
DDK: The Outsourced Kinase of Chromosome Maintenance


Simple Summary
Abstract
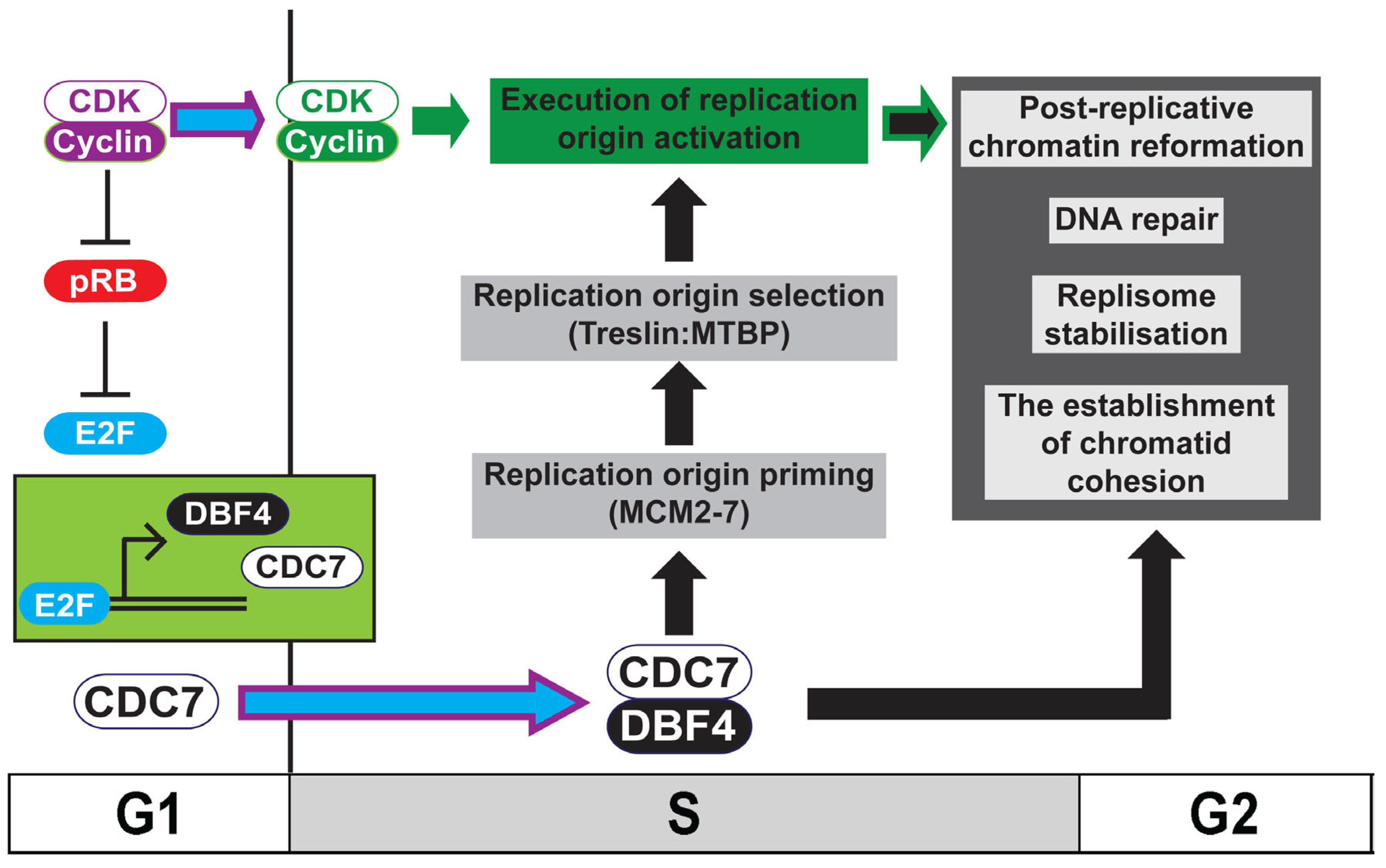
1. Introduction: Ensuring Chromosome Maintenance during the Mitotic Cell Division Cycle
2. Introducing the Dbf4-Dependent Kinase: DDK
3. Cell-Cycle Regulation of DDK Activity
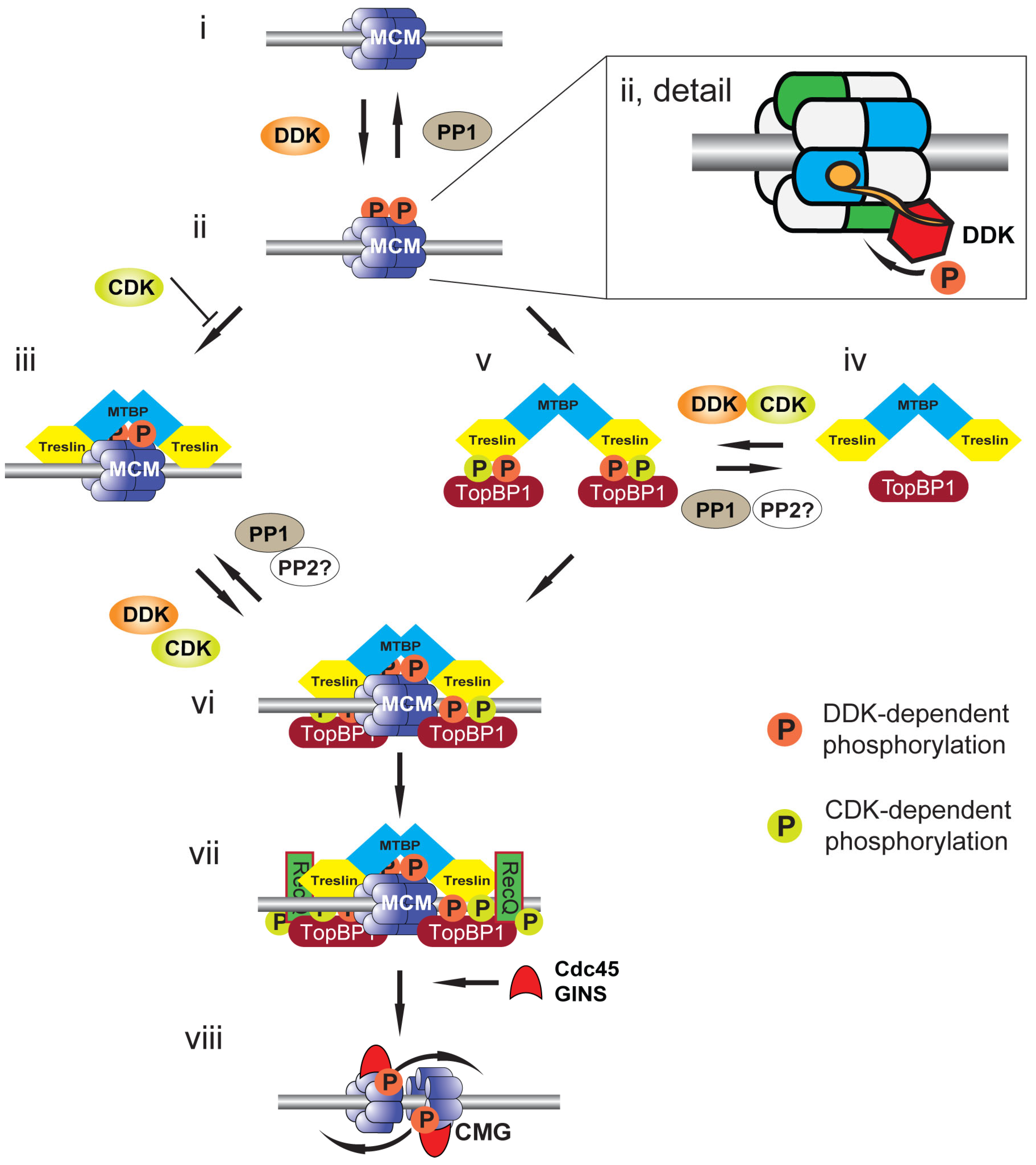
4. Replication Origin Priming: DDK-Mediated Phosphorylation of MCM2-7
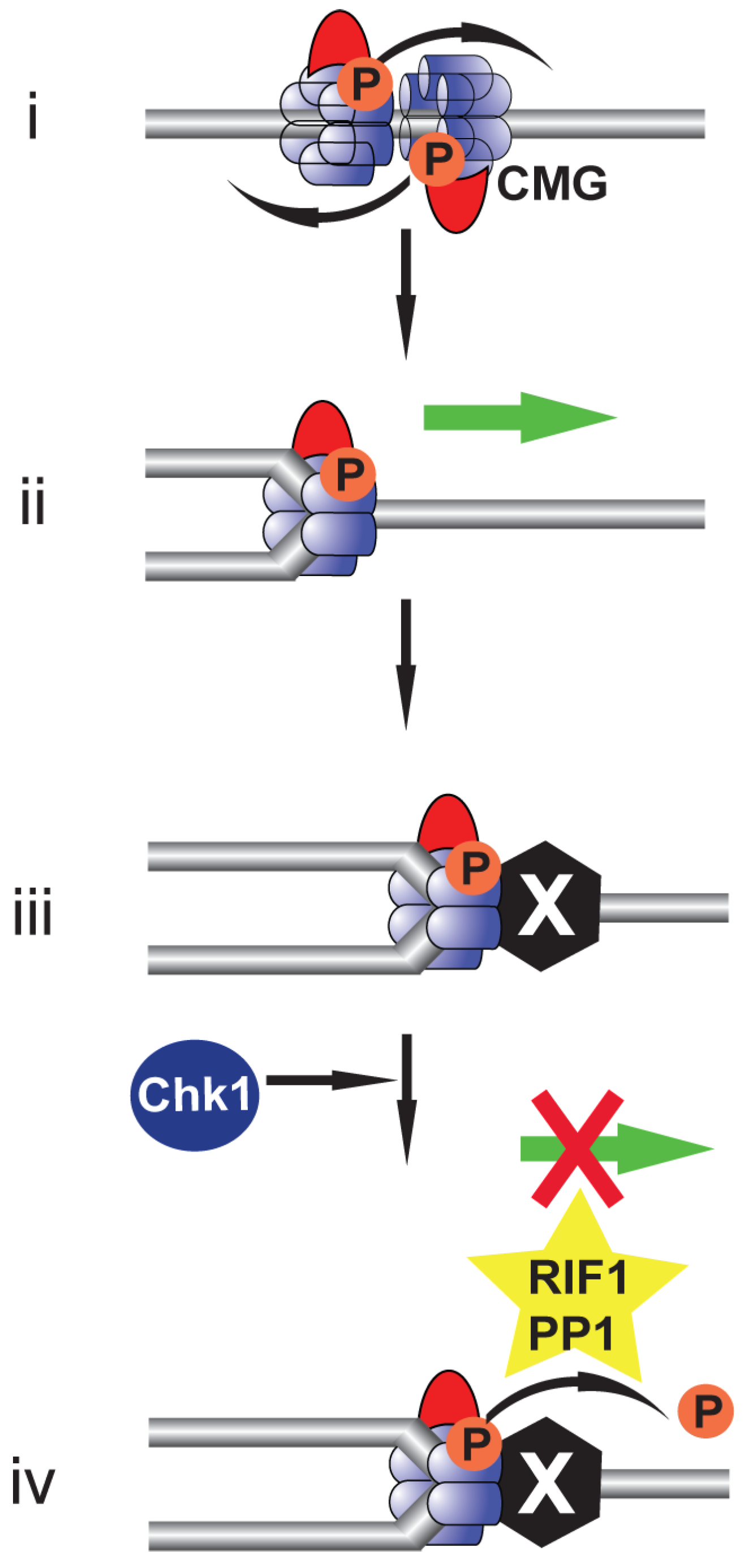
5. Rif1: Antagonising DDK-Mediated MCM4 Phosphorylation
6. Replication Origin Selection and Activation: DDK-Mediated Phosphorylation of Treslin:MTBP
7. Which Acts First: CDK or DDK?
8. Replication Origin Selection
9. Beyond Replication Initiation: The Role of DDK in Replication Fork Stability during Replication Stress
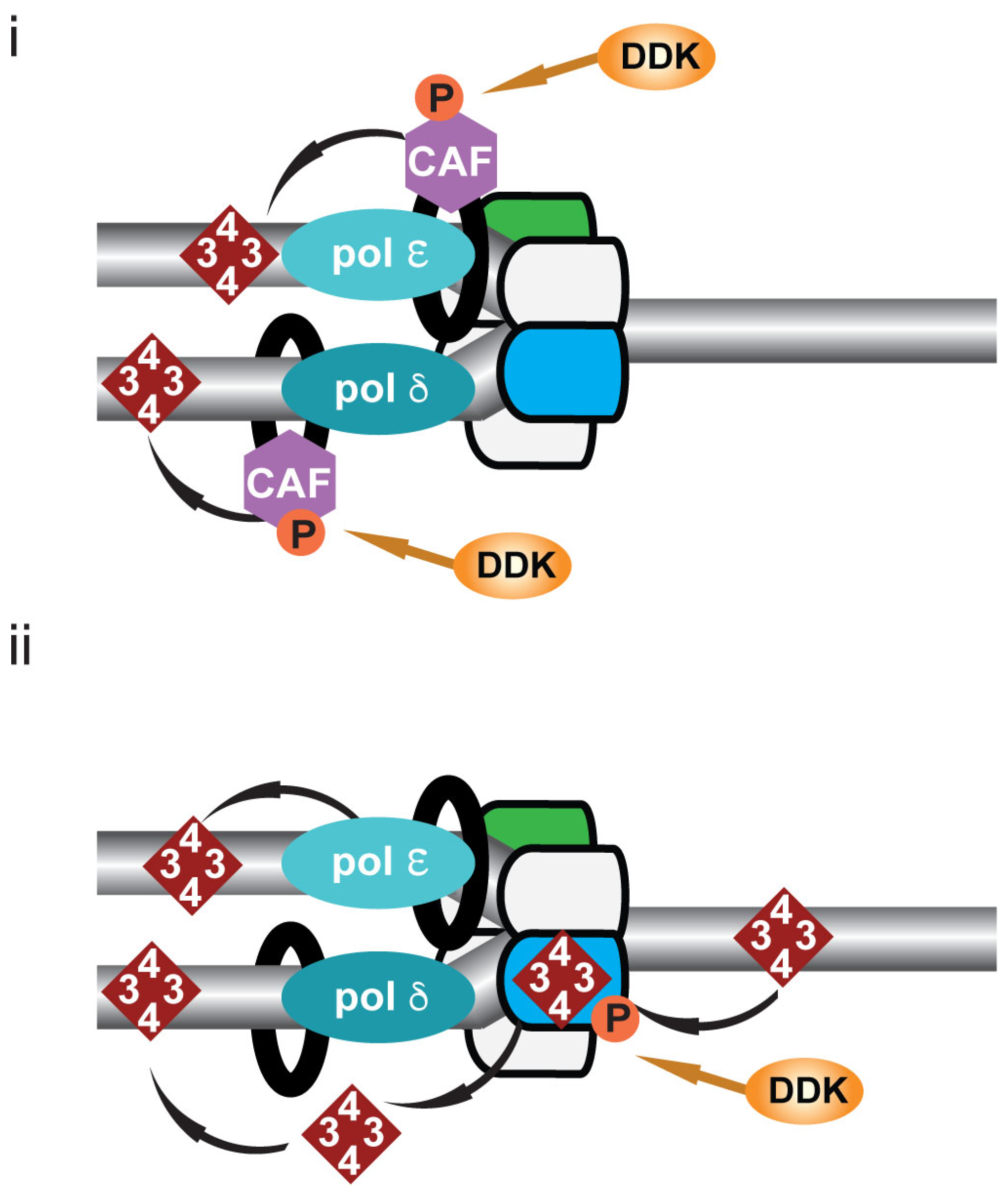
10. DDK and Post-Replicative Chromatin Formation
11. DDK and The Establishment of Replicated Chromatid Cohesion
12. Therapeutic Potential of Targeting DDK
13. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, S.P.; Labib, K. Chromosome Duplication in Saccharomyces cerevisiae. Genetics 2016, 203, 1027–1067. [Google Scholar] [CrossRef] [PubMed]
- Riera, A.; Barbon, M.; Noguchi, Y.; Reuter, L.M.; Schneider, S.; Speck, C. From structure to mechanism-understanding initiation of DNA replication. Genes Dev. 2017, 31, 1073–1088. [Google Scholar] [CrossRef]
- Li, H.; O’Donnell, M.E. The Eukaryotic CMG Helicase at the Replication Fork: Emerging Architecture Reveals an Unexpected Mechanism. Bioessays 2018, 40, 1700208, 1700201–1700209. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Diffley, J.F.X. The Initiation of Eukaryotic DNA Replication. Annu. Rev. Biochem. 2022, 91, 26.21–26.25. [Google Scholar] [CrossRef] [PubMed]
- Volpi, I.; Gillespie, P.J.; Chadha, G.S.; Blow, J.J. The role of DDK and Treslin-MTBP in coordinating replication licensing and pre-initiation complex formation. Open Biol. 2021, 11, 210121. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Sato, N.; Yamada, M.; Arai, K.; Masai, H. Growth regulation of the expression of mouse cDNA and gene encoding a serine/threonine kinase related to Saccharomyces cerevisiae CDC7 essential for G1/S transition. Structure, chromosomal localization, and expression of mouse gene for s. cerevisiae Cdc7-related kinase. J. Biol. Chem. 1998, 273, 23248–23257. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sato, N.; Taniyama, C.; Ohtani, K.; Arai, K.; Masai, H. A 63-base pair DNA segment containing an Sp1 site but not a canonical E2F site can confer growth-dependent and E2F-mediated transcriptional stimulation of the human ASK gene encoding the regulatory subunit for human Cdc7-related kinase. J. Biol. Chem. 2002, 277, 27668–27681. [Google Scholar] [CrossRef] [PubMed]
- Sasi, N.K.; Bhutkar, A.; Lanning, N.J.; MacKeigan, J.P.; Weinreich, M. DDK Promotes Tumor Chemoresistance and Survival via Multiple Pathways. Neoplasia 2017, 19, 439–450. [Google Scholar] [CrossRef]
- Gerard, A.; Koundrioukoff, S.; Ramillon, V.; Sergere, J.C.; Mailand, N.; Quivy, J.P.; Almouzni, G. The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferating cell nuclear antigen. EMBO Rep. 2006, 7, 817–823. [Google Scholar] [CrossRef]
- Jeffery, D.C.; Kakusho, N.; You, Z.; Gharib, M.; Wyse, B.; Drury, E.; Weinreich, M.; Thibault, P.; Verreault, A.; Masai, H.; et al. CDC28 phosphorylates Cac1p and regulates the association of chromatin assembly factor I with chromatin. Cell Cycle 2015, 14, 74–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Young, T.J.; Cui, Y.; Irudayaraj, J.; Kirchmaier, A.L. Modulation of Gene Silencing by Cdc7p via H4 K16 Acetylation and Phosphorylation of Chromatin Assembly Factor CAF-1 in Saccharomyces cerevisiae. Genetics 2019, 211, 1219–1237. [Google Scholar] [CrossRef] [PubMed]
- Alver, R.C.; Chadha, G.S.; Gillespie, P.J.; Blow, J.J. Reversal of DDK-Mediated MCM Phosphorylation by Rif1-PP1 Regulates Replication Initiation and Replisome Stability Independently of ATR/Chk1. Cell Rep. 2017, 18, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Ones, M.J.K.; Gelot, C.; Munk, S.; Koren, A.; Kawasoe, Y.; George, K.A.; Santos, R.E.; Olsen, J.V.; McCarroll, S.A.; Frattini, M.G.; et al. Human DDK rescues stalled forks and counteracts checkpoint inhibition at unfired origins to complete DNA replication. Mol. Cell 2021, 81, 426–441.e428. [Google Scholar] [CrossRef]
- Sasi, N.K.; Coquel, F.; Lin, Y.L.; MacKeigan, J.P.; Pasero, P.; Weinreich, M. DDK Has a Primary Role in Processing Stalled Replication Forks to Initiate Downstream Checkpoint Signaling. Neoplasia 2018, 20, 985–995. [Google Scholar] [CrossRef]
- Rainey, M.D.; Quinlan, A.; Cazzaniga, C.; Mijic, S.; Martella, O.; Krietsch, J.; Goder, A.; Lopes, M.; Santocanale, C. CDC7 kinase promotes MRE11 fork processing, modulating fork speed and chromosomal breakage. EMBO Rep. 2020, 21, e48920. [Google Scholar] [CrossRef]
- Takahashi, T.S.; Basu, A.; Bermudez, V.; Hurwitz, J.; Walter, J.C. Cdc7-Drf1 kinase links chromosome cohesion to the initiation of DNA replication in Xenopus egg extracts. Genes Dev. 2008, 22, 1894–1905. [Google Scholar] [CrossRef]
- Zheng, G.; Kanchwala, M.; Xing, C.; Yu, H. MCM2-7-dependent cohesin loading during S phase promotes sister-chromatid cohesion. Elife 2018, 7, e33920. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Pahl, P.M.; Harrison, K.; Rosamond, J.; Sclafani, R.A. Cell cycle regulation of the yeast Cdc7 protein kinase by association with the Dbf4 protein. Mol. Cell Biol. 1993, 13, 2899–2908. [Google Scholar] [CrossRef]
- Nasmyth, K. Viewpoint: Putting the cell cycle in order. Science 1996, 274, 1643–1645. [Google Scholar] [CrossRef]
- Torres-Zelada, E.F.; Stephenson, R.E.; Alpsoy, A.; Anderson, B.D.; Swanson, S.K.; Florens, L.; Dykhuizen, E.C.; Washburn, M.P.; Weake, V.M. The Drosophila Dbf4 ortholog Chiffon forms a complex with Gcn5 that is necessary for histone acetylation and viability. J. Cell Sci. 2019, 132, jcs.214072. [Google Scholar] [CrossRef]
- Montagnoli, A.; Bosotti, R.; Villa, F.; Rialland, M.; Brotherton, D.; Mercurio, C.; Berthelsen, J.; Santocanale, C. Drf1, a novel regulatory subunit for human Cdc7 kinase. EMBO J. 2002, 21, 3171–3181. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa-Sugata, N.; Ishii, A.; Taniyama, C.; Matsui, E.; Arai, K.; Masai, H. A second human Dbf4/ASK-related protein, Drf1/ASKL1, is required for efficient progression of S and M phases. J. Biol. Chem. 2005, 280, 13062–13070. [Google Scholar] [CrossRef]
- Yanow, S.K.; Gold, D.A.; Yoo, H.Y.; Dunphy, W.G. Xenopus Drf1, a regulator of Cdc7, displays checkpoint-dependent accumulation on chromatin during an S-phase arrest. J. Biol. Chem. 2003, 278, 41083–41092. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.S.; Walter, J.C. Cdc7-Drf1 is a developmentally regulated protein kinase required for the initiation of vertebrate DNA replication. Genes Dev. 2005, 19, 2295–2300. [Google Scholar] [CrossRef]
- Newport, J.; Kirschner, M. A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell 1982, 30, 675–686. [Google Scholar] [CrossRef]
- Newport, J.; Kirschner, M. A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell 1982, 30, 687–696. [Google Scholar] [CrossRef]
- Gotoh, T.; Kishimoto, T.; Sible, J.C. Phosphorylation of Claspin is triggered by the nucleocytoplasmic ratio at the Xenopus laevis midblastula transition. Dev. Biol. 2011, 353, 302–308. [Google Scholar] [CrossRef]
- Collart, C.; Smith, J.C.; Zegerman, P. Chk1 Inhibition of the Replication Factor Drf1 Guarantees Cell-Cycle Elongation at the Xenopus laevis Mid-blastula Transition. Dev. Cell 2017, 42, 82–96.e83. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Mortimer, R.K.; Culotti, J.; Culotti, M. Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics 1973, 74, 267–286. [Google Scholar] [CrossRef]
- Hereford, L.M.; Hartwell, L.H. Sequential gene function in the initiation of Saccharomyces cerevisiae DNA synthesis. J. Mol. Biol. 1974, 84, 445–461. [Google Scholar] [CrossRef]
- Yoon, H.J.; Loo, S.; Campbell, J.L. Regulation of Saccharomyces cerevisiae CDC7 function during the cell cycle. Mol. Biol. Cell 1993, 4, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Collyer, T.; Hardy, C.F. Cell cycle regulation of DNA replication initiator factor Dbf4p. Mol. Cell Biol. 1999, 19, 4270–4278. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, G.; Owens, J.C.; Shellman, Y.; Sclafani, R.A.; Li, J.J. Cell cycle control of Cdc7p kinase activity through regulation of Dbf4p stability. Mol. Cell Biol. 1999, 19, 4888–4896. [Google Scholar] [CrossRef]
- Weinreich, M.; Stillman, B. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 1999, 18, 5334–5346. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.F.; Santocanale, C.; Drury, L.S.; Diffley, J.F. Dbf4p, an essential S phase-promoting factor, is targeted for degradation by the anaphase-promoting complex. Mol. Cell Biol. 2000, 20, 242–248. [Google Scholar] [CrossRef]
- Sato, N.; Sato, M.; Nakayama, M.; Saitoh, R.; Arai, K.; Masai, H. Cell cycle regulation of chromatin binding and nuclear localization of human Cdc7-ASK kinase complex. Genes Cells 2003, 8, 451–463. [Google Scholar] [CrossRef]
- Masai, H.; Taniyama, C.; Ogino, K.; Matsui, E.; Kakusho, N.; Matsumoto, S.; Kim, J.M.; Ishii, A.; Tanaka, T.; Kobayashi, T.; et al. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 2006, 281, 39249–39261. [Google Scholar] [CrossRef]
- Jiang, W.; McDonald, D.; Hope, T.J.; Hunter, T. Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 1999, 18, 5703–5713. [Google Scholar] [CrossRef]
- Kumagai, H.; Sato, N.; Yamada, M.; Mahony, D.; Seghezzi, W.; Lees, E.; Arai, K.; Masai, H. A novel growth- and cell cycle-regulated protein, ASK, activates human Cdc7-related kinase and is essential for G1/S transition in mammalian cells. Mol. Cell Biol. 1999, 19, 5083–5095. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Watanabe, K.; Mistrik, M.; Vesela, E.; Protivankova, I.; Mailand, N.; Lee, M.; Masai, H.; Lukas, J.; Bartek, J. ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes Dev. 2013, 27, 2459–2472. [Google Scholar] [CrossRef]
- Hardy, C.F.; Dryga, O.; Seematter, S.; Pahl, P.M.; Sclafani, R.A. mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc. Natl. Acad. Sci. USA 1997, 94, 3151–3155. [Google Scholar] [CrossRef]
- Dowell, S.J.; Romanowski, P.; Diffley, J.F. Interaction of Dbf4, the Cdc7 protein kinase regulatory subunit, with yeast replication origins in vivo. Science 1994, 265, 1243–1246. [Google Scholar] [CrossRef]
- Sato, N.; Arai, K.; Masai, H. Human and Xenopus cDNAs encoding budding yeast Cdc7-related kinases: In vitro phosphorylation of MCM subunits by a putative human homologue of Cdc7. EMBO J. 1997, 16, 4340–4351. [Google Scholar] [CrossRef] [PubMed]
- Jares, P.; Blow, J.J. Xenopus cdc7 function is dependent on licensing but not on XORC, XCdc6, or CDK activity and is required for XCdc45 loading. Genes Dev. 2000, 14, 1528–1540. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef]
- Sheu, Y.J.; Kinney, J.B.; Lengronne, A.; Pasero, P.; Stillman, B. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc. Natl. Acad. Sci. USA 2014, 111, E1899–E1908. [Google Scholar] [CrossRef]
- Poh, W.T.; Chadha, G.S.; Gillespie, P.J.; Kaldis, P.; Blow, J.J. Xenopus Cdc7 executes its essential function early in S phase and is counteracted by checkpoint-regulated protein phosphatase 1. Open Biol. 2014, 4, 130138. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Yeeles, J.T.; Diffley, J.F. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016, 35, 961–973. [Google Scholar] [CrossRef]
- Rainey, M.D.; Quachthithu, H.; Gaboriau, D.; Santocanale, C. DNA Replication Dynamics and Cellular Responses to ATP Competitive CDC7 Kinase Inhibitors. ACS Chem. Biol. 2017, 12, 1893–1902. [Google Scholar] [CrossRef]
- Greiwe, J.F.; Miller, T.C.R.; Locke, J.; Martino, F.; Howell, S.; Schreiber, A.; Nans, A.; Diffley, J.F.X.; Costa, A. Structural mechanism for the selective phosphorylation of DNA-loaded MCM double hexamers by the Dbf4-dependent kinase. Nat. Struct. Mol. Biol. 2022, 29, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Li, N.; Huo, Y.; Dang, S.; Tye, B.K.; Gao, N.; Zhai, Y. Structural Insight into the MCM double hexamer activation by Dbf4-Cdc7 kinase. Nat. Commun. 2022, 13, 1396. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Noguchi, Y.; Aramayo, R.; Ivanova, M.E.; Stevens, K.M.; Montoya, A.; Sunidhi, S.; Carranza, N.L.; Skwark, M.J.; Speck, C. The structural basis of Cdc7-Dbf4 kinase dependent targeting and phosphorylation of the MCM2-7 double hexamer. Nat. Commun. 2022, 13, 2915. [Google Scholar] [CrossRef]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef]
- Boos, D.; Yekezare, M.; Diffley, J.F. Identification of a heteromeric complex that promotes DNA replication origin firing in human cells. Science 2013, 340, 981–984. [Google Scholar] [CrossRef]
- Sansam, C.G.; Goins, D.; Siefert, J.C.; Clowdus, E.A.; Sansam, C.L. Cyclin-dependent kinase regulates the length of S phase through TICRR/TRESLIN phosphorylation. Genes Dev. 2015, 29, 555–566. [Google Scholar] [CrossRef]
- Hiraga, S.; Alvino, G.M.; Chang, F.; Lian, H.Y.; Sridhar, A.; Kubota, T.; Brewer, B.J.; Weinreich, M.; Raghuraman, M.K.; Donaldson, A.D. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 2014, 28, 372–383. [Google Scholar] [CrossRef]
- Dave, A.; Cooley, C.; Garg, M.; Bianchi, A. Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep 2014, 7, 53–61. [Google Scholar] [CrossRef]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thoma, N.H.; Hardy, C.F.; Shore, D. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef]
- Hiraga, S.I.; Ly, T.; Garzon, J.; Horejsi, Z.; Ohkubo, Y.N.; Endo, A.; Obuse, C.; Boulton, S.J.; Lamond, A.I.; Donaldson, A.D. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 2017, 18, 403–419. [Google Scholar] [CrossRef]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef]
- Moorhead, G.B.; Trinkle-Mulcahy, L.; Nimick, M.; De Wever, V.; Campbell, D.G.; Gourlay, R.; Lam, Y.W.; Lamond, A.I. Displacement affinity chromatography of protein phosphatase one (PP1) complexes. BMC Biochem 2008, 9, 28. [Google Scholar] [CrossRef]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef]
- Gnan, S.; Flyamer, I.M.; Klein, K.N.; Castelli, E.; Rapp, A.; Maiser, A.; Chen, N.; Weber, P.; Enervald, E.; Cardoso, M.C.; et al. Nuclear organisation and replication timing are coupled through RIF1-PP1 interaction. Nat. Commun. 2021, 12, 2910. [Google Scholar] [CrossRef]
- Daley, J.M.; Sung, P. RIF1 in DNA break repair pathway choice. Mol. Cell 2013, 49, 840–841. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Ratnayeke, N.; Braun, M.; Zhang, T.; Strmiska, V.; Michowski, W.; Can, G.; Simoneau, A.; Snioch, K.; Cup, M.; et al. CDC7-independent G1/S transition revealed by targeted protein degradation. Nature 2022, 605, 357–365. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 13374–13383. [Google Scholar] [CrossRef]
- Rainey, M.D.; Bennett, D.; O’Dea, R.; Zanchetta, M.E.; Voisin, M.; Seoighe, C.; Santocanale, C. ATR Restrains DNA Synthesis and Mitotic Catastrophe in Response to CDC7 Inhibition. Cell Rep. 2020, 32, 108096. [Google Scholar] [CrossRef] [PubMed]
- Ciardo, D.; Haccard, O.; Narassimprakash, H.; Cornu, D.; Guerrera, I.C.; Goldar, A.; Marheineke, K. Polo-like kinase 1 (Plk1) regulates DNA replication origin firing and interacts with Rif1 in Xenopus. Nucleic Acids Res. 2021, 49, 9851–9869. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, M.; Albergante, L.; Moreno, A.; Carrington, J.T.; Blow, J.J.; Newman, T.J. Inevitability and containment of replication errors for eukaryotic genome lengths spanning megabase to gigabase. Proc. Natl. Acad. Sci. USA 2016, 113, E5765–E5774. [Google Scholar] [CrossRef]
- Sansam, C.G.; Pietrzak, K.; Majchrzycka, B.; Kerlin, M.A.; Chen, J.; Rankin, S.; Sansam, C.L. A mechanism for epigenetic control of DNA replication. Genes Dev. 2018, 32, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Dunphy, W.G. Binding of the Treslin-MTBP Complex to Specific Regions of the Human Genome Promotes the Initiation of DNA Replication. Cell Rep. 2020, 32, 108178. [Google Scholar] [CrossRef]
- Fang, D.; Lengronne, A.; Shi, D.; Forey, R.; Skrzypczak, M.; Ginalski, K.; Yan, C.; Wang, X.; Cao, Q.; Pasero, P.; et al. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. 2017, 31, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Kohler, K.; Sanchez-Pulido, L.; Hofer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-cyclin C transcription regulator functions in genome replication through metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef]
- Marheineke, K.; Hyrien, O. Control of replication origin density and firing time in Xenopus egg extracts: Role of a caffeine-sensitive, ATR-dependent checkpoint. J. Biol. Chem. 2004, 279, 28071–28081. [Google Scholar] [CrossRef]
- Woodward, A.M.; Gohler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Janska, A.; Early, A.; Diffley, J.F.X. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef]
- Lewis, J.S.; Spenkelink, L.M.; Schauer, G.D.; Hill, F.R.; Georgescu, R.E.; O’Donnell, M.E.; van Oijen, A.M. Single-molecule visualization of Saccharomyces cerevisiae leading-strand synthesis reveals dynamic interaction between MTC and the replisome. Proc. Natl. Acad. Sci. USA 2017, 114, 10630–10635. [Google Scholar] [CrossRef] [PubMed]
- McClure, A.W.; Diffley, J.F. Rad53 checkpoint kinase regulation of DNA replication fork rate via Mrc1 phosphorylation. Elife 2021, 10, e69726. [Google Scholar] [CrossRef]
- Hodgson, B.; Calzada, A.; Labib, K. Mrc1 and Tof1 regulate DNA replication forks in different ways during normal S phase. Mol. Biol. Cell 2007, 18, 3894–3902. [Google Scholar] [CrossRef]
- Petermann, E.; Helleday, T.; Caldecott, K.W. Claspin promotes normal replication fork rates in human cells. Mol. Biol. Cell 2008, 19, 2373–2378. [Google Scholar] [CrossRef]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef]
- Osborn, A.J.; Elledge, S.J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol. Cell 2003, 11, 329–340. [Google Scholar] [CrossRef]
- Kim, J.M.; Kakusho, N.; Yamada, M.; Kanoh, Y.; Takemoto, N.; Masai, H. Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene 2008, 27, 3475–3482. [Google Scholar] [CrossRef]
- Rainey, M.D.; Harhen, B.; Wang, G.N.; Murphy, P.V.; Santocanale, C. Cdc7-dependent and -independent phosphorylation of Claspin in the induction of the DNA replication checkpoint. Cell Cycle 2013, 12, 1560–1568. [Google Scholar] [CrossRef]
- Yang, C.C.; Suzuki, M.; Yamakawa, S.; Uno, S.; Ishii, A.; Yamazaki, S.; Fukatsu, R.; Fujisawa, R.; Sakimura, K.; Tsurimoto, T.; et al. Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat. Commun. 2016, 7, 12135. [Google Scholar] [CrossRef]
- Meng, Z.; Capalbo, L.; Glover, D.M.; Dunphy, W.G. Role for casein kinase 1 in the phosphorylation of Claspin on critical residues necessary for the activation of Chk1. Mol. Biol. Cell 2011, 22, 2834–2847. [Google Scholar] [CrossRef]
- Yang, C.C.; Kato, H.; Shindo, M.; Masai, H. Cdc7 activates replication checkpoint by phosphorylating the Chk1-binding domain of Claspin in human cells. Elife 2019, 8, e50796. [Google Scholar] [CrossRef] [PubMed]
- Njagi, G.D.; Kilbey, B.J. cdc7-1 a temperature sensitive cell-cycle mutant which interferes with induced mutagenesis in Saccharomyces cerevisiae. Mol. Gen. Genet. 1982, 186, 478–481. [Google Scholar] [CrossRef]
- Ostroff, R.M.; Sclafani, R.A. Cell cycle regulation of induced mutagenesis in yeast. Mutat. Res. 1995, 329, 143–152. [Google Scholar] [CrossRef]
- Princz, L.N.; Wild, P.; Bittmann, J.; Aguado, F.J.; Blanco, M.G.; Matos, J.; Pfander, B. Dbf4-dependent kinase and the Rtt107 scaffold promote Mus81-Mms4 resolvase activation during mitosis. EMBO J. 2017, 36, 664–678. [Google Scholar] [CrossRef]
- Day, T.A.; Palle, K.; Barkley, L.R.; Kakusho, N.; Zou, Y.; Tateishi, S.; Verreault, A.; Masai, H.; Vaziri, C. Phosphorylated Rad18 directs DNA polymerase eta to sites of stalled replication. J. Cell Biol. 2010, 191, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.I.; Monerawela, C.; Katou, Y.; Shaw, S.; Clark, K.R.; Shirahige, K.; Donaldson, A.D. Budding yeast Rif1 binds to replication origins and protects DNA at blocked replication forks. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Garzon, J.; Ursich, S.; Lopes, M.; Hiraga, S.I.; Donaldson, A.D. Human RIF1-Protein Phosphatase 1 Prevents Degradation and Breakage of Nascent DNA on Replication Stalling. Cell Rep. 2019, 27, 2558–2566.e2554. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.; Ray Chaudhuri, A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Andreani, M.; Andrade, J.G.; Saha, T.; Sundaravinayagam, D.; Garzon, J.; Zhang, W.; Popp, O.; Hiraga, S.I.; Rahjouei, A.; et al. Protection of nascent DNA at stalled replication forks is mediated by phosphorylation of RIF1 intrinsically disordered region. Elife 2022, 11, e75047. [Google Scholar] [CrossRef]
- Zegerman, P.; Diffley, J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chiba, T.; Truong, L.N.; Cheng, A.N.; Do, J.; Cho, M.J.; Chen, L.; Wu, X. Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) protein to regulate intra-S-phase checkpoint. J. Biol. Chem. 2012, 287, 2531–2543. [Google Scholar] [CrossRef] [PubMed]
- Abd Wahab, S.; Remus, D. Antagonistic control of DDK binding to licensed replication origins by Mcm2 and Rad53. Elife 2020, 9, e58571. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, V.; Shechter, D.; Lupardus, P.J.; Cimprich, K.A.; Gottesman, M.; Gautier, J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell 2003, 11, 203–213. [Google Scholar] [CrossRef]
- Serra-Cardona, A.; Zhang, Z. Replication-Coupled Nucleosome Assembly in the Passage of Epigenetic Information and Cell Identity. Trends BioChem. Sci. 2018, 43, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Escobar, T.M.; Loyola, A.; Reinberg, D. Parental nucleosome segregation and the inheritance of cellular identity. Nat. Rev. Genet. 2021, 22, 379–392. [Google Scholar] [CrossRef]
- Hwang, Y.; Futran, M.; Hidalgo, D.; Pop, R.; Iyer, D.R.; Scully, R.; Rhind, N.; Socolovsky, M. Global increase in replication fork speed during a p57(KIP2)-regulated erythroid cell fate switch. Sci. Adv. 2017, 3, e1700298. [Google Scholar] [CrossRef]
- Nakatani, T.; Lin, J.; Ji, F.; Ettinger, A.; Pontabry, J.; Tokoro, M.; Altamirano-Pacheco, L.; Fiorentino, J.; Mahammadov, E.; Hatano, Y.; et al. DNA replication fork speed underlies cell fate changes and promotes reprogramming. Nat. Genet. 2022, 54, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Verreault, A.; Kaufman, P.D.; Kobayashi, R.; Stillman, B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 1996, 87, 95–104. [Google Scholar] [CrossRef]
- Shibahara, K.; Stillman, B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999, 96, 575–585. [Google Scholar] [CrossRef]
- Zhang, Z.; Shibahara, K.; Stillman, B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 2000, 408, 221–225. [Google Scholar] [CrossRef]
- Ishimi, Y.; Ichinose, S.; Omori, A.; Sato, K.; Kimura, H. Binding of human minichromosome maintenance proteins with histone H3. J. Biol. Chem. 1996, 271, 24115–24122. [Google Scholar] [CrossRef]
- Groth, A.; Corpet, A.; Cook, A.J.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef]
- Foltman, M.; Evrin, C.; De Piccoli, G.; Jones, R.C.; Edmondson, R.D.; Katou, Y.; Nakato, R.; Shirahige, K.; Labib, K. Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep. 2013, 3, 892–904. [Google Scholar] [CrossRef]
- Huang, H.; Stromme, C.B.; Saredi, G.; Hodl, M.; Strandsby, A.; Gonzalez-Aguilera, C.; Chen, S.; Groth, A.; Patel, D.J. A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat. Struct. Mol. Biol. 2015, 22, 618–626. [Google Scholar] [CrossRef]
- Evrin, C.; Maman, J.D.; Diamante, A.; Pellegrini, L.; Labib, K. Histone H2A-H2B binding by Pol alpha in the eukaryotic replisome contributes to the maintenance of repressive chromatin. EMBO J. 2018, 37, e99021. [Google Scholar] [CrossRef]
- Gan, H.; Serra-Cardona, A.; Hua, X.; Zhou, H.; Labib, K.; Yu, C.; Zhang, Z. The Mcm2-Ctf4-Polalpha Axis Facilitates Parental Histone H3-H4 Transfer to Lagging Strands. Mol. Cell 2018, 72, 140–151.e143. [Google Scholar] [CrossRef]
- Petryk, N.; Dalby, M.; Wenger, A.; Stromme, C.B.; Strandsby, A.; Andersson, R.; Groth, A. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 2018, 361, 1389–1392. [Google Scholar] [CrossRef]
- Yu, C.; Gan, H.; Serra-Cardona, A.; Zhang, L.; Gan, S.; Sharma, S.; Johansson, E.; Chabes, A.; Xu, R.M.; Zhang, Z. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 2018, 361, 1386–1389. [Google Scholar] [CrossRef]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Guacci, V.; Koshland, D.; Strunnikov, A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 1997, 91, 47–57. [Google Scholar] [CrossRef]
- Losada, A.; Hirano, M.; Hirano, T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998, 12, 1986–1997. [Google Scholar] [CrossRef]
- Ciosk, R.; Shirayama, M.; Shevchenko, A.; Tanaka, T.; Toth, A.; Shevchenko, A.; Nasmyth, K. Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol. Cell 2000, 5, 243–254. [Google Scholar] [CrossRef]
- Gillespie, P.J.; Hirano, T. Scc2 couples replication licensing to sister chromatid cohesion in Xenopus egg extracts. Curr. Biol. 2004, 14, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.S.; Yiu, P.; Chou, M.F.; Gygi, S.; Walter, J.C. Recruitment of Xenopus Scc2 and cohesin to chromatin requires the pre-replication complex. Nat. Cell Biol. 2004, 6, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.P.; Farina, A.; Higashi, T.L.; Du, F.; Tappin, I.; Takahashi, T.S.; Hurwitz, J. In vitro loading of human cohesin on DNA by the human Scc2-Scc4 loader complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9366–9371. [Google Scholar] [CrossRef] [PubMed]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Mendez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef]
- Bonte, D.; Lindvall, C.; Liu, H.; Dykema, K.; Furge, K.; Weinreich, M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 2008, 10, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef]
- Koltun, E.S.; Tsuhako, A.L.; Brown, D.S.; Aay, N.; Arcalas, A.; Chan, V.; Du, H.; Engst, S.; Ferguson, K.; Franzini, M.; et al. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 3727–3731. [Google Scholar] [CrossRef]
- Iwai, K.; Nambu, T.; Dairiki, R.; Ohori, M.; Yu, J.; Burke, K.; Gotou, M.; Yamamoto, Y.; Ebara, S.; Shibata, S.; et al. Molecular mechanism and potential target indication of TAK-931, a novel CDC7-selective inhibitor. Sci. Adv. 2019, 5, eaav3660. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human & Xenopus laevis | S. cerevisiae, S. pombe & D melanogaster |
|---|---|
| Cdc7 | Cdc7 (Sc), Hsk1 (Sp), Cdc7 (Dm) |
| Dbf4, ASK (Hs) | Dbf4 (Sc), dfp1, him1, rad35 (Sp), Chiffon A (Dm) |
| Drf1, Dbf4B (Hs) | - |
| Treslin | Sld3 (Sc) |
| MTBP | Sld7 (Sc) |
| RecQ4 | Sld2 (Sc) |
| Claspin | Mrc1 (Sc) |
| Timeless | Tof1 (Sc) |
| Tipin | Csm3 (Sc) |
| Chk1 | Rad53 (Sc) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillespie, P.J.; Blow, J.J. DDK: The Outsourced Kinase of Chromosome Maintenance. Biology 2022, 11, 877. https://doi.org/10.3390/biology11060877
Gillespie PJ, Blow JJ. DDK: The Outsourced Kinase of Chromosome Maintenance. Biology. 2022; 11(6):877. https://doi.org/10.3390/biology11060877
Chicago/Turabian StyleGillespie, Peter J., and J. Julian Blow. 2022. "DDK: The Outsourced Kinase of Chromosome Maintenance" Biology 11, no. 6: 877. https://doi.org/10.3390/biology11060877
APA StyleGillespie, P. J., & Blow, J. J. (2022). DDK: The Outsourced Kinase of Chromosome Maintenance. Biology, 11(6), 877. https://doi.org/10.3390/biology11060877