
Prediction of Major Histocompatibility Complex Binding with Bilateral and Variable Long Short Term Memory Networks




Abstract
:Simple Summary
Abstract
1. Introduction
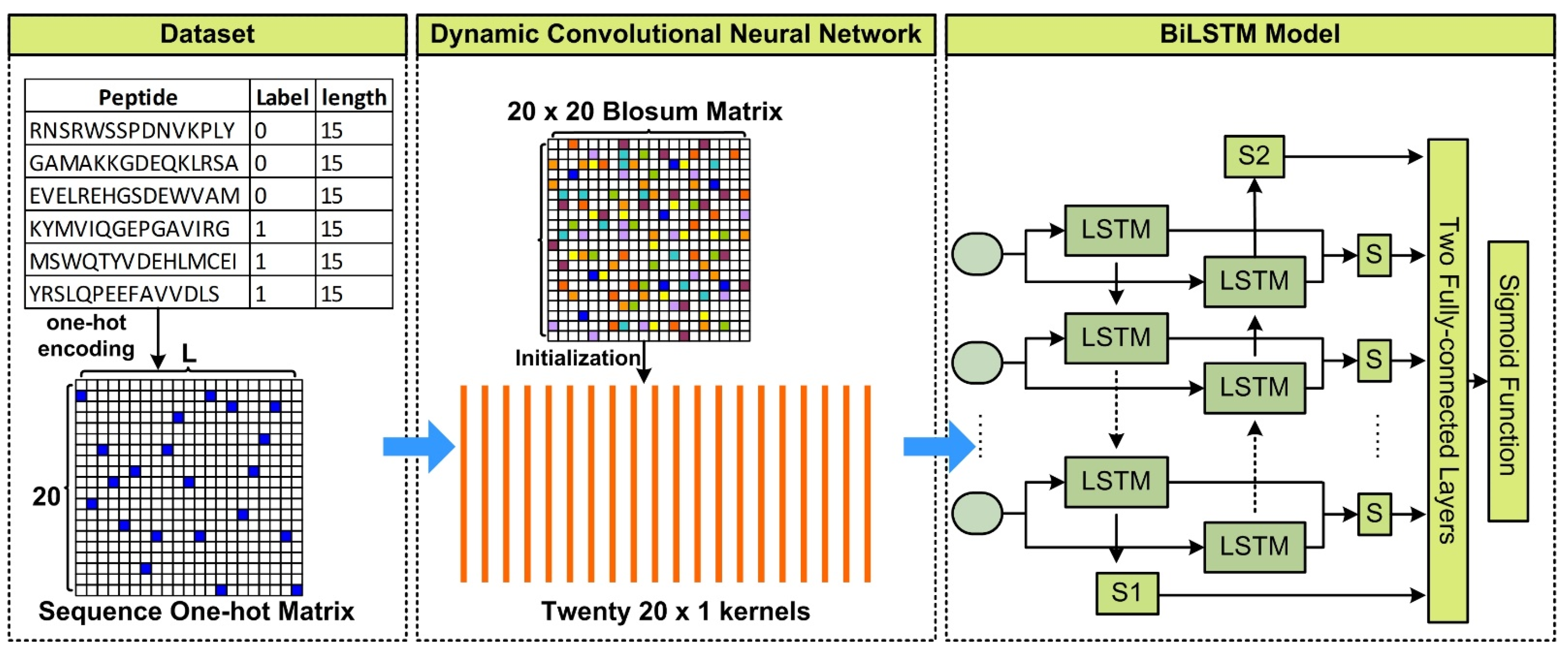
2. Materials and Methods
2.1. Training and Validation Datasets
2.2. Feature Representation at Evolutionary Level
2.3. Feature Representation at Sequential Level
2.4. Evaluation Criteria
3. Results
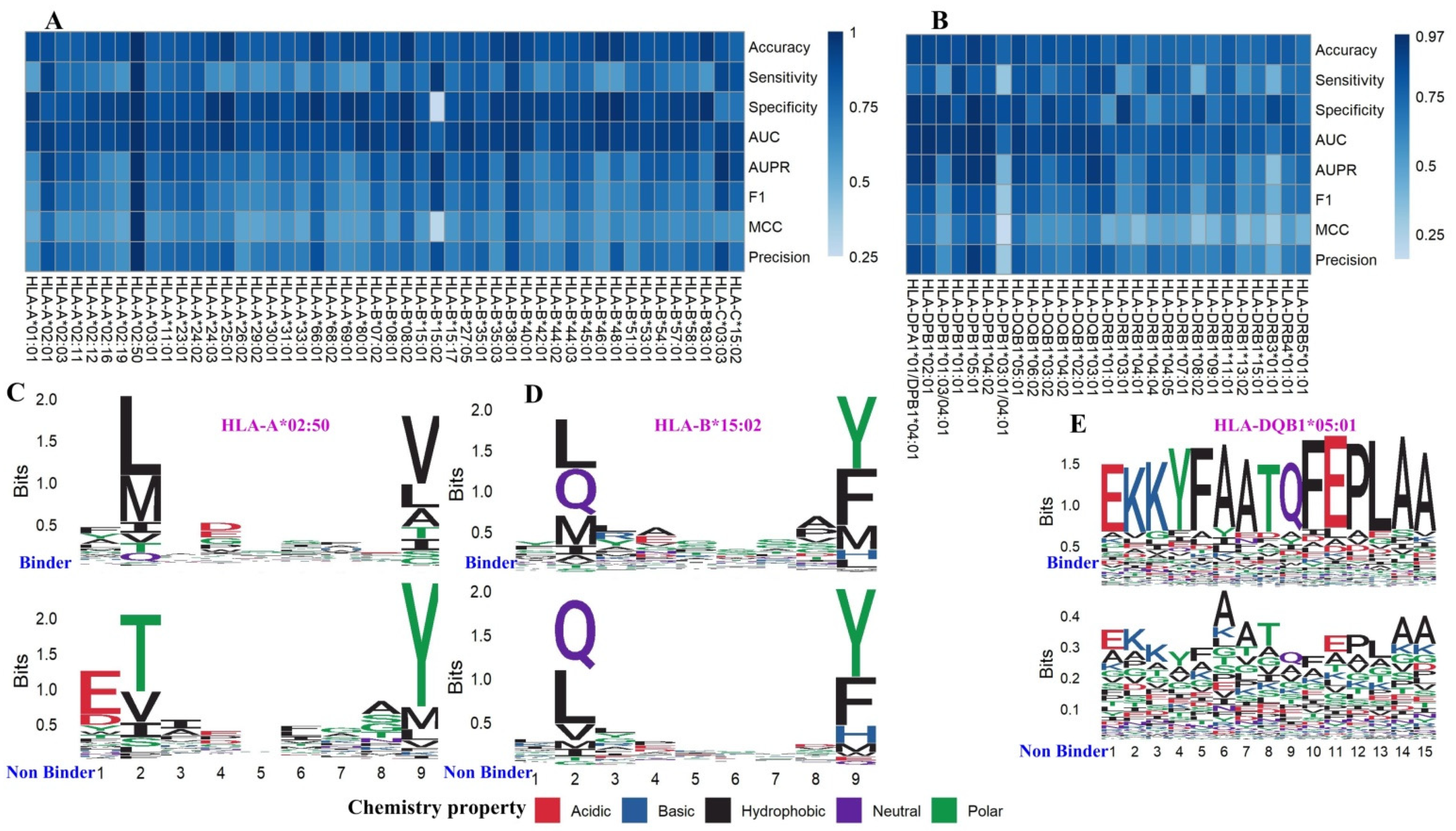
3.1. Human Dataset Description
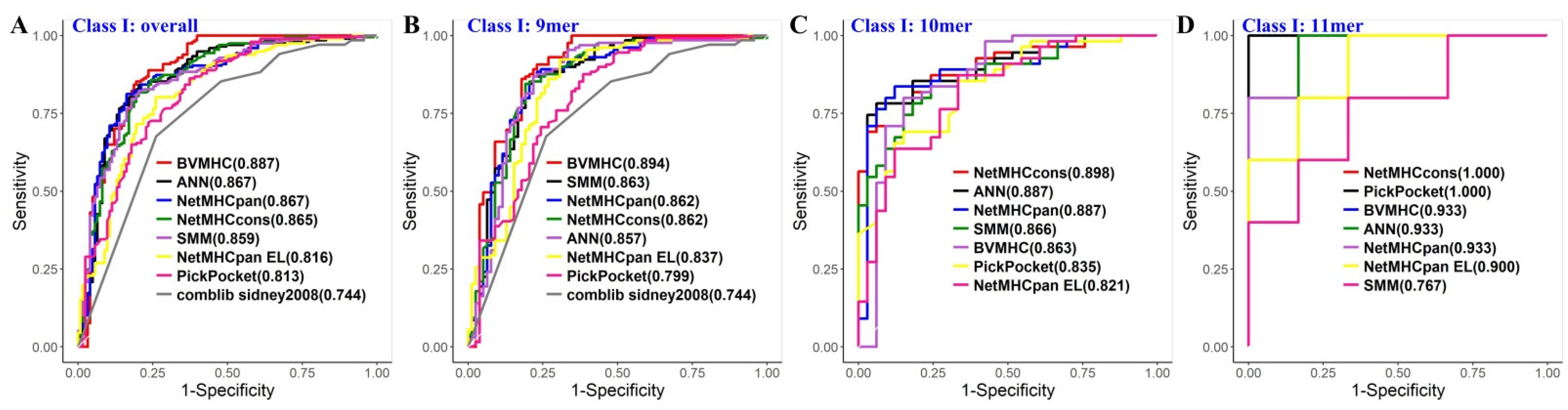
3.2. Independent Validation and Comparison with Other MHC Binding Predictors
3.3. Performance of Non-Human Species
3.4. Web Server Implementation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kasahara, M. The MHC class I ligand-generating system: Roles of immunoproteasomes and the interferon-gamma-inducible proteasome activator PA28. Immunol. Rev. 1998, 163, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Schott, G.; Garcia-Blanco, M.A. MHC Class III RNA Binding Proteins and Immunity. RNA Biol. 2021, 18, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced beta M-2/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, F.; Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, K.; Rotzschke, O.; Stevanovie, S.; Jung, G.; Rammensee, H.-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 1991, 351, 290–296. [Google Scholar] [CrossRef]
- Hobohm, U.; Meyerhans, A. A pattern search method for putative anchor residues in T cell epitopes. Eur. J. Immunol. 1993, 23, 1271–1276. [Google Scholar] [CrossRef]
- Kessler, J.H.; Benckhuijsen, W.E.; Mutis, T.; Melief, C.J.; van der Burg, S.H.; Drijfhout, J.W. Competition-based cellular peptide binding assay for HLA class I. Curr. Protoc. Immunol. 2004, 18. [Google Scholar] [CrossRef]
- Jiang, L.; Yu, H.; Li, J.; Tang, J.; Guo, Y.; Guo, F. Predicting MHC class I binder: Existing approaches and a novel recurrent neural network solution. Brief Bioinform. 2021, 22, bbab216. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, M.; Singh, H.; Raghava, G.P. MHCBN: A comprehensive database of MHC binding and non-binding peptides. Bioinformatics 2003, 19, 665–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Wang, Z.; Hu, H.; Wan, F.; Chen, L.; Xiong, Y.; Wang, X.; Zhao, D.; Huang, W.; Zeng, J. ACME: Pan-specific peptide-MHC class I binding prediction through attention-based deep neural networks. Bioinformatics 2019, 35, 4946–4954. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Gifford, D.K. DeepLigand: Accurate prediction of MHC class I ligands using peptide embedding. Bioinformatics 2019, 35, i278–i283. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.A.; Krishna, S.; Anderson, K.S. A Random Forest based approach to MHC class I epitope prediction and analysis. J. Immunol. 2018, 200. [Google Scholar]
- Jensen, K.K.; Andreatta, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Karosiene, E.; Lundegaard, C.; Lund, O.; Nielsen, M. NetMHCcons: A consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012, 64, 177–186. [Google Scholar] [CrossRef]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1. [Google Scholar] [CrossRef] [Green Version]
- Lundegaard, C.; Lund, O.; Nielsen, M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 2008, 24, 1397–1398. [Google Scholar] [CrossRef]
- Zhang, H.; Lund, O.; Nielsen, M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: Application to MHC-peptide binding. Bioinformatics 2009, 25, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Sidney, J.; Assarsson, E.; Moore, C.; Ngo, S.; Pinilla, C.; Sette, A.; Peters, B. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreatta, M.; Karosiene, E.; Rasmussen, M.; Stryhn, A.; Buus, S.; Nielsen, M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015, 67, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanessa, J.; Sinu, P.; Massimo, A.; Paolo, M.; Bjoern, P.; Morten, N. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Singh, H.; Raghava, G.P. ProPred1: Prediction of promiscuous MHC Class-I binding sites. Bioinformatics 2003, 19, 1009–1014. [Google Scholar] [CrossRef]
- Dönnes, P.; Elofsson, A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinform. 2002, 3, 25. [Google Scholar] [CrossRef]
- Rasmussen, M.; Fenoy, E. Pan-Specific Prediction of Peptide-MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016, 197, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Li, D.; Li, Z.; Qiu, S.; Li, W.; Chao, C.C.; Yang, N.; Li, H.; Cheng, Z.; Song, X.; et al. PSSMHCpan: A novel PSSM-based software for predicting class I peptide-HLA binding affinity. GigaScience 2017, 6, gix017. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Systems 2018, 7, 129–132.e4. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Cheng, L.; Zang, T.; Hu, Y. Peptide-Major Histocompatibility Complex Class I Binding Prediction Based on Deep Learning With Novel Feature. Front. Genet. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Lata, S.; Bhasin, M.; Raghava, G.P.S. MHCBN 4.0: A database of MHC/TAP binding peptides and T-cell epitopes. BMC Res. Notes 2009, 2, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammensee, H.G.; Bachmann, J.; Emmerich, N.P.N.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J. Contrast Coding in Multiple Regression Analysis: Strengths, Weaknesses, and Utility of Popular Coding Structures. J. Data Sci. 2010, 8, 61–73. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Paco, L.; Romero, I.; Berruguilla, E.; Stefansky, J.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis 2012, 33, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Sun, Z.; Chen, F.; Meng, F.; Wei, J.; Liu, B. MHC class II restricted neoantigen: A promising target in tumor immunotherapy. Cancer Lett. 2017, 392, 17–25. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Length | Accuracy | AUC | F1 | MCC | Specificity | Sensitivity | Precision | AUPR | Positive 1 | Negative 2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Class I | 8 mer | 0.891 | 0.924 | 0.783 | 0.531 | 0.887 | 0.677 | 0.525 | 0.785 | 229 | 1879 |
| 9 mer | 0.883 | 0.915 | 0.745 | 0.650 | 0.902 | 0.735 | 0.760 | 0.800 | 23,000 | 72,963 | |
| 10 mer | 0.813 | 0.850 | 0.693 | 0.527 | 0.842 | 0.690 | 0.661 | 0.725 | 7263 | 14,024 | |
| 11 mer | 0.879 | 0.905 | 0.768 | 0.608 | 0.881 | 0.756 | 0.651 | 0.755 | 310 | 1604 | |
| Others | 0.986 | 1.000 | 0.992 | 0.564 | 0.750 | 1.000 | 0.985 | 1.000 | 54 | 803 | |
| Class II | 13 mer | 0.857 | 0.879 | 0.883 | 0.700 | 0.833 | 0.872 | 0.895 | 0.923 | 232 | 205 |
| 14 mer | 0.898 | 0.907 | 0.880 | 0.792 | 0.912 | 0.880 | 0.880 | 0.873 | 131 | 239 | |
| 15 mer | 0.868 | 0.906 | 0.781 | 0.687 | 0.912 | 0.769 | 0.794 | 0.840 | 16,743 | 25,683 | |
| 16 mer | 0.776 | 0.846 | 0.802 | 0.545 | 0.718 | 0.823 | 0.782 | 0.878 | 563 | 569 | |
| 17 mer | 0.680 | 0.673 | 0.429 | 0.312 | 0.933 | 0.300 | 0.750 | 0.643 | 106 | 257 | |
| 18 mer | 0.643 | 0.939 | 0.706 | 0.452 | 1.000 | 0.545 | 1.000 | 0.986 | 71 | 40 | |
| 19 mer | 0.875 | 0.938 | 0.857 | 0.775 | 1.000 | 0.750 | 1.000 | 0.950 | 55 | 75 | |
| 20 mer | 0.750 | 0.900 | 0.500 | 0.488 | 1.000 | 0.333 | 1.000 | 0.886 | 65 | 66 | |
| Other | 0.690 | 0.640 | 0.381 | 0.183 | 0.758 | 0.444 | 0.333 | 0.566 | 81 | 259 |
| Methods | Accuracy | Sensitivity | Specificity | AUC | AUPR | F1 | MCC | Precision | Positive 1 | Negative 2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Class I | BVMHC | 0.597 | 0.371 | 0.959 | 0.887 | 0.866 | 0.531 | 0.374 | 0.936 | 197 | 123 |
| NetMHCcons [16] | 0.600 | 0.386 | 0.943 | 0.865 | 0.890 | 0.543 | 0.365 | 0.916 | 197 | 123 | |
| SMM [17] | 0.584 | 0.350 | 0.959 | 0.859 | 0.891 | 0.509 | 0.357 | 0.932 | 197 | 123 | |
| NetMHCpan [18] | 0.566 | 0.330 | 0.943 | 0.867 | 0.886 | 0.483 | 0.318 | 0.903 | 197 | 123 | |
| ANN [19] | 0.563 | 0.325 | 0.943 | 0.867 | 0.880 | 0.478 | 0.314 | 0.901 | 197 | 123 | |
| PickPocket [20] | 0.563 | 0.345 | 0.911 | 0.813 | 0.833 | 0.493 | 0.289 | 0.861 | 197 | 123 | |
| NetMHCpan EL [24] | 0.553 | 0.335 | 0.902 | 0.816 | 0.856 | 0.480 | 0.269 | 0.846 | 197 | 123 | |
| comblib_sidney2008 [21] | NAN § | NAN § | NAN § | 0.744 | NAN § | NAN § | NAN § | NAN § | 68 | 46 | |
| Class II | BVMHC | 0.878 | 0.333 | 0.965 | 0.718 | 0.417 | 0.429 | 0.386 | 0.600 | 18 | 113 |
| NN-align [15] | 0.863 | 0.278 | 0.956 | 0.866 | 0.484 | 0.357 | 0.303 | 0.500 | 18 | 113 | |
| NETMHCIIPan [23] | 0.870 | 0.111 | 0.991 | 0.795 | 0.423 | 0.190 | 0.235 | 0.667 | 18 | 113 | |
| SMM-align [22] | 0.840 | 0.000 | 0.973 | 0.787 | 0.319 | NA § | −0.061 | 0.000 | 18 | 113 |
| Alleles | Accuracy | AUC | F1 | MCC | Specificity | Sensitivity | Precision | AUPR | |
|---|---|---|---|---|---|---|---|---|---|
| Class I | H-2-Db | 0.829 | 0.855 | 0.573 | 0.466 | 0.897 | 0.564 | 0.583 | 0.602 |
| H-2-Dd | 0.924 | 0.870 | 0.696 | 0.660 | 0.975 | 0.615 | 0.800 | 0.751 | |
| H-2-Ld | 0.814 | 0.852 | 0.698 | 0.564 | 0.875 | 0.682 | 0.714 | 0.779 | |
| Mamu-A07 | 0.905 | 0.949 | 0.854 | 0.783 | 0.929 | 0.854 | 0.854 | 0.902 | |
| Mamu-A11 | 0.822 | 0.899 | 0.726 | 0.595 | 0.880 | 0.707 | 0.747 | 0.805 | |
| Mamu-A2201 | 0.908 | 0.957 | 0.854 | 0.789 | 0.955 | 0.814 | 0.897 | 0.943 | |
| Mamu-B01 | 0.942 | 0.865 | 0.667 | 0.654 | 0.988 | 0.550 | 0.846 | 0.767 | |
| Mamu-B03 | 0.857 | 0.921 | 0.769 | 0.666 | 0.903 | 0.758 | 0.781 | 0.843 | |
| Mamu-B08 | 0.852 | 0.911 | 0.690 | 0.600 | 0.875 | 0.769 | 0.625 | 0.776 | |
| Mamu-B17 | 0.822 | 0.882 | 0.717 | 0.592 | 0.838 | 0.782 | 0.662 | 0.710 | |
| Mamu-B52 | 0.827 | 0.870 | 0.870 | 0.617 | 0.677 | 0.912 | 0.832 | 0.884 | |
| Patr-A0101 | 0.816 | 0.838 | 0.619 | 0.520 | 0.935 | 0.520 | 0.765 | 0.688 | |
| Patr-A0401 | 0.881 | 0.904 | 0.636 | 0.565 | 0.929 | 0.636 | 0.636 | 0.616 | |
| Patr-A0701 | 0.825 | 0.820 | 0.545 | 0.438 | 0.901 | 0.522 | 0.571 | 0.682 | |
| Patr-B0101 | 0.911 | 0.947 | 0.794 | 0.759 | 0.991 | 0.675 | 0.964 | 0.894 | |
| Patr-B1301 | 0.875 | 0.917 | 0.903 | 0.727 | 0.824 | 0.903 | 0.903 | 0.951 | |
| RT1A | 0.893 | 0.923 | 0.400 | 0.352 | 0.923 | 0.500 | 0.333 | 0.667 | |
| Class II | H-2-IAb | 0.826 | 0.797 | 0.489 | 0.394 | 0.925 | 0.423 | 0.579 | 0.627 |
| H-2-IAd | 0.810 | 0.810 | 0.571 | 0.452 | 0.896 | 0.533 | 0.615 | 0.632 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Tang, J.; Guo, F.; Guo, Y. Prediction of Major Histocompatibility Complex Binding with Bilateral and Variable Long Short Term Memory Networks. Biology 2022, 11, 848. https://doi.org/10.3390/biology11060848
Jiang L, Tang J, Guo F, Guo Y. Prediction of Major Histocompatibility Complex Binding with Bilateral and Variable Long Short Term Memory Networks. Biology. 2022; 11(6):848. https://doi.org/10.3390/biology11060848
Chicago/Turabian StyleJiang, Limin, Jijun Tang, Fei Guo, and Yan Guo. 2022. "Prediction of Major Histocompatibility Complex Binding with Bilateral and Variable Long Short Term Memory Networks" Biology 11, no. 6: 848. https://doi.org/10.3390/biology11060848
APA StyleJiang, L., Tang, J., Guo, F., & Guo, Y. (2022). Prediction of Major Histocompatibility Complex Binding with Bilateral and Variable Long Short Term Memory Networks. Biology, 11(6), 848. https://doi.org/10.3390/biology11060848