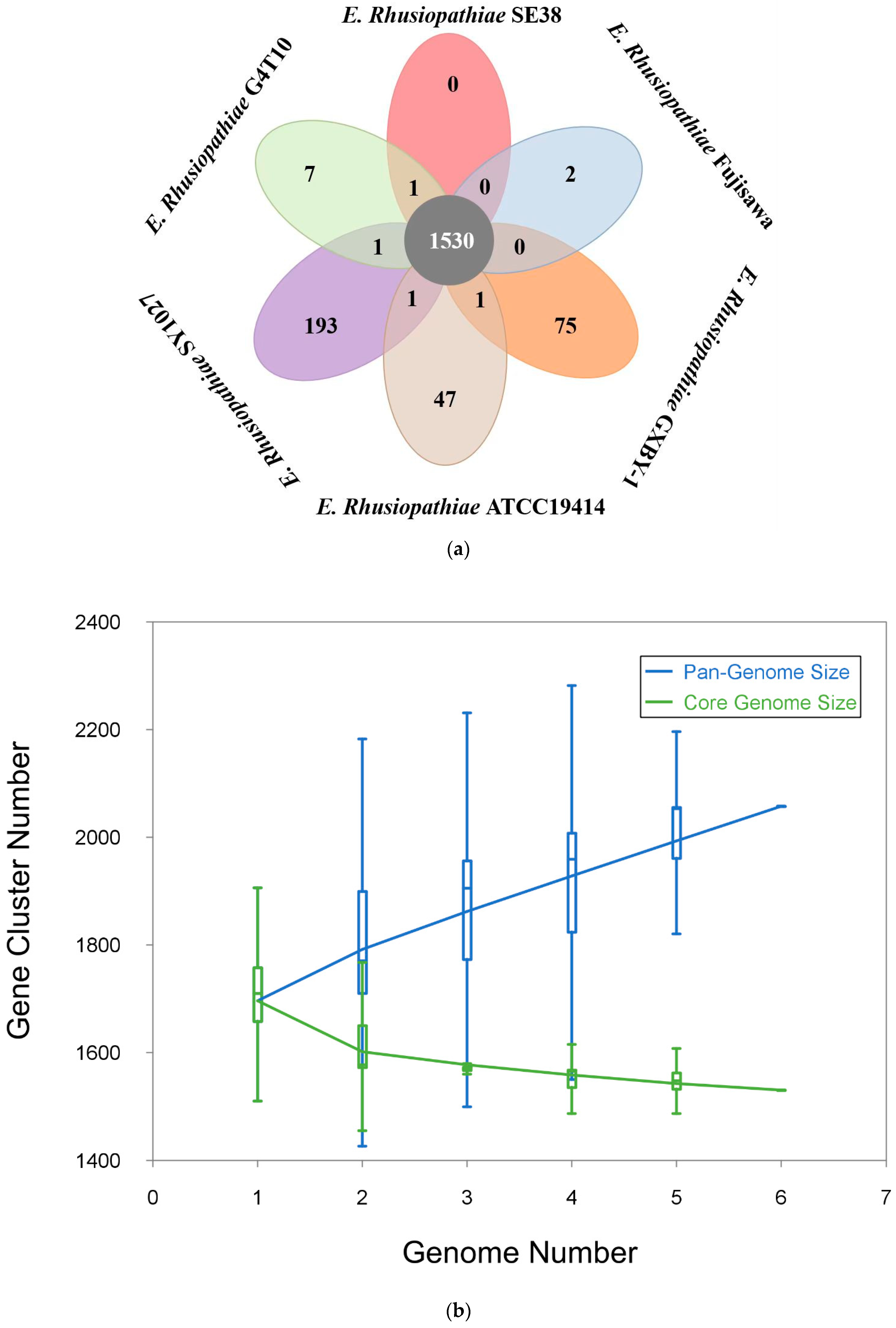{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Bacterial Strains and Cultivation
2.2. Genome Sequencing and Assembly
2.3. Genome Annotation and Analysis
2.4. Pan-Genomic Analyses and Genome Comparison
2.5. Phylogenetic Analyses
2.6. Gene Gain and Loss Analyses
2.7. RNA-Seq Library Construction and Sequencing
2.8. Read Mapping and Gene Expression Analysis
2.9. Construction of the ΔSpaA Mutant
2.10. Bacterial Cell Adhesion Assay
2.11. Evaluation of ΔSpaA Pathogenicity in Mice
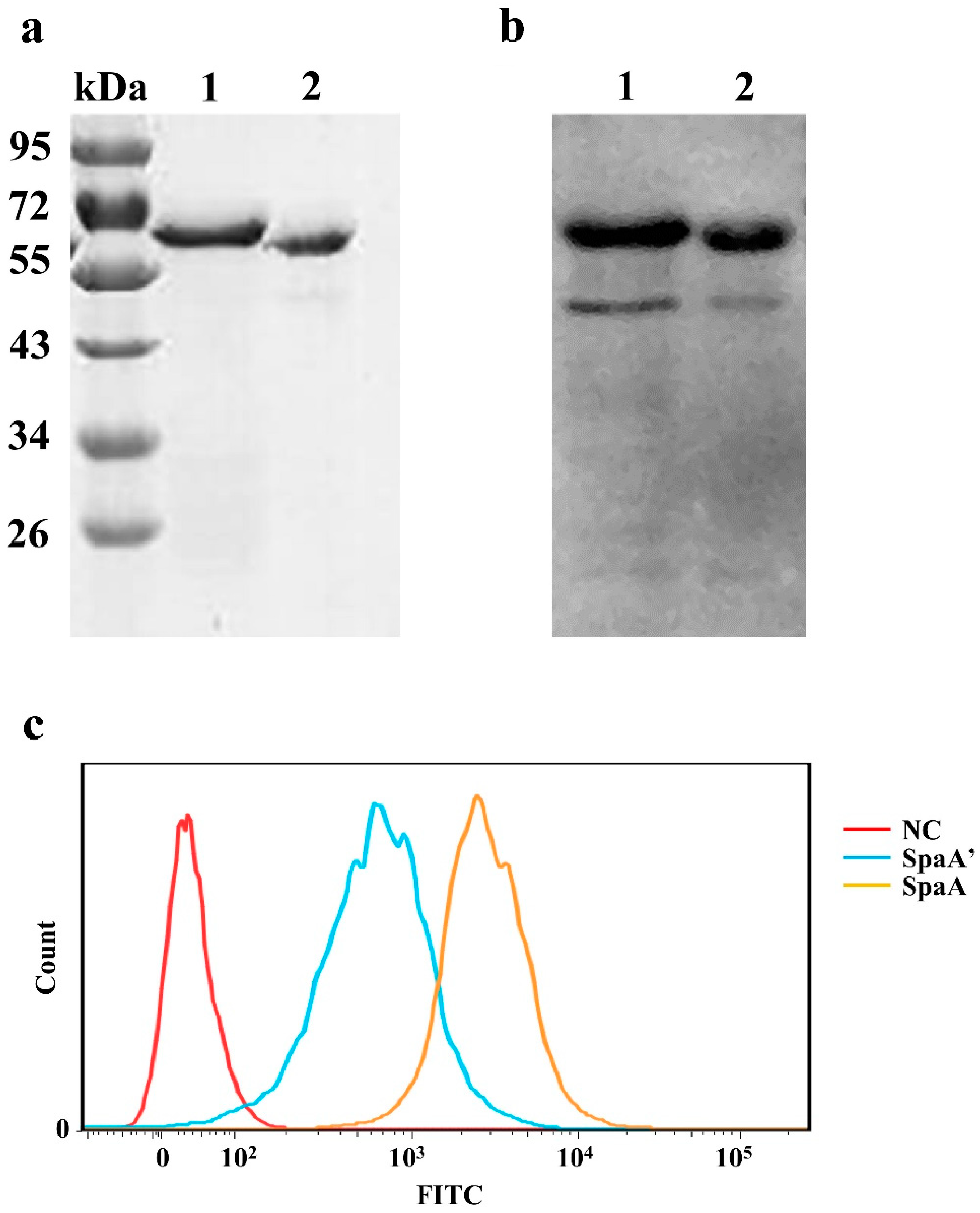
2.12. Expression of Recombinant Proteins
2.13. Analysis of Recombinant Protein Adhesion to PIECs
2.14. Data Accessibility
3. Results
3.1. General Genomic Features of Strains G4T10 and SE38
3.2. Comparative Genomic Analysis Results
3.3. Potential Virulence Factors
3.4. Characteristics of the Mutant Strain
3.5. ΔSpaA Displays Attenuated Virulence in Mice, and Decreased Adhesion to PIECs
3.6. The Deleted Sequence Influences the Adhesive Ability of SpaA in PIECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, Y.; Zhu, D.; Zhang, J.; Yang, L.; Wang, X.; Chen, H.; Tan, C. Virulence determinants, antimicrobial susceptibility, and molecular profiles of Erysipelothrix rhusiopathiae strains isolated from China. Emerg. Microbes Infect. 2015, 4, e69. [Google Scholar] [CrossRef] [PubMed]
- Shimoji, Y.; Ogawa, Y.; Osaki, M.; Kabeya, H.; Maruyama, S.; Mikami, T.; Sekizaki, T. Adhesive surface proteins of Erysipelothrix rhusiopathiae bind to polystyrene, fibronectin, and type I and IV collagens. J. Bacteriol. 2003, 185, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Borrathybay, E.; Gong, F.J.; Zhang, L.; Nazierbieke, W. Role of surface protective antigen A in the pathogenesis of Erysipelothrix rhusiopathiae strain C43065. J. Microbiol. Biotechnol. 2015, 25, 206–216. [Google Scholar] [CrossRef]
- Zhu, W.; Cai, C.; Wang, Y.; Li, J.; Wu, C.; Kang, C.; Sun, X.; Jin, M. Characterization of roles of SpaA in Erysipelothrix rhusiopathiae adhesion to porcine endothelial cells. Microb. Pathog. 2017, 113, 176–180. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Zhang, A.; Zhu, W.; Zhang, Q.; Xu, Z.; Yan, S.; Sun, X.; Chen, H.; Jin, M. iTRAQ-based quantitative proteomic analysis reveals potential virulence factors of Erysipelothrix rhusiopathiae. J. Proteom. 2017, 160, 28–37. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, Y.; Cai, C.; Li, J.; Wu, C.; Kang, C.; Jin, M. Erysipelothrix rhusiopathiae recruits host plasminogen via the major protective antigen SpaA. FEMS Microbiol. Lett. 2017, 364, fnx036. [Google Scholar] [CrossRef]
- Floristan, U.; Feltes, R.A.; Gomez, A.; Vidaurrazaga, C. Erythematous-violaceous lesion on the leg of a researcher in contact with swine. Enferm. Infecc. Y Microbiol. Clin. 2009, 27, 365–366. [Google Scholar]
- Wang, Q.; Chang, B.J.; Riley, T.V. Erysipelothrix rhusiopathiae. Vet. Microbiol. 2010, 140, 405–417. [Google Scholar] [CrossRef]
- Zhu, W.; Cai, C.; Huang, J.; Liu, L.; Xu, Z.; Sun, X.; Jin, M. Characterization of pathogenic roles of two Erysipelothrix rhusiopathiae surface proteins. Microb. Pathog. 2018, 114, 166–168. [Google Scholar] [CrossRef]
- Kang, C.; Zhang, Q.; Zhu, W.; Cai, C.; Sun, X.; Jin, M. Transcription analysis of the responses of porcine heart to Erysipelothrix rhusiopathiae. PLoS ONE 2017, 12, e0185548. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Chen, L.H.; Zheng, D.D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Guy, L.; Roat Kultima, J.; Andersson, S.G.E. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2010, 26, 2334–2335. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Zhao, Y.B.; Wu, J.Y.; Yang, J.H.; Sun, S.X.; Xiao, J.F.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef]
- Zhao, Y.B.; Jia, X.M.; Yang, J.H.; Ling, Y.C.; Zhang, Z.; Yu, J.; Wu, J.Y.; Xiao, J.F. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Richards, V.P.; Palmer, S.R.; Bitar, P.D.P.; Qin, X.; Weinstock, G.M.; Highlander, S.K.; Town, C.D.; Burne, R.A.; Stanhope, M.J. Phylogenomics and the Dynamic Genome Evolution of the Genus Streptococcus. Genome Biol. Evol. 2014, 6, 741–753. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tjaden, B. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 2015, 16, 1. [Google Scholar] [CrossRef]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Alm, E.J. Rapid evolutionary innovation during an Archaean genetic expansion. Nature 2011, 469, 93–96. [Google Scholar] [CrossRef]
- Kamneva, O.K.; Knight, S.J.; Liberles, D.A.; Ward, N.L. Analysis of genome content evolution in pvc bacterial super-phylum: Assessment of candidate genes associated with cellular organization and lifestyle. Genome Biol. Evol. 2012, 4, 1375–1390. [Google Scholar] [CrossRef]
- Georgiades, K.; Merhej, V.; El Karkouri, K.; Raoult, D.; Pontarotti, P. Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct 2011, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A.; Wernegreen, J.J. Lifestyle evolution in symbiotic bacteria: Insights from genomics. Trends Ecol. Evol. 2000, 15, 321–326. [Google Scholar] [CrossRef]
- Moran, N.A.; Mira, A. The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biol. 2001, 2, research0054.1. [Google Scholar] [CrossRef]
- Li, Y.F.; Zou, Y.; Xia, Y.T.; Bai, J.; Wang, X.W.; Jiang, P. Proteomic and Transcriptomic Analyses of Swine Pathogen Erysipelothrix rhusiopathiae Reveal Virulence Repertoire. PLoS ONE 2016, 11, e0159462. [Google Scholar] [CrossRef]
- Imada, Y.; Goji, N.; Ishikawa, H.; Kishima, M.; Sekizaki, T. Truncated surface protective antigen (SpaA) of Erysipelothrix rhusiopathiae serotype 1a elicits protection against challenge with serotypes 1a and 2b in pigs. Infect. Immun. 1999, 67, 4376–4382. [Google Scholar] [CrossRef]
- Makino, S.; Yamamoto, K.; Murakami, S.; Shirahata, T.; Uemura, K.; Sawada, T.; Wakamoto, H.; Morita, Y. Properties of repeat domain found in a novel protective antigen, SpaA, of Erysipelothrix rhusiopathiae. Microb. Pathog. 1998, 25, 101–109. [Google Scholar] [CrossRef]
- Ogawa, Y.; Ooka, T.; Shi, F.; Ogura, Y.; Nakayama, K.; Hayashi, T.; Shimoji, Y. The genome of Erysipelothrix rhusiopathiae, the causative agent of swine erysipelas, reveals new insights into the evolution of firmicutes and the organism’s intracellular adaptations. J. Bacteriol. 2011, 193, 2959–2971. [Google Scholar] [CrossRef]
- Yother, J.; White, J.M. Novel Surface Attachment Mechanism of the Streptococcus-Pneumoniae Protein Pspa. J. Bacteriol. 1994, 176, 2976–2985. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Ayanbule, K.; Smedinghoff, M.; Salzberg, S.L. OperonDB: A comprehensive database of predicted operons in microbial genomes. Nucleic Acids Res. 2009, 37, D479–D482. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shi, F.; Ogawa, Y.; Sano, A.; Harada, T.; Hirota, J.; Eguchi, M.; Oishi, E.; Shimoji, Y. Characterization and Identification of a Novel Candidate Vaccine Protein through Systematic Analysis of Extracellular Proteins of Erysipelothrix rhusiopathiae. Infect. Immun. 2013, 81, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Yamamoto, K.; Asakura, H.; Shirahata, T. Surface antigen, SpaA, of Erysipelothrix rhusiopathiae binds to Gram-positive bacterial cell surfaces. FEMS Microbiol. Lett. 2000, 186, 313–317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harada, T.; Ogawa, Y.; Eguchi, M.; Shi, F.; Sato, M.; Uchida, K.; Nakayama, H.; Shimoji, Y. Phosphorylcholine and SpaA, a choline-binding protein, are involved in the adherence of Erysipelothrix rhusiopathiae to porcine endothelial cells, but this adherence is not mediated by the PAF receptor. Vet. Microbiol. 2014, 172, 216–222. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Zhang, Z.; Kang, C.; Zhang, Q.; Zhu, W.; Zhang, Y.; Zhang, H.; Xiao, J.; Jin, M. The C-Terminal Repeat Units of SpaA Mediate Adhesion of Erysipelothrix rhusiopathiae to Host Cells and Regulate Its Virulence. Biology 2022, 11, 1010. https://doi.org/10.3390/biology11071010
Wu C, Zhang Z, Kang C, Zhang Q, Zhu W, Zhang Y, Zhang H, Xiao J, Jin M. The C-Terminal Repeat Units of SpaA Mediate Adhesion of Erysipelothrix rhusiopathiae to Host Cells and Regulate Its Virulence. Biology. 2022; 11(7):1010. https://doi.org/10.3390/biology11071010
Chicago/Turabian StyleWu, Chao, Zhewen Zhang, Chao Kang, Qiang Zhang, Weifeng Zhu, Yadong Zhang, Hao Zhang, Jingfa Xiao, and Meilin Jin. 2022. "The C-Terminal Repeat Units of SpaA Mediate Adhesion of Erysipelothrix rhusiopathiae to Host Cells and Regulate Its Virulence" Biology 11, no. 7: 1010. https://doi.org/10.3390/biology11071010
APA StyleWu, C., Zhang, Z., Kang, C., Zhang, Q., Zhu, W., Zhang, Y., Zhang, H., Xiao, J., & Jin, M. (2022). The C-Terminal Repeat Units of SpaA Mediate Adhesion of Erysipelothrix rhusiopathiae to Host Cells and Regulate Its Virulence. Biology, 11(7), 1010. https://doi.org/10.3390/biology11071010