
Comparative Transcriptome Analysis Reveals Candidate Genes and Pathways for Potential Branch Growth in Elm (Ulmus pumila) Cultivars


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Anatomical Observation
2.2. RNA Extraction
2.3. Illumina Library Construction and Sequencing
2.4. De Novo Assembly of Transcriptome
2.5. Calculation of Gene Expression Levels
2.6. Gene Ontology (GO) and KEGG Annotation and Enrichment
2.7. qRT-PCR Verification
3. Results
3.1. Branches Phenotypic Traits
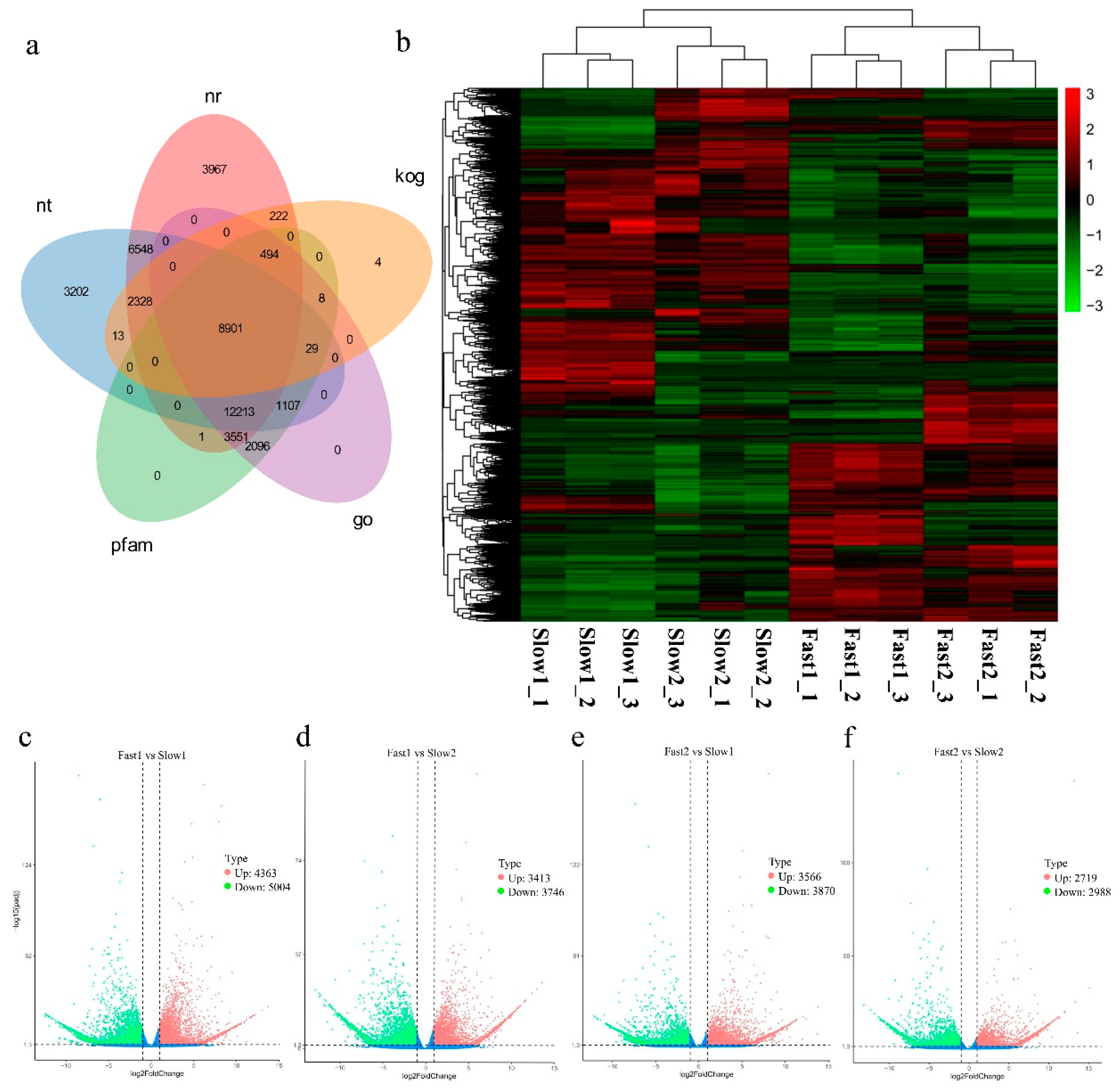
3.2. Transcriptome Profiling of U. pumila
3.3. Functional Annotations of Unigenes
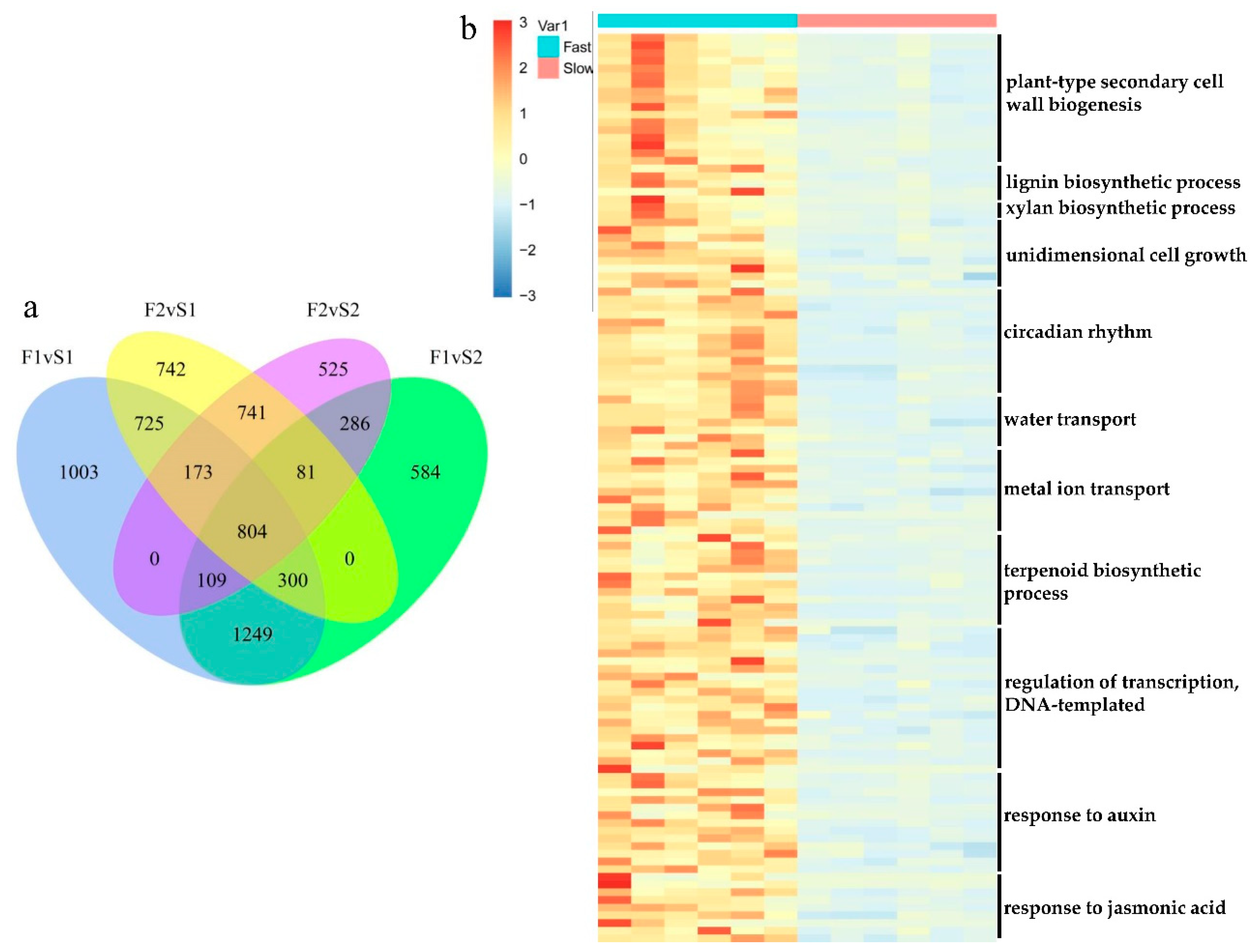
3.4. Differentially Expressed Genes (DEGs) Calculation
3.5. Gene Ontology (GO) and KEGG Enrichment Result of DEGs
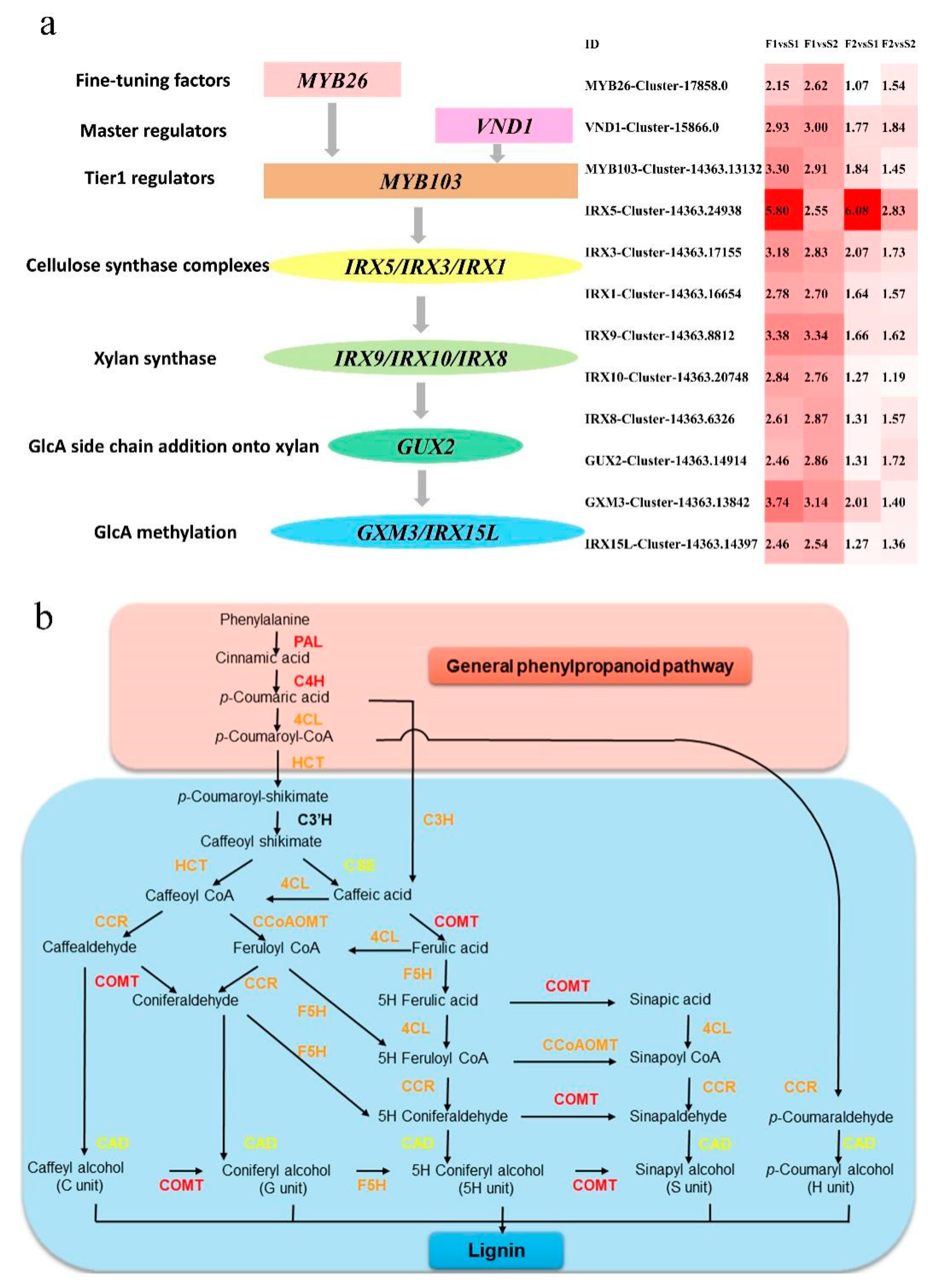
3.6. Gene Expression Pattern and Functional Transition of Fast- and Slow-Growing Genotypes
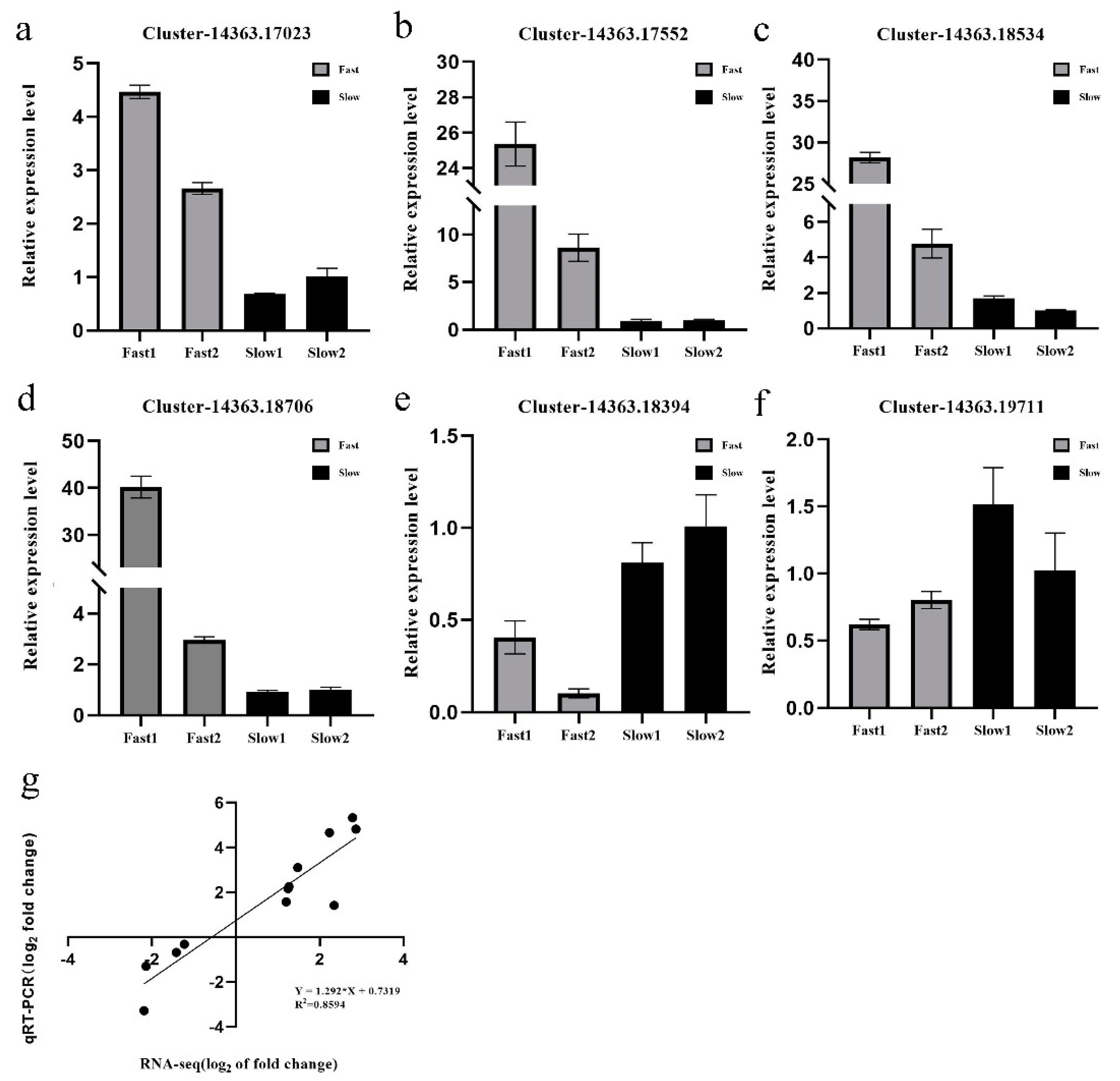
3.7. Real-Time Quantitative PCR Validation
3.8. qRT-PCR Analysis for Secondary Walls and Lignin Biosynthesis-Associated Genes in Different Seasons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fischer, U.; Kucukoglu, M.; Helariutta, Y.; Bhalerao, R.P. The Dynamics of Cambial Stem Cell Activity. Annu. Rev. Plant Biol. 2019, 70, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Escamez, S.; Latha Gandla, M.; Derba-Maceluch, M.; Lundqvist, S.O.; Mellerowicz, E.J.; Jonsson, L.J.; Tuominen, H. A collection of genetically engineered Populus trees reveals wood biomass traits that predict glucose yield from enzymatic hydrolysis. Sci. Rep. 2017, 7, 15798. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; McCarthy, R.L.; Haghighat, M.; Ye, Z.H. The poplar MYB master switches bind to the SMRE site and activate the secondary wall biosynthetic program during wood formation. PLoS ONE 2013, 8, e69219. [Google Scholar] [CrossRef] [PubMed]
- Demura, T.; Ye, Z.H. Regulation of plant biomass production. Curr. Opin. Plant Biol. 2010, 13, 299–304. [Google Scholar] [CrossRef]
- De Rybel, B.; Adibi, M.; Breda, A.S.; Wendrich, J.R.; Smit, M.E.; Novak, O.; Yamaguchi, N.; Yoshida, S.; Van Isterdael, G.; Palovaara, J.; et al. Plant development. Integration of growth and patterning during vascular tissue formation in Arabidopsis. Science 2014, 345, 1255215. [Google Scholar] [CrossRef]
- Yang, J.H.; Wang, H. Molecular Mechanisms for Vascular Development and Secondary Cell Wall Formation. Front. Plant Sci. 2016, 7, 356. [Google Scholar] [CrossRef]
- Ye, Z.H.; Freshour, G.; Hahn, M.G.; Burk, D.H.; Zhong, R. Vascular development in Arabidopsis. Int. Rev. Cytol. 2002, 220, 225–256. [Google Scholar]
- Agusti, J.; Blazquez, M.A. Plant vascular development: Mechanisms and environmental regulation. Cell. Mol. Life Sci. 2020, 77, 3711–3728. [Google Scholar] [CrossRef]
- Li, W.F.; Ding, Q.; Chen, J.J.; Cui, K.M.; He, X.Q. Induction of PtoCDKB and PtoCYCB transcription by temperature during cambium reactivation in Populus tomentosa Carr. J. Exp. Bot. 2009, 60, 2621–2630. [Google Scholar] [CrossRef]
- Kim, H.; Zhou, J.; Kumar, D.; Jang, G.; Ryu, K.H.; Sebastian, J.; Miyashima, S.; Helariutta, Y.; Lee, J.Y. SHORTROOT-Mediated Intercellular Signals Coordinate Phloem Development in Arabidopsis Roots. Plant Cell 2020, 32, 1519–1535. [Google Scholar] [CrossRef]
- Zhong, R.; Ye, Z.H. Secondary cell walls: Biosynthesis, patterned deposition and transcriptional regulation. Plant Cell Physiol. 2015, 56, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Wang, L.Y.; Immanen, J.; Nieminen, K.; Spicer, R.; Helariutta, Y.; Zhang, J.; He, X.Q. Differential regulation of auxin and cytokinin during the secondary vascular tissue regeneration in Populus trees. New Phytol. 2019, 224, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Endler, A.; Persson, S. Cellulose synthases and synthesis in Arabidopsis. Mol. Plant 2011, 4, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.R.; Somerville, C.R. Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant Cell 1997, 9, 689–701. [Google Scholar]
- Taylor, N.G.; Scheible, W.R.; Cutler, S.; Somerville, C.R.; Turner, S.R. The irregular xylem3 locus of Arabidopsis encodes a cellulose synthase required for secondary cell wall synthesis. Plant Cell 1999, 11, 769–780. [Google Scholar] [CrossRef]
- Taylor, N.G.; Laurie, S.; Turner, S.R. Multiple cellulose synthase catalytic subunits are required for cellulose synthesis in Arabidopsis. Plant Cell 2000, 12, 2529–2540. [Google Scholar] [CrossRef]
- Taylor, N.G.; Howells, R.M.; Huttly, A.K.; Vickers, K.; Turner, S.R. Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1450–1455. [Google Scholar] [CrossRef]
- Brown, D.M.; Zhang, Z.; Stephens, E.; Dupree, P.; Turner, S.R. Characterization of IRX10 and IRX10-like reveals an essential role in glucuronoxylan biosynthesis in Arabidopsis. Plant J. 2009, 57, 732–746. [Google Scholar] [CrossRef]
- Lee, C.; Teng, Q.; Huang, W.; Zhong, R.; Ye, Z.H. The Arabidopsis family GT43 glycosyltransferases form two functionally nonredundant groups essential for the elongation of glucuronoxylan backbone. Plant Physiol. 2010, 153, 526–541. [Google Scholar] [CrossRef]
- Wu, A.M.; Rihouey, C.; Seveno, M.; Hornblad, E.; Singh, S.K.; Matsunaga, T.; Ishii, T.; Lerouge, P.; Marchant, A. The Arabidopsis IRX10 and IRX10-LIKE glycosyltransferases are critical for glucuronoxylan biosynthesis during secondary cell wall formation. Plant J. 2009, 57, 718–731. [Google Scholar] [CrossRef]
- Ratke, C.; Terebieniec, B.K.; Winestrand, S.; Derba-Maceluch, M.; Grahn, T.; Schiffthaler, B.; Ulvcrona, T.; Ozparpucu, M.; Ruggeberg, M.; Lundqvist, S.O.; et al. Downregulating aspen xylan biosynthetic GT43 genes in developing wood stimulates growth via reprograming of the transcriptome. New Phytol. 2018, 219, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, K.; Immanen, J.; Laxell, M.; Kauppinen, L.; Tarkowski, P.; Dolezal, K.; Tahtiharju, S.; Elo, A.; Decourteix, M.; Ljung, K.; et al. Cytokinin signaling regulates cambial development in poplar. Proc. Natl. Acad. Sci. USA 2008, 105, 20032–20037. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, T.; Bolu-Arianto, W.H.; Olbrich, A.; Langenfeld-Heyser, R.; Gobel, C.; Grzeganek, P.; Feussner, I.; Hansch, R.; Polle, A. GH3::GUS reflects cell-specific developmental patterns and stress-induced changes in wood anatomy in the poplar stem. Tree Physiol. 2008, 28, 1305–1315. [Google Scholar] [CrossRef]
- Immanen, J.; Nieminen, K.; Smolander, O.P.; Kojima, M.; Alonso Serra, J.; Koskinen, P.; Zhang, J.; Elo, A.; Mahonen, A.P.; Street, N.; et al. Cytokinin and Auxin Display Distinct but Interconnected Distribution and Signaling Profiles to Stimulate Cambial Activity. Curr. Biol. 2016, 26, 1990–1997. [Google Scholar] [CrossRef]
- Uggla, C.; Mellerowicz, E.J.; Sundberg, B. Indole-3-acetic acid controls cambial growth in scots pine by positional signaling. Plant Physiol. 1998, 117, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Garces, M.; Le Provost, G.; Lalanne, C.; Claverol, S.; Barre, A.; Plomion, C.; Herrera, R. Proteomic analysis during ontogenesis of secondary xylem in maritime pine. Tree Physiol. 2014, 34, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, H.X.; Southerton, S.G. Identification of putative candidate genes for juvenile wood density in Pinus radiata. Tree Physiol. 2012, 32, 1046–1057. [Google Scholar] [CrossRef]
- Wang, H.Z.; Dixon, R.A. On-off switches for secondary cell wall biosynthesis. Mol. Plant 2012, 5, 297–303. [Google Scholar] [CrossRef]
- Zhong, R.; Ye, Z.H. MYB46 and MYB83 bind to the SMRE sites and directly activate a suite of transcription factors and secondary wall biosynthetic genes. Plant Cell Physiol. 2012, 53, 368–380. [Google Scholar] [CrossRef]
- Zhong, R.; Demura, T.; Ye, Z.H. SND1, a NAC domain transcription factor, is a key regulator of secondary wall synthesis in fibers of Arabidopsis. Plant Cell 2006, 18, 3158–3170. [Google Scholar] [CrossRef]
- Zhou, J.; Zhong, R.; Ye, Z.H. Arabidopsis NAC domain proteins, VND1 to VND5, are transcriptional regulators of secondary wall biosynthesis in vessels. PLoS ONE 2014, 9, e105726. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, N.; Iwase, A.; Yamamoto, H.; Yoshida, M.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. NAC transcription factors, NST1 and NST3, are key regulators of the formation of secondary walls in woody tissues of Arabidopsis. Plant Cell 2007, 19, 270–280. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.L.; Zhong, R.; Ye, Z.H. Secondary wall NAC binding element (SNBE), a key cis-acting element required for target gene activation by secondary wall NAC master switches. Plant Signal. Behav. 2011, 6, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.-Q.; Lin, H.-X. Contribution of phenylpropanoid metabolism to plant development and plant–environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Spear, M.J.; Broda, M. Comparison of Contemporary Elm (Ulmus spp.) and Degraded Archaeological Elm: The Use of Dynamic Mechanical Analysis Under Ambient Moisture Conditions. Materials 2020, 13, 5026. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Li, M.; Wang, N.; Qu, X.; Fan, S. Transcriptome Analysis of Elm (Ulmus pumila) Fruit to Identify Phytonutrients Associated Genes and Pathways. Forests 2019, 10, 738. [Google Scholar] [CrossRef]
- Wiegrefe, S.J.; Sytsma, K.J.; Guries, R.P. Phylogeny of elms (Ulmus, Ulmaceae): Molecular evidence for a sectional classification. Syst. Bot. 1994, 19, 590–612. [Google Scholar] [CrossRef]
- Solla, A.; Martin, J.A.; Corral, P.; Gil, L. Seasonal changes in wood formation of Ulmus pumila and U. minor and its relation with Dutch elm disease. New Phytol. 2005, 166, 1025–1034. [Google Scholar] [CrossRef]
- Chen, P.; Liu, P.; Zhang, Q.; Bu, C.; Lu, C.; Srivastava, S.; Zhang, D.; Song, Y. Gene Coexpression Network Analysis Indicates that Hub Genes Related to Photosynthesis and Starch Synthesis Modulate Salt Stress Tolerance in Ulmus pumila. Int. J. Mol. Sci. 2021, 22, 4410. [Google Scholar] [CrossRef]
- Wang, C.-S.; Tang, C.; Hein, S.; Guo, J.-J.; Zhao, Z.-G.; Zeng, J. Branch Development of Five-Year-Old Betula alnoides Plantations in Response to Planting Density. Forests 2018, 9, 42. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Lee, C.; Zhou, J.; McCarthy, R.L.; Ye, Z.H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell 2008, 20, 2763–2782. [Google Scholar] [CrossRef]
- Kubo, M.; Udagawa, M.; Nishikubo, N.; Horiguchi, G.; Yamaguchi, M.; Ito, J.; Mimura, T.; Fukuda, H.; Demura, T. Transcription switches for protoxylem and metaxylem vessel formation. Genes Dev. 2005, 19, 1855–1860. [Google Scholar] [CrossRef]
- Yang, C.; Song, J.; Ferguson, A.C.; Klisch, D.; Simpson, K.; Mo, R.; Taylor, B.; Mitsuda, N.; Wilson, Z.A. Transcription Factor MYB26 Is Key to Spatial Specificity in Anther Secondary Thickening Formation. Plant Physiol. 2017, 175, 333–350. [Google Scholar] [CrossRef]
- Ko, J.H.; Kim, W.C.; Han, K.H. Ectopic expression of MYB46 identifies transcriptional regulatory genes involved in secondary wall biosynthesis in Arabidopsis. Plant J. 2009, 60, 649–665. [Google Scholar] [CrossRef]
- Newman, R.H.; Hill, S.J.; Harris, P.J. Wide-angle x-ray scattering and solid-state nuclear magnetic resonance data combined to test models for cellulose microfibrils in mung bean cell walls. Plant Physiol. 2013, 163, 1558–1567. [Google Scholar] [CrossRef]
- Pear, J.R.; Kawagoe, Y.; Schreckengost, W.E.; Delmer, D.P.; Stalker, D.M. Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 12637–12642. [Google Scholar] [CrossRef]
- Simmons, T.J.; Mortimer, J.C.; Bernardinelli, O.D.; Poppler, A.C.; Brown, S.P.; de Azevedo, E.R.; Dupree, R.; Dupree, P. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat. Commun. 2016, 7, 13902. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book 2011, 9, e0152. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.; Besseau, S.; Geoffroy, P.; Ritzenthaler, C.; Meyer, D.; Lapierre, C.; Pollet, B.; Legrand, M. Silencing of hydroxycinnamoyl-coenzyme A shikimate/quinate hydroxycinnamoyltransferase affects phenylpropanoid biosynthesis. Plant Cell 2004, 16, 1446–1465. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Geisler, M.; Springer, P.S. LATERAL ORGAN FUSION1 and LATERAL ORGAN FUSION2 function in lateral organ separation and axillary meristem formation in Arabidopsis. Development 2009, 136, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- He, X.J.; Mu, R.L.; Cao, W.H.; Zhang, Z.G.; Zhang, J.S.; Chen, S.Y. AtNAC2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J. 2005, 44, 903–916. [Google Scholar] [CrossRef]
- Scholten, T.; Goebes, P.; Kühn, P.; Seitz, S.; Assmann, T.; Bauhus, J.; Bruelheide, H.; Buscot, F.; Erfmeier, A.; Fischer, M.; et al. On the combined effect of soil fertility and topography on tree growth in subtropical forest ecosystems—A study from SE China. J. Plant Ecol. 2017, 10, 111–127. [Google Scholar] [CrossRef]
- Kwasna, H.; Szewczyk, W.; Baranowska, M.; Gallas, E.; Wisniewska, M.; Behnke-Borowczyk, J. Mycobiota Associated with the Vascular Wilt of Poplar. Plants 2021, 10, 892. [Google Scholar] [CrossRef]
- Llado, S.; Lopez-Mondejar, R.; Baldrian, P. Forest Soil Bacteria: Diversity, Involvement in Ecosystem Processes, and Response to Global Change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Total Reads | Total Mapped | Mapping Ratio |
|---|---|---|---|
| Fast1_1 | 49,014,302 | 39,324,430 | 80.23% |
| Fast1_2 | 41,483,608 | 33,367,760 | 80.44% |
| Fast1_3 | 41,955,680 | 33,679,770 | 80.27% |
| Fast2_1 | 41,945,930 | 33,784,144 | 80.54% |
| Fast2_2 | 40,140,204 | 32,087,136 | 79.94% |
| Fast2_3 | 42,014,884 | 33,631,348 | 80.05% |
| Slow1_1 | 44,732,848 | 35,697,784 | 79.80% |
| Slow1_2 | 42,697,528 | 34,428,142 | 80.63% |
| Slow1_3 | 40,034,844 | 32,328,242 | 80.75% |
| Slow2_1 | 42,918,790 | 34,843,800 | 81.19% |
| Slow2_2 | 41,464,616 | 33,715,910 | 81.31% |
| Slow2_3 | 41,702,398 | 34,017,546 | 81.57% |
| KEGG Pathway | F1VS1 | F1VS2 | F2VS1 | F2VS2 |
|---|---|---|---|---|
| Phenylpropanoid biosynthesis | 1.27 × 10−11 | 9.28 × 10−9 | 6.83 × 10−9 | 7.54 × 10−4 |
| Flavonoid biosynthesis | 6.02 × 10−10 | 3.52 × 10−11 | 2.25 × 10−3 | 1.73 × 10−4 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | 2.83 × 10−6 | 6.41 × 10−6 | 2.46 × 10−2 | - |
| Photosynthesis-antenna proteins | 4.91 × 10−4 | - | 4.42 × 10−6 | 2.01 × 10−4 |
| DNA replication | 6.62 × 10−4 | 5.46 × 10−5 | 1.16 × 10−3 | 3.95 × 10−3 |
| Starch and sucrose metabolism | 2.47 × 10−3 | - | 4.54 × 10−3 | |
| Diterpenoid biosynthesis | 2.89 × 10−3 | 2.62 × 10−2 | 1.85 × 10−4 | 1.10 × 10−2 |
| Linoleic acid metabolism | 3.87 × 10−3 | 1.97 × 10−3 | 3.21 × 10−2 | 2.82 × 10−2 |
| Plant hormone signal transduction | 4.89 × 10−3 | 3.52 × 10−5 | - | |
| Phenylalanine metabolism | 4.90 × 10−3 | 7.28 × 10−5 | 3.42 × 10−3 | - |
| Process | Speculative Function | Unigene ID | F1vS1 | F1vS2 | F2vS1 | F2vS2 |
|---|---|---|---|---|---|---|
| plant-type secondary cell wall biogenesis | cellulose synthase | Cluster-14363.16654 | 2.781 | 2.704 | 1.643 | 1.570 |
| Cluster-14363.17155 | 3.181 | 2.834 | 2.070 | 1.726 | ||
| Cluster-14363.24938 | 5.799 | 2.547 | 6.081 | 2.832 | ||
| Exostosin | Cluster-14363.20748 | 2.841 | 2.757 | 1.268 | 1.185 | |
| fasciclin-like arabinogalactan protein | Cluster-14363.18562 | 2.603 | 3.123 | 1.765 | 2.289 | |
| Cluster-14363.14849 | 2.962 | 3.642 | 1.753 | 2.433 | ||
| Cluster-14363.25117 | 2.656 | 2.678 | 1.524 | 1.547 | ||
| Cluster-14363.6739 | 4.230 | 4.432 | 2.344 | 2.552 | ||
| glucuronoxylan 4-O-methyltransferase | Cluster-14363.13842 | 3.744 | 3.140 | 2.008 | 1.404 | |
| glucuronyltransferase | Cluster-14363.14914 | 2.455 | 2.865 | 1.309 | 1.720 | |
| glycosyl transferase | Cluster-14363.8812 | 3.382 | 3.340 | 1.658 | 1.617 | |
| laccase | Cluster-14363.9138 | 3.477 | 3.514 | 1.736 | 1.774 | |
| MYB transcription factor | Cluster-14363.13132 | 3.301 | 2.909 | 1.843 | 1.452 | |
| Cluster-17858.0 | 2.152 | 2.620 | 1.069 | 1.538 | ||
| NAC transcription factor | Cluster-15866.0 | 2.925 | 2.997 | 1.766 | 1.845 | |
| phytochelatin synthetase | Cluster-14363.17226 | 3.718 | 3.903 | 3.050 | 3.245 | |
| xylan synthesis | Cluster-14363.14397 | 2.455 | 2.537 | 1.274 | 1.359 | |
| lignin biosynthetic process | chitinase | Cluster-14363.19471 | 3.163 | 2.963 | 1.626 | 1.427 |
| interfascicular fibers synthesis | Cluster-14363.13437 | 2.402 | 3.274 | 2.872 | 3.745 | |
| laccase | Cluster-14363.7596 | 4.162 | 3.920 | 2.297 | 2.055 | |
| O-methyltransferase | Cluster-14363.16741 | 3.258 | 2.758 | 2.537 | 2.039 | |
| xylan biosynthetic process | galacturonosyltransferase | Cluster-14363.6326 | 2.609 | 2.866 | 1.306 | 1.566 |
| synthesis and deposition of secondary wall cellulose | Cluster-14363.15500 | 3.517 | 3.539 | 2.184 | 2.206 | |
| unidimensional cell growth | actin monomer-binding protein | Cluster-14363.20463 | 1.884 | 2.078 | 1.727 | 1.920 |
| Alpha-Expansin | Cluster-14363.17330 | 4.839 | 2.400 | 4.704 | 2.264 | |
| cell differentiation | Cluster-14363.13311 | 2.983 | 2.661 | 1.934 | 1.618 | |
| expansin A6 | Cluster-14363.28593 | 1.493 | 1.715 | 1.113 | 1.336 | |
| Immunoglobulin E-set | Cluster-14363.18373 | 4.244 | 2.924 | 2.466 | 1.144 | |
| key turgor pressure regulator in plant cells | Cluster-14363.35635 | 1.745 | 1.599 | 1.174 | 1.027 | |
| plasma-membrane associated cation-binding protein | Cluster-14363.13070 | 2.431 | 2.956 | 1.436 | 1.961 | |
| water transport | ABC transporter | Cluster-14363.32034 | 1.416 | 2.427 | 1.937 | 2.949 |
| plasma membrane intrinsic protein | Cluster-14363.15480 | 1.787 | 1.351 | 1.987 | 1.553 | |
| Cluster-14363.16016 | 2.476 | 2.960 | 2.677 | 3.161 | ||
| Cluster-14363.19969 | 2.378 | 2.143 | 2.477 | 2.242 | ||
| Cluster-14363.20258 | 1.136 | 1.286 | 1.258 | 1.410 | ||
| water and urea channels | Cluster-14363.8769 | 4.830 | 3.982 | 4.473 | 3.620 | |
| regulation of transcription, DNA-templated | Auxin inducible protein | Cluster-13847.0 | 1.559 | 1.218 | 1.797 | 1.455 |
| Auxin-responsive gene | Cluster-14363.35167 | 1.811 | 1.168 | 2.146 | 1.509 | |
| B-box type zinc finger protein | Cluster-14363.31847 | 3.759 | 2.030 | 3.805 | 2.075 | |
| bHLH DNA-binding protein | Cluster-14363.10371 | 3.195 | 2.068 | 3.374 | 2.250 | |
| Cluster-14363.388 | 1.248 | 2.707 | 1.614 | 3.076 | ||
| C2H2 transcription factor | Cluster-14363.17522 | 2.245 | 1.967 | 1.666 | 1.391 | |
| Cluster-14363.19420 | 2.797 | 3.476 | 2.111 | 2.800 | ||
| Cluster-14363.25933 | 1.883 | 1.732 | 1.247 | 1.098 | ||
| Dof transcription factor | Cluster-14363.5357 | 1.185 | 1.278 | 1.471 | 1.570 | |
| multiprotein bridging factor | Cluster-14363.21135 | 1.831 | 2.109 | 2.962 | 3.240 | |
| MYB transcription factor | Cluster-14363.31703 | 5.646 | 3.215 | 5.811 | 3.389 | |
| Cluster-14363.50 | 2.631 | 2.067 | 2.778 | 2.216 | ||
| Cluster-16574.0 | 5.926 | 3.140 | 6.629 | 3.849 | ||
| Cluster-17031.0 | 1.934 | 1.959 | 1.801 | 1.827 | ||
| Cluster-17693.0 | 5.932 | 5.833 | 5.440 | 5.353 | ||
| zinc knuckle protein | Cluster-14363.23170 | 1.345 | 1.180 | 1.644 | 1.483 | |
| response to auxin | Auxin efflux carrier protein | Cluster-14363.23375 | 2.303 | 1.763 | 2.336 | 1.793 |
| Cluster-14363.23376 | 1.570 | 1.270 | 1.796 | 1.501 | ||
| modulator of auxin signaling | Cluster-14363.24574 | 1.570 | 2.781 | 1.064 | 2.279 | |
| regulates vesicle trafficking | Cluster-14363.13767 | 1.350 | 1.129 | 2.224 | 2.006 | |
| regulator of cellular auxin efflux | Cluster-14363.4717 | 2.327 | 1.505 | 2.275 | 1.457 | |
| response to jasmonic acid | controls the balance between salicylic acid and jasmonic acid signaling | Cluster-14363.16305 | 2.509 | 2.540 | 1.252 | 1.294 |
| NAC transcription factor | Cluster-18724.0 | 2.013 | 1.488 | 2.351 | 1.834 | |
| required for wound-induced jasmonic acid accumulation | Cluster-14363.17181 | 2.753 | 2.264 | 2.969 | 2.478 | |
| response to abscisisc acid and jasmonic acid | Cluster-16707.0 | 7.905 | 7.813 | 4.921 | 4.835 | |
| vacuole formation | Cluster-14363.19730 | 3.595 | 3.708 | 2.602 | 2.710 | |
| wound- and methyl jasmonate-induced secondary metabolism | Cluster-14363.28007 | 2.998 | 1.359 | 2.978 | 1.338 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Xie, S.; Yang, C.; Cao, D.; Fan, S.; Zhang, X. Comparative Transcriptome Analysis Reveals Candidate Genes and Pathways for Potential Branch Growth in Elm (Ulmus pumila) Cultivars. Biology 2022, 11, 711. https://doi.org/10.3390/biology11050711
Zhang L, Xie S, Yang C, Cao D, Fan S, Zhang X. Comparative Transcriptome Analysis Reveals Candidate Genes and Pathways for Potential Branch Growth in Elm (Ulmus pumila) Cultivars. Biology. 2022; 11(5):711. https://doi.org/10.3390/biology11050711
Chicago/Turabian StyleZhang, Luoyan, Shaoqiu Xie, Cheng Yang, Dongling Cao, Shoujin Fan, and Xuejie Zhang. 2022. "Comparative Transcriptome Analysis Reveals Candidate Genes and Pathways for Potential Branch Growth in Elm (Ulmus pumila) Cultivars" Biology 11, no. 5: 711. https://doi.org/10.3390/biology11050711
APA StyleZhang, L., Xie, S., Yang, C., Cao, D., Fan, S., & Zhang, X. (2022). Comparative Transcriptome Analysis Reveals Candidate Genes and Pathways for Potential Branch Growth in Elm (Ulmus pumila) Cultivars. Biology, 11(5), 711. https://doi.org/10.3390/biology11050711