
ITRAQ-Based Proteomic Analysis of Wheat (Triticum aestivum) Spikes in Response to Tilletia controversa Kühn and Tilletia foetida Kühn Infection, Causal Organisms of Dwarf Bunt and Common Bunt of Wheat

 , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Spore Preparation
2.2. Plant Material and Spore Inoculation
2.3. Molecular Detection of T. controversa and T. foetida in Wheat Spikes Using Conventional PCR
2.4. Protein Extraction and Sample Preparation
2.5. The Protein Digestion and iTRAQ Labelling
2.6. High pH Reversed Phase Separation
2.7. Ultra-High-Performance Liquid Chromatography (UHPLC)-MS/MS Analysis
2.8. Proteomic Data Analysis
2.9. Protein Function Annotation
2.10. Parallel Reaction Monitoring (PRM) Verification Analysis
3. Results
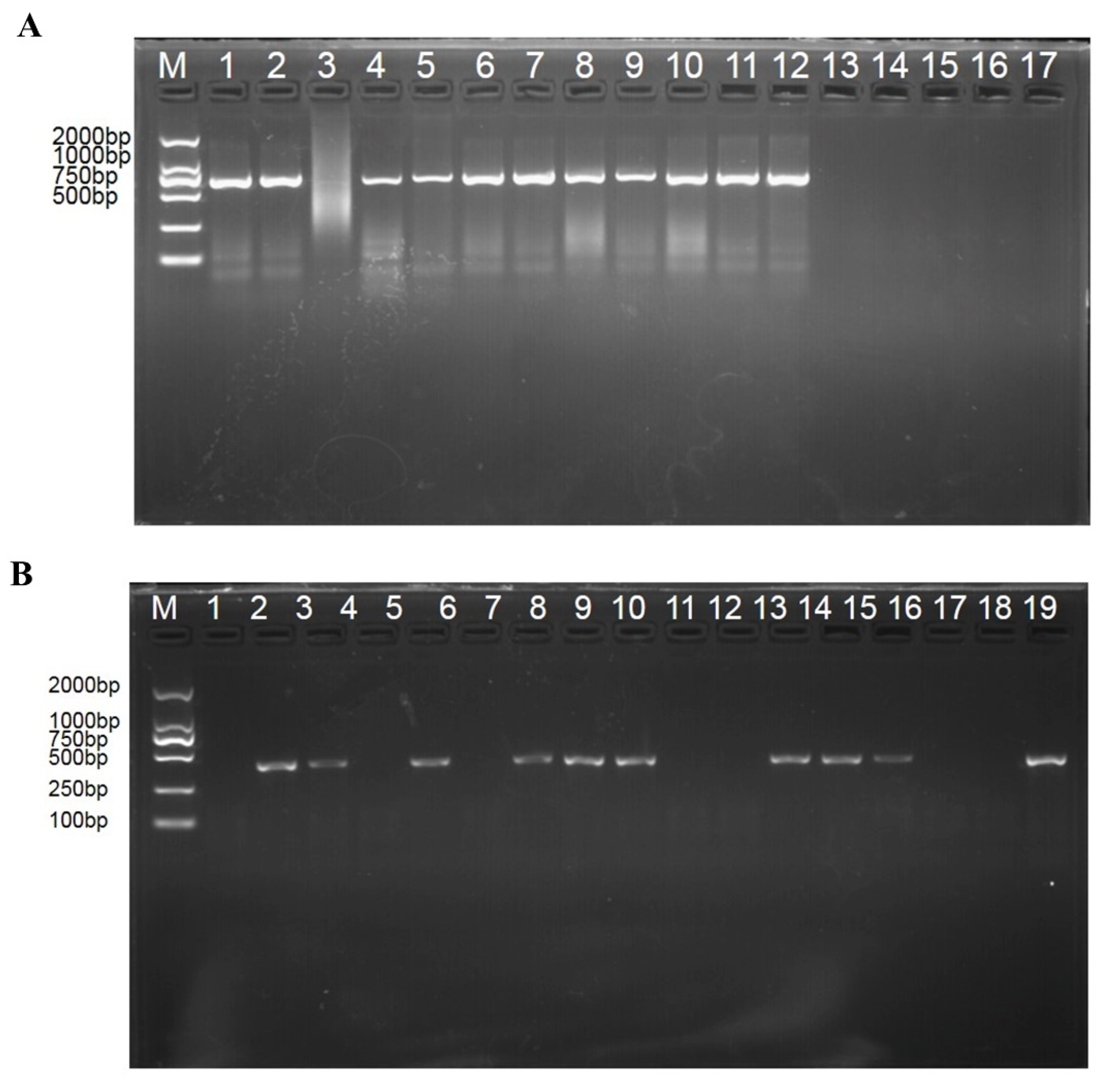
3.1. Pathogen Identification
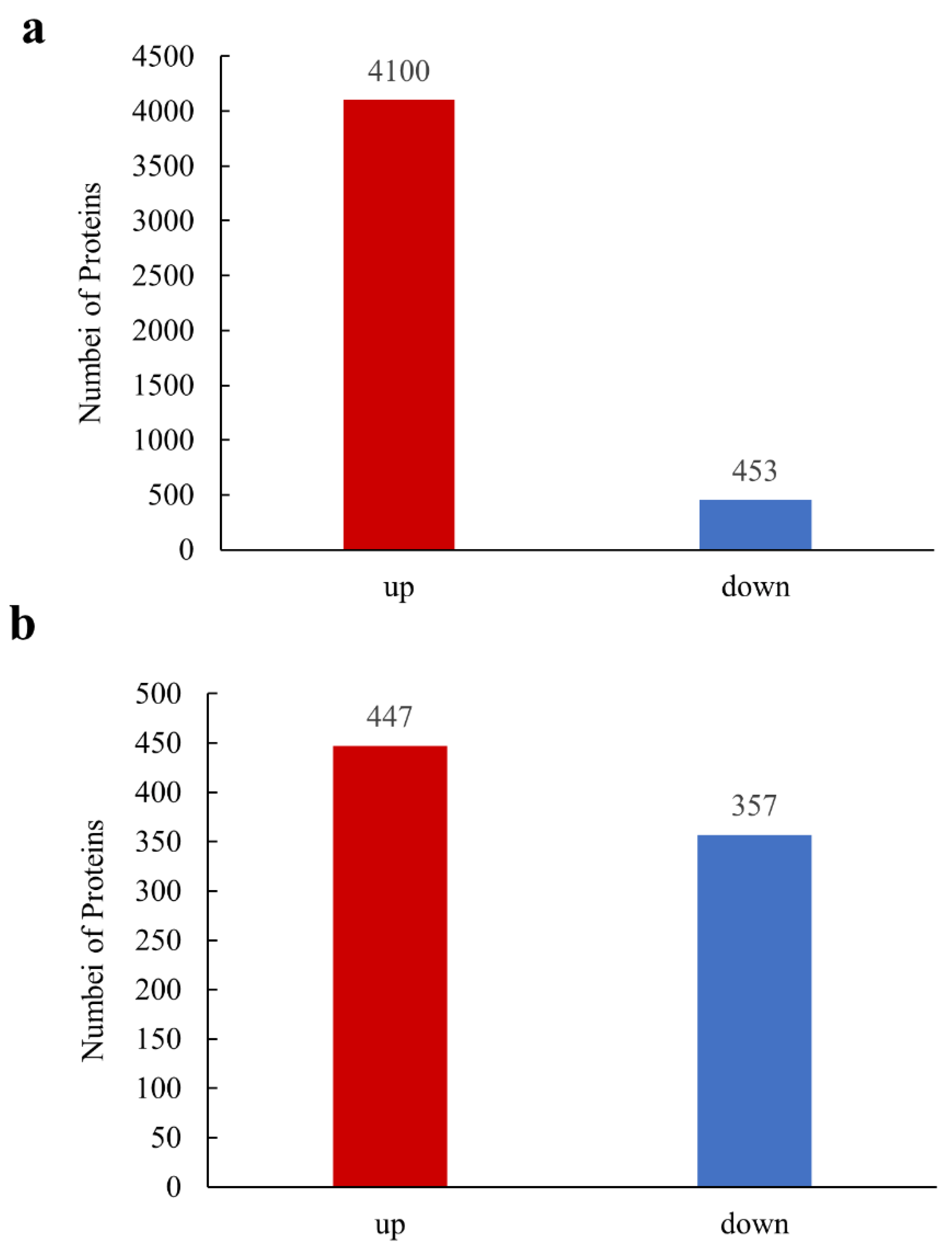
3.2. Identification of Significantly Differently Expressed Proteins (DEPs) on iTRAQ Technology
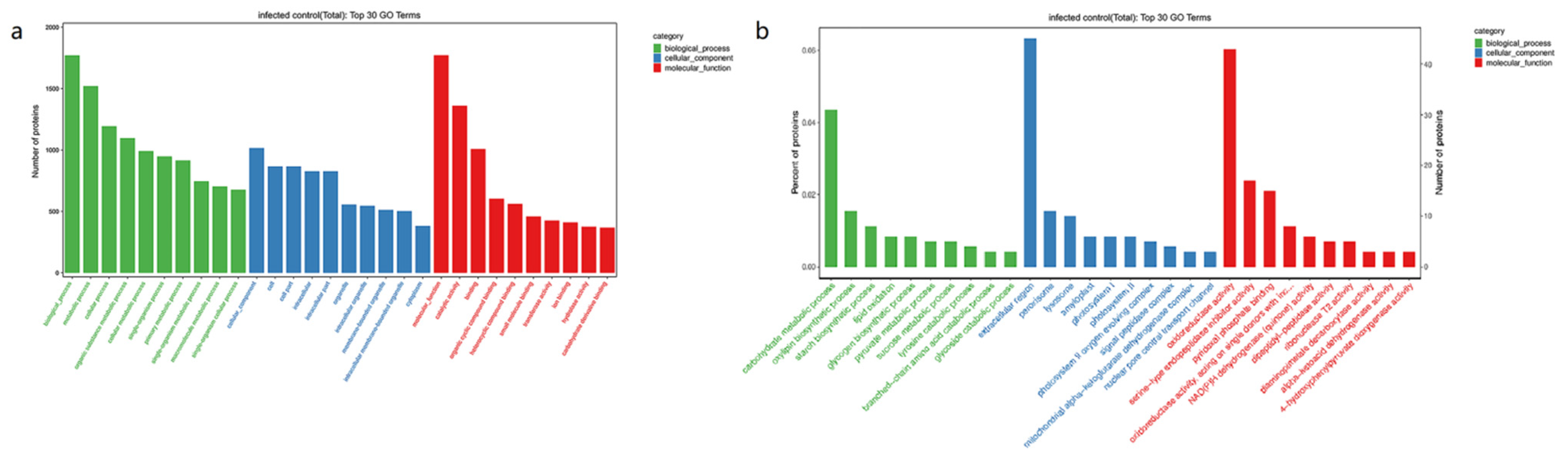
3.3. GO Enrichment Analysis of DEPs
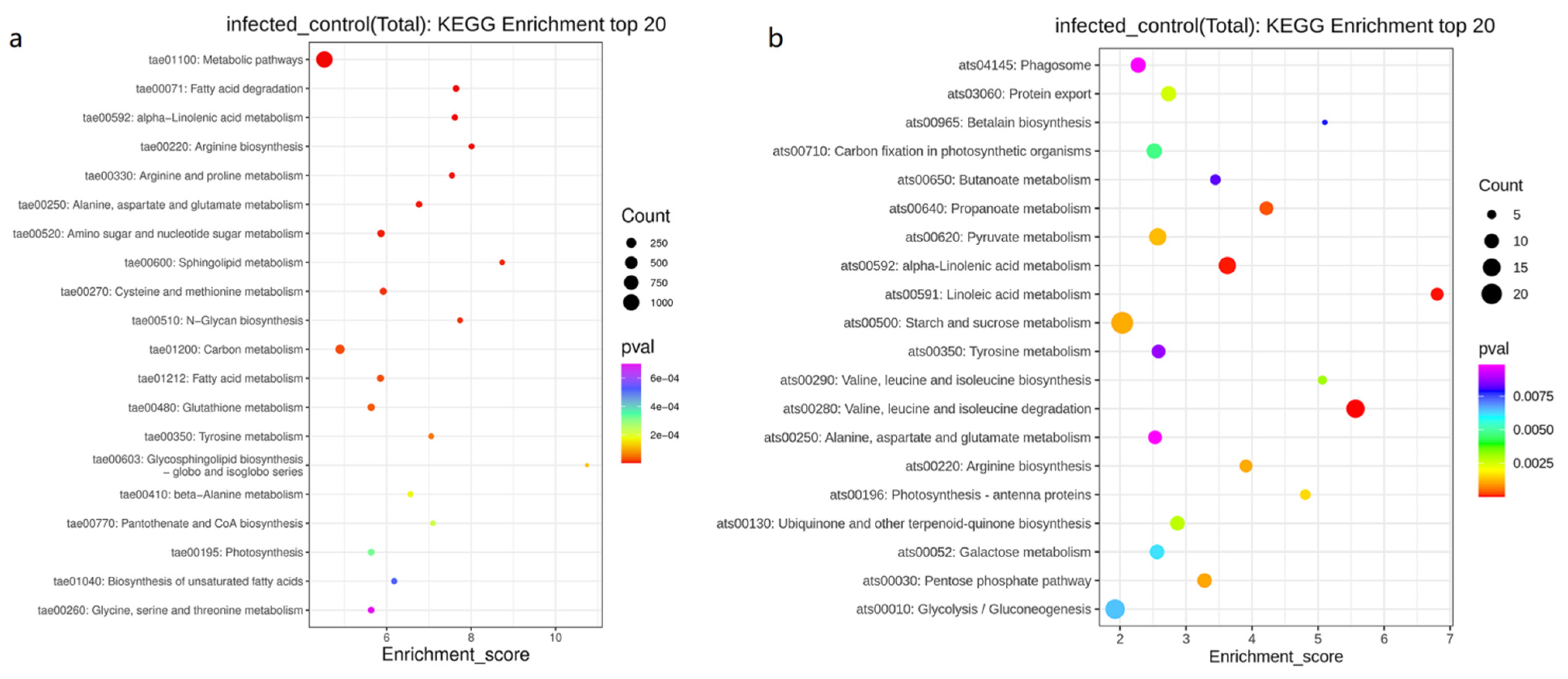
3.4. KEGG Pathway Enrichment Analysis of DEPs
3.5. Differentially Expressed Proteins (DEPs) in Response to T. controversa and T. foetida Infection
3.6. PRM Analysis to Verify DEPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Fang, S.; Zhu, Y.; Wu, D. Risk analysis of wheat yield losses at the county level in mainland china. Front. Environ. Sci. 2021, 9, 141. [Google Scholar] [CrossRef]
- Goates, B.J. Identification of new pathogenic races of common bunt and dwarf bunt fungi, and evaluation of known races using an expanded set of differential wheat lines. Plant Dis. 2012, 96, 361–369. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, W.; Zhou, Y.; Duan, X.; Yong, L.U.O.; Ding, S.; Cao, X.; Bruce, D.L.F. Assessment of risk of establishment of wheat dwarf bunt (Tilletia controversa) in China. J. Integr. Agric. 2013, 12, 87–94. [Google Scholar] [CrossRef]
- Forster, M.K.; Sedaghatjoo, S.; Maier, W.; Killermann, B.; Niessen, L. Discrimination of Tilletia controversa from the T. caries/T. laevis complex by MALDI-TOF MS analysis of teliospores. Appl. Microbiol. Biotechnol. 2022, 106, 1257–1278. [Google Scholar] [CrossRef]
- Gaudet, D.A.; Lu, Z.X.; Leggett, F.; Puchalski, B.; Laroche, A. Compatible and incompatible interactions in wheat involving the Bt-10 gene for resistance to Tilletia tritici, the common bunt pathogen. Phytopathology 2007, 97, 1397–1405. [Google Scholar] [CrossRef][Green Version]
- Mathre, D.E. Dwarf bunt: Politics, identification, and biology. Annu. Rev. Phytopathol. 1996, 34, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.X.; Gaudet, D.A.; Frick, M.; Puchalski, B.; Genswein, B.; Laroche, A. Identification and characterization of genes differentially expressed in the resistance reaction in wheat infected with Tilletia tritici, the common bunt pathogen. J. Biochem. Mol. Biol. 2005, 38, 420–431. [Google Scholar] [CrossRef]
- Chen, D.; Muhae-Ud-din, G.; Liu, T.; Chen, W.; Liu, C.; Gao, L. Wheat varietal response to Tilletia controversa J. G. Kühn using qRT-PCR and laser confocal microscopy. Genes 2021, 12, 425. [Google Scholar] [CrossRef]
- Muhae-Ud-Din, G.; Chen, D.; Liu, T.; Chen, W.; Gao, L. Characterization of the wheat cultivars against Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 2020, 10, 9029. [Google Scholar] [CrossRef]
- Ren, Z.; Liu, J.; Din, G.M.U.; Zhang, H.; Du, Z.; Chen, W.; Liu, T.; Zhang, J.; Zhao, S.; Gao, L. Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci. Rep. 2020, 10, 21567. [Google Scholar] [CrossRef]
- Bonman, J.M.; Bockelman, H.E.; Goates, B.J.; Obert, D.E.; McGuire, P.E.; Qualset, C.O.; Hijmans, R.J. Geographic distribution of common and dwarf bunt resistance in landraces of Triticum aestivum subsp. aestivum. Crop Sci. 2006, 46, 1622–1629. [Google Scholar] [CrossRef]
- Dou, D.; Zhou, J.M. Phytopathogen effectors subverting host immunity: Different foes, similar battleground. Cell Host Microbe 2012, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Howden, A.J.M.; Huitema, E. Effector-triggered post-translational modifications and their role in suppression of plant immunity. Front. Plant Sci. 2012, 3, 160. [Google Scholar] [CrossRef] [PubMed]
- Quentin, M.; Abad, P.; Favery, B. Plant parasitic nematode effectors target host defense and nuclear functions to establish feeding cells. Front. Plant Sci. 2013, 4, 53. [Google Scholar] [CrossRef]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.R.; Pan, H.; et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [Google Scholar] [CrossRef]
- Mogga, V.; Delventhal, R.; Weidenbach, D.; Langer, S.; Bertram, P.M.; Andresen, K.; Thines, E.; Kroj, T.; Schaffrath, U. Magnaporthe oryzae effectors MoHEG13 and MoHEG16 interfere with host infection and MoHEG13 counteracts cell death caused by Magnaporthe-NLPs in tobacco. Plant Cell Rep. 2016, 35, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.N.; Ziemann, S.; Treitschke, S.; Aßmann, D.; Doehlemann, G. Compatibility in the Ustilago maydis-maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog. 2013, 9, e1003177. [Google Scholar] [CrossRef]
- Redkar, A.; Villajuana-Bonequi, M.; Doehlemann, G. Conservation of the Ustilago maydis effector see1 in related smuts. Plant Signal. Behav. 2015, 10, e1086855. [Google Scholar] [CrossRef][Green Version]
- Hemetsberger, C.; Herrberger, C.; Zechmann, B.; Hillmer, M.; Doehlemann, G. The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog. 2012, 8, e1002684. [Google Scholar] [CrossRef]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef]
- Tanaka, S.; Brefort, T.; Neidig, N.; Djamei, A.; Kahnt, J.; Vermerris, W.; Koenig, S.; Feussner, K.; Feussner, I.; Kahmann, R. A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. Elife 2014, 3, e01355. [Google Scholar] [CrossRef]
- Luderer, R.; Takken, F.L.W.; De Wit, P.J.G.M.; Joosten, M.H.A.J. Cladosporium fulvum overcomes Cf-2-mediated resistance by producing truncated AVR2 elicitor proteins. Mol. Microbiol. 2002, 45, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Van Der Hoorn, R.A.L.; Laurent, F.; Roth, R.; De Wit, P.J.G.M. Agroinfiltration is a versatile tool that facilitates comparative analyses of Avr9/Cf-9-induced and Avr4/Cf-4-induced necrosis. Mol. Plant-Microbe Interact. 2000, 13, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Han, Y.; Zhang, N.; Zhang, M.; Liu, L.; Li, S.; Lu, F.; Sun, W. Identification and characterization of plant cell death-inducing secreted proteins from Ustilaginoidea virens. Mol. Plant-Microbe Interact. 2016, 29, 405–416. [Google Scholar] [CrossRef]
- Li, N.; Han, X.; Feng, D.; Yuan, D.; Huang, L.J. Signaling crosstalk between salicylic acid and ethylene/Jasmonate in plant defense: Do we understand what they are whispering? Int. J. Mol. Sci. 2019, 20, 671. [Google Scholar] [CrossRef]
- Wang, A.; Pang, L.; Wang, N.; Ai, P.; Yin, D.; Li, S.; Deng, Q.; Zhu, J.; Liang, Y.; Zhu, J.; et al. The pathogenic mechanisms of Tilletia horrida as revealed by comparative and functional genomics. Sci. Rep. 2018, 8, 15413. [Google Scholar] [CrossRef]
- Wang, A.; Pan, L.; Niu, X.; Shu, X.; Yi, X.; Yamamoto, N.; Li, S. Comparative secretome analysis of different smut fungi and identification of plant cell death-inducing secreted proteins from Tilletia horrida. BMC Plant Biol. 2019, 19, 360. [Google Scholar] [CrossRef]
- Wu, X.; Yan, J.; Wu, Y.; Zhang, H.; Mo, S.; Xu, X.; Zhou, F.; Ding, H. Proteomic analysis by iTRAQ-PRM provides integrated insight into mechanisms of resistance in pepper to Bemisia tabaci (Gennadius). BMC Plant Biol. 2019, 19, 270. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Luo, Y.M.; Ye, Z.Q.; Cao, X.; Liang, J.N.; Wang, Q.; Wu, Y.; Wu, J.H.; Wang, H.Y.; Zhang, M.; et al. iTRAQ-based proteomics analysis of autophagy-mediated immune responses against the vascular fungal pathogen Verticillium dahliae in Arabidopsis. Autophagy 2018, 14, 598–618. [Google Scholar] [CrossRef]
- Din, G.M.U.; Du, Z.; Zhang, H.; Zhao, S.; Liu, T.; Chen, W.; Gao, L. Effects of Tilletia foetida on microbial communities in the rhizosphere soil of wheat seeds coated with different concentrations of Jianzhuang. Microb. Ecol. 2021, 82, 736–745. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, W.; Wang, M.; Gao, H.; Shen, H.; Wang, C.; Liu, T.; Chen, W.; Gao, L. Characteristics of the infection of Tilletia laevis Kühn (syn. Tilletia foetida (Wallr.) Liro.) in compatible wheat. Plant Pathol. J. 2021, 37, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yu, H.; Han, W.; Gao, F.; Liu, T.; Liu, B.; Kang, X.; Gao, J.; Chen, W. Development of a SCAR marker for molecular detection and diagnosis of Tilletia controversa Kühn, the causal fungus of wheat dwarf bunt. World J. Microbiol. Biotechnol. 2014, 30, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Qin, D.; Chen, D.; Liu, C.; Chen, W.; Liu, T.; Liu, B.; Gao, L. Development of ISSR-derived SCAR marker and SYBR Green I real-time PCR method for detection of teliospores of Tilletia laevis Kühn. Sci. Rep. 2019, 9, 17651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Jiang, W.; Liu, X.; Duan, Y.; Xiang, L.; Wang, Y.; Jiang, Y.; Shen, X.; Chen, X.; Yin, C.; et al. ITRAQ-based quantitative proteomic analysis of Fusarium moniliforme (Fusarium verticillioides) in response to Phloridzin inducers. Proteome Sci. 2021, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteom. 2012, 11, 1–8. [Google Scholar] [CrossRef]
- Ma, Q.; Shi, C.; Su, C.; Liu, Y. Complementary analyses of the transcriptome and iTRAQ proteome revealed mechanism of ethylene dependent salt response in bread wheat (Triticum aestivum L.). Food Chem. 2020, 325, 126866. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Giraldo, M.C.; Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Microbiol. 2013, 11, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Zetzsche, H.; Friedt, W.; Ordon, F. Breeding progress for pathogen resistance is a second major driver for yield increase in German winter wheat at contrasting N levels. Sci. Rep. 2020, 10, 20374. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Yan, F.; Ji, J.; Li, Y.; Wang, R.; Xu, C. Proteomic analysis of Arabidopsis thaliana leaves infested by tobacco whitefly Bemisia tabaci (Gennadius) B biotype. Plant Mol. Biol. Rep. 2012, 30, 379–390. [Google Scholar] [CrossRef]
- Huang, H.; Ullah, F.; Zhou, D.X.; Yi, M.; Zhao, Y. Mechanisms of ROS regulation of plant development and stress responses. Front. Plant Sci. 2019, 10, 800. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Williams, C.E.; Nemacheck, J.A.; Wang, H.; Subramanyam, S.; Zheng, C.; Chen, M.S. Reactive oxygen species are involved in plant defense against a gall midge. Plant Physiol. 2010, 152, 985–999. [Google Scholar] [CrossRef]
- Medina, E.; Kim, S.H.; Yun, M.; Choi, W.G. Recapitulation of the function and role of ros generated in response to heat stress in plants. Plants 2021, 10, 371. [Google Scholar] [CrossRef]
- Phua, S.Y.; De Smet, B.; Remacle, C.; Chan, K.X.; Van Breusegem, F. Reactive oxygen species and organellar signaling. J. Exp. Bot. 2021, 72, 5807–5824. [Google Scholar] [CrossRef]
- El-Beltagi, H.S.; Sofy, M.R.; Aldaej, M.I.; Mohamed, H.I. Silicon alleviates copper toxicity in flax plants by up-regulating antioxidant defense and secondary metabolites and decreasing oxidative damage. Sustainability 2020, 12, 4732. [Google Scholar] [CrossRef]
- Masamba, P.; Kappo, A.P. Immunological and biochemical interplay between cytokines, oxidative stress and schistosomiasis. Int. J. Mol. Sci. 2021, 22, 7216. [Google Scholar] [CrossRef]
- Zeng, W.; Sun, Z.; Cai, Z.; Chen, H.; Lai, Z.; Yang, S.; Tang, X. Proteomic analysis by iTRAQ-MRM of soybean resistance to Lamprosema Indicate. BMC Genom. 2017, 18, 444. [Google Scholar] [CrossRef]
- Pasin, F.; Simón-Mateo, C.; García, J.A. The Hypervariable amino-terminus of P1 protease modulates potyviral replication and host defense responses. PLoS Pathog. 2014, 10, e1003985. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Bhuyan, M.H.M.B.; Zulfiqar, F.; Raza, A.; Mohsin, S.M.; Al Mahmud, J.; Fujita, M.; Fotopoulos, V. Reactive oxygen species and antioxidant defense in plants under abiotic stress: Revisiting the crucial role of a universal defense regulator. Antioxidants 2020, 9, 681. [Google Scholar] [CrossRef]
- Polidoros, A.N.; Mylona, P.V.; Scandalios, J.G. Transgenic tobacco plants expressing the maize Cat2 gene have altered catalase levels that affect plant-pathogen interactions and resistance to oxidative stress. Transgenic Res. 2001, 10, 555–569. [Google Scholar] [CrossRef]
- Pittner, E.; Marek, J.; Bortuli, D.; Santos, L.A.; Knob, A.; Faria, C.M.D.R. Defense responses of wheat plants (Triticum aestivum L.) against brown spot as a result of possible elicitors application. Arq. Inst. Biol. 2019, 86, 1–16. [Google Scholar] [CrossRef]
- Foyer, C.H.; Noctor, G. Ascorbate and glutathione: The heart of the redox hub. Plant Physiol. 2011, 155, 2–18. [Google Scholar] [CrossRef]
- Park, S.J.; Huang, Y.; Ayoubi, P. Identification of expression profiles of sorghum genes in response to greenbug phloem-feeding using cDNA subtraction and microarray analysis. Planta 2006, 223, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ariza, J.; Campo, S.; Rufat, M.; Estopà, M.; Messeguer, J.; San Segundo, B.; Coca, M. Sucrose-mediated priming of plant defense responses and broad-spectrum disease resistance by overexpression of the maize pathogenesis-related PRms protein in rice plants. Mol. Plant-Microbe Interact. 2007, 20, 832–842. [Google Scholar] [CrossRef]
- Mackey, D.; Belkhadir, Y.; Alonso, J.M.; Ecker, J.R.; Dangl, J.L. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 2003, 112, 379–389. [Google Scholar] [CrossRef]
- Kankanala, P.; Nandety, R.S.; Mysore, K.S. Genomics of plant disease resistance in legumes. Front. Plant Sci. 2019, 10, 1345. [Google Scholar] [CrossRef] [PubMed]
- van Loon, L.C.; Rep, M.; Pieterse, C.M.J. Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 2006, 44, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Rouhier, N.; Lemaire, S.D.; Jacquot, J.P. The role of glutathione in photosynthetic organisms: Emerging functions for glutaredoxins and glutathionylation. Annu. Rev. Plant Biol. 2008, 59, 143–166. [Google Scholar] [CrossRef]
- Fengming, S.; Xiuchun, G.; Zhong, Z. Changes of glutathione contents in cotton seedlings infected by Fusarium oxysporum f. sp. vasinfectum and its relationship to disease resistance. J. Zhejiang Univ. Life Sci. 2001, 27, 615–618. [Google Scholar]
- Aliyeva, D.R.; Aydinli, L.M.; Zulfugarov, I.S.; Huseynova, I.M. Diurnal changes of the ascorbate-glutathione cycle components in wheat genotypes exposed to drought. Funct. Plant Biol. 2020, 47, 998–1006. [Google Scholar] [CrossRef]
- Zechmann, B. Subcellular roles of glutathione in mediating plant defense during biotic stress. Plants 2020, 9, 1067. [Google Scholar] [CrossRef]
- Duan, Y.H.; Guo, J.; Ding, K.; Wang, S.J.; Zhang, H.; Dai, X.W.; Chen, Y.Y.; Govers, F.; Huang, L.L.; Kang, Z.S. Characterization of a wheat HSP70 gene and its expression in response to stripe rust infection and abiotic stresses. Mol. Biol. Rep. 2011, 38, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Lu, Y.; Li, K.; Lin, L.; Zheng, H.; Yan, F.; Chen, J. Heat shock protein 70 is necessary for Rice stripe virus infection in plants. Mol. Plant Pathol. 2014, 15, 907–917. [Google Scholar] [CrossRef]
- Andreasson, E.; Jenkins, T.; Brodersen, P.; Thorgrimsen, S.; Petersen, N.H.T.; Zhu, S.; Qiu, J.; Micheelsen, P.; Rocher, A.; Petersen, M.; et al. The MAP kinase substrate MKS1 is a regulator of plant defense responses. EMBO J. 2005, 24, 2579–2589. [Google Scholar] [CrossRef]
- Ehness, R.; Ecker, M.; Godt, D.E.; Roitsch, T. Glucose and stress independently regulate source and sink metabolism and defense mechanisms via signal transduction pathways involving protein phosphorylation. Plant Cell 1997, 9, 1825–1841. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Sinha, A.K.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactions. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef]
- Siemens, J.; Keller, I.; Sarx, J.; Kunz, S.; Schuller, A.; Nagel, W.; Schmülling, T.; Parniske, M.; Ludwig-Müller, J. Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol. Plant-Microbe Interact. 2006, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein id. | Symbol | FC (iTRAQ) | p-Value | FC (PRM) | p-Value |
|---|---|---|---|---|---|
| Reference protein | GST4 | 1.48 | 0.00003950 | 2.00 | 0.00000004 |
| TraesCS1A02G355300.1 | PR4A | 2.20 | 0.01852322 | 7.48 | 0.00089088 |
| TraesCS2A02G320400.1 | G6PDH | 1.64 | 2.61 × 10−14 | 1.10 | 0.00005540 |
| TraesCS2D02G123300.1 | SODCC.3 | 1.23 | 0.00422247 | 0.88 | 0.29967867 |
| TraesCS4A02G106400.1 | MPK5 | 1.76 | 0.00196922 | 2.52 | 0.00144951 |
| TraesCS5A02G018200.1.cds1 | RASTL-4 | 5.79 | 0.00210438 | 40.01 | 0.00000303 |
| TraesCS5D02G364600.1 | PER2 | 2.05 | 0.00019080 | 17.69 | 0.00000019 |
| TraesCS7A02G198900.1.cds1 | PRMS | 2.28 | 0.00033301 | 31.00 | 0.00000006 |
| TraesCS7A02G424100.1 | PER22 | 1.34 | 0.00002050 | 1.26 | 0.00412683 |
| TraesCS7D02G299500.1 | MDAR5 | 1.23 | 0.00000550 | 1.42 | 0.16694490 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, T.; Xu, T.; Muhae-Ud-Din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. ITRAQ-Based Proteomic Analysis of Wheat (Triticum aestivum) Spikes in Response to Tilletia controversa Kühn and Tilletia foetida Kühn Infection, Causal Organisms of Dwarf Bunt and Common Bunt of Wheat. Biology 2022, 11, 865. https://doi.org/10.3390/biology11060865
He T, Xu T, Muhae-Ud-Din G, Guo Q, Liu T, Chen W, Gao L. ITRAQ-Based Proteomic Analysis of Wheat (Triticum aestivum) Spikes in Response to Tilletia controversa Kühn and Tilletia foetida Kühn Infection, Causal Organisms of Dwarf Bunt and Common Bunt of Wheat. Biology. 2022; 11(6):865. https://doi.org/10.3390/biology11060865
Chicago/Turabian StyleHe, Ting, Tongshuo Xu, Ghulam Muhae-Ud-Din, Qingyun Guo, Taiguo Liu, Wanquan Chen, and Li Gao. 2022. "ITRAQ-Based Proteomic Analysis of Wheat (Triticum aestivum) Spikes in Response to Tilletia controversa Kühn and Tilletia foetida Kühn Infection, Causal Organisms of Dwarf Bunt and Common Bunt of Wheat" Biology 11, no. 6: 865. https://doi.org/10.3390/biology11060865
APA StyleHe, T., Xu, T., Muhae-Ud-Din, G., Guo, Q., Liu, T., Chen, W., & Gao, L. (2022). ITRAQ-Based Proteomic Analysis of Wheat (Triticum aestivum) Spikes in Response to Tilletia controversa Kühn and Tilletia foetida Kühn Infection, Causal Organisms of Dwarf Bunt and Common Bunt of Wheat. Biology, 11(6), 865. https://doi.org/10.3390/biology11060865