Whole Genome Sequencing Contributions and Challenges in Disease Reduction Focused on Malaria

 , ,
, ,
Simple Summary
Abstract
1. Introduction
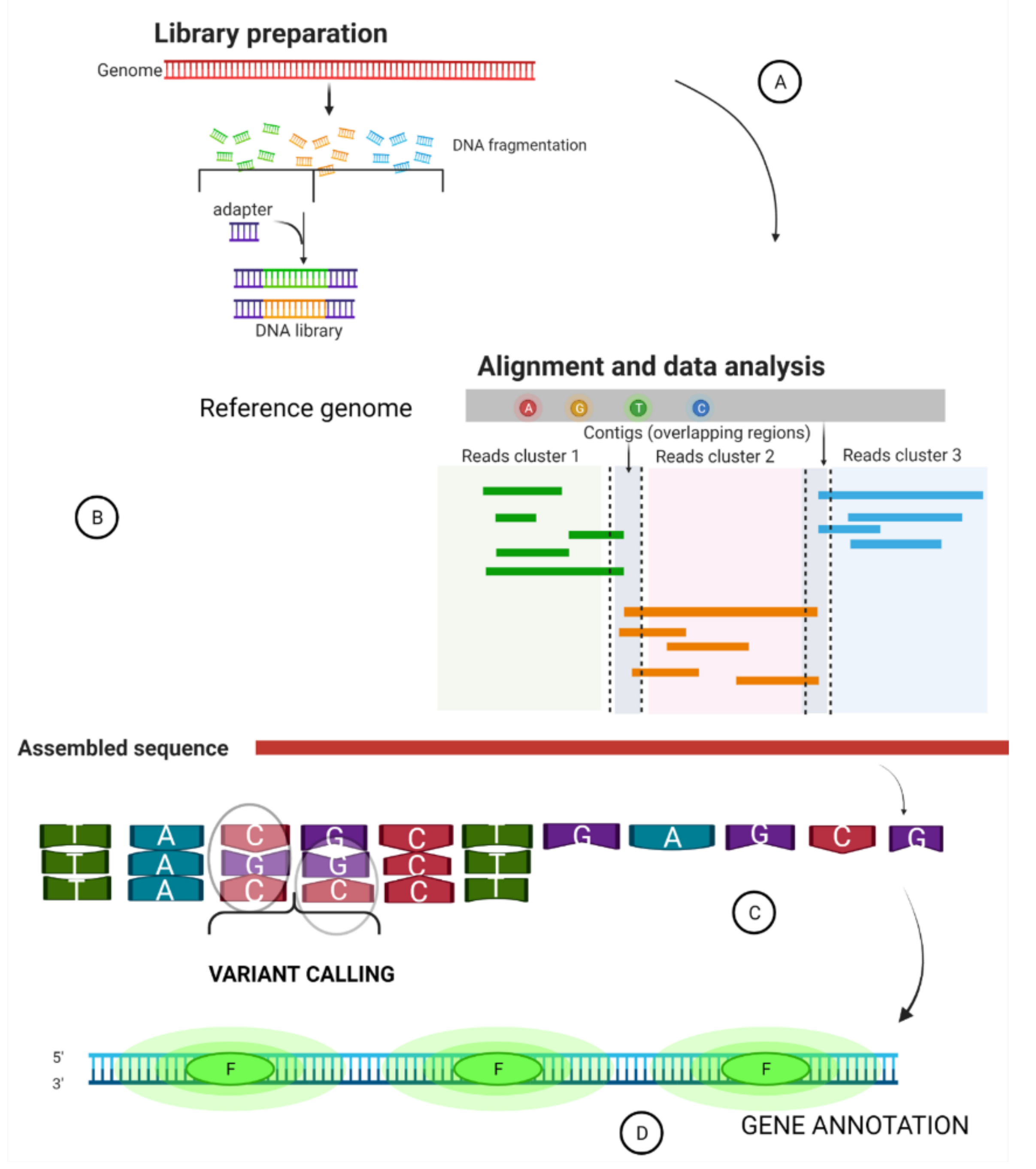
2. Summary of a Whole Genome Sequence Pipeline
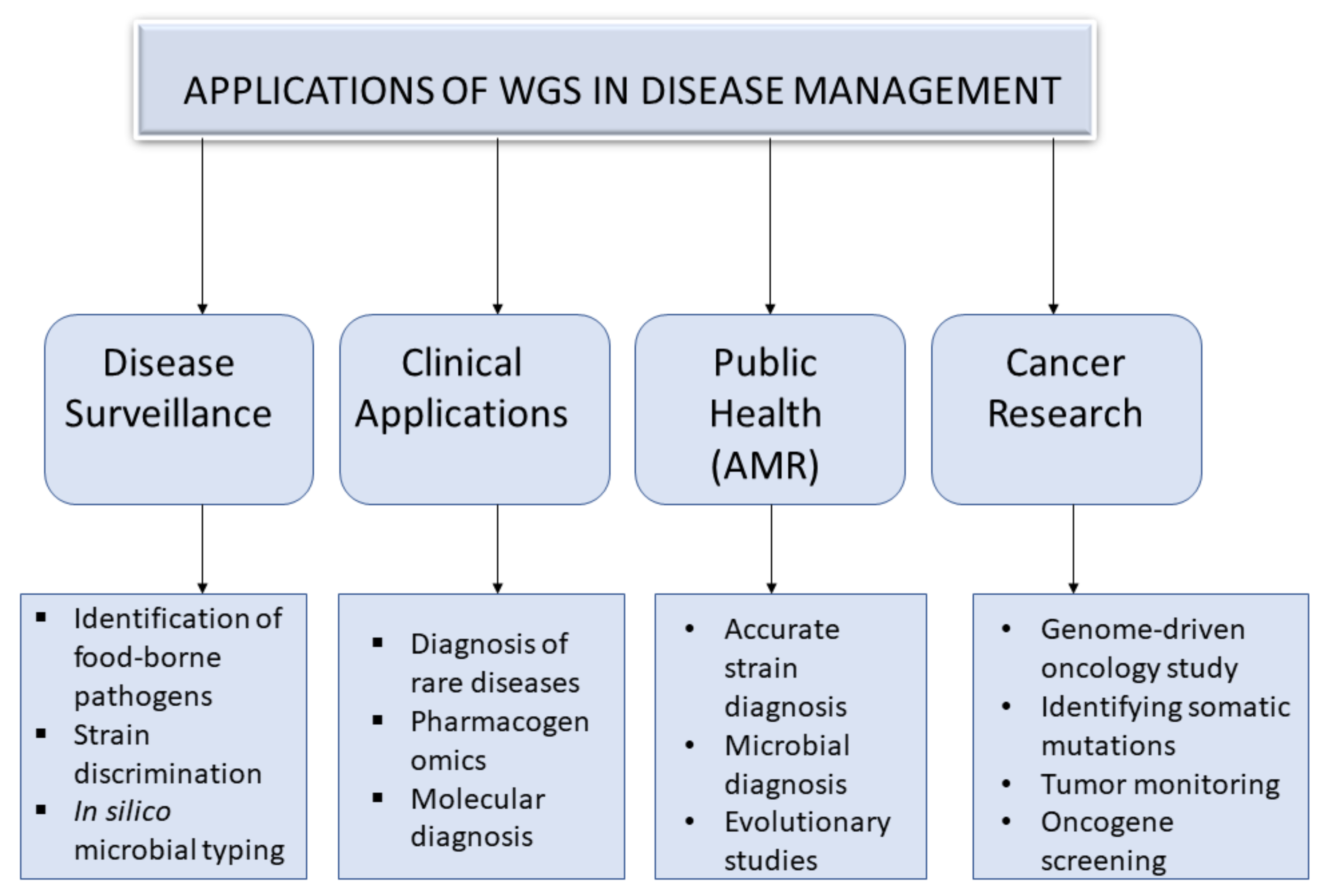
3. An Overview of Applications of Whole Genome Sequencing in Disease Management
3.1. Disease Surveillance
3.2. Public Health
3.2.1. Strain Diagnosis in Antimicrobial Resistance
3.2.2. Microbial Diagnosis and Evolutionary Studies
4. Different Whole Genome Sequencing Platforms
4.1. Roche 454 Pyrosequencing
4.2. Illumina (Solexa)
4.3. Sequencing by Oligonucleotide Ligation and Detection (SOLiD)
4.4. BGI Retrovelocity
4.5. The Ion Torrent
4.6. Single-Molecule Real-Time (SMRT) Sequencing by Pacific Biosciences
4.7. Helicos Sequencing by the Genetic Analysis System
4.8. Nanopore Sequencing by Oxford Nanopore Technologies (MinION and PromethION)
5. Sequencing of Plasmodium falciparum 3D7 Genome
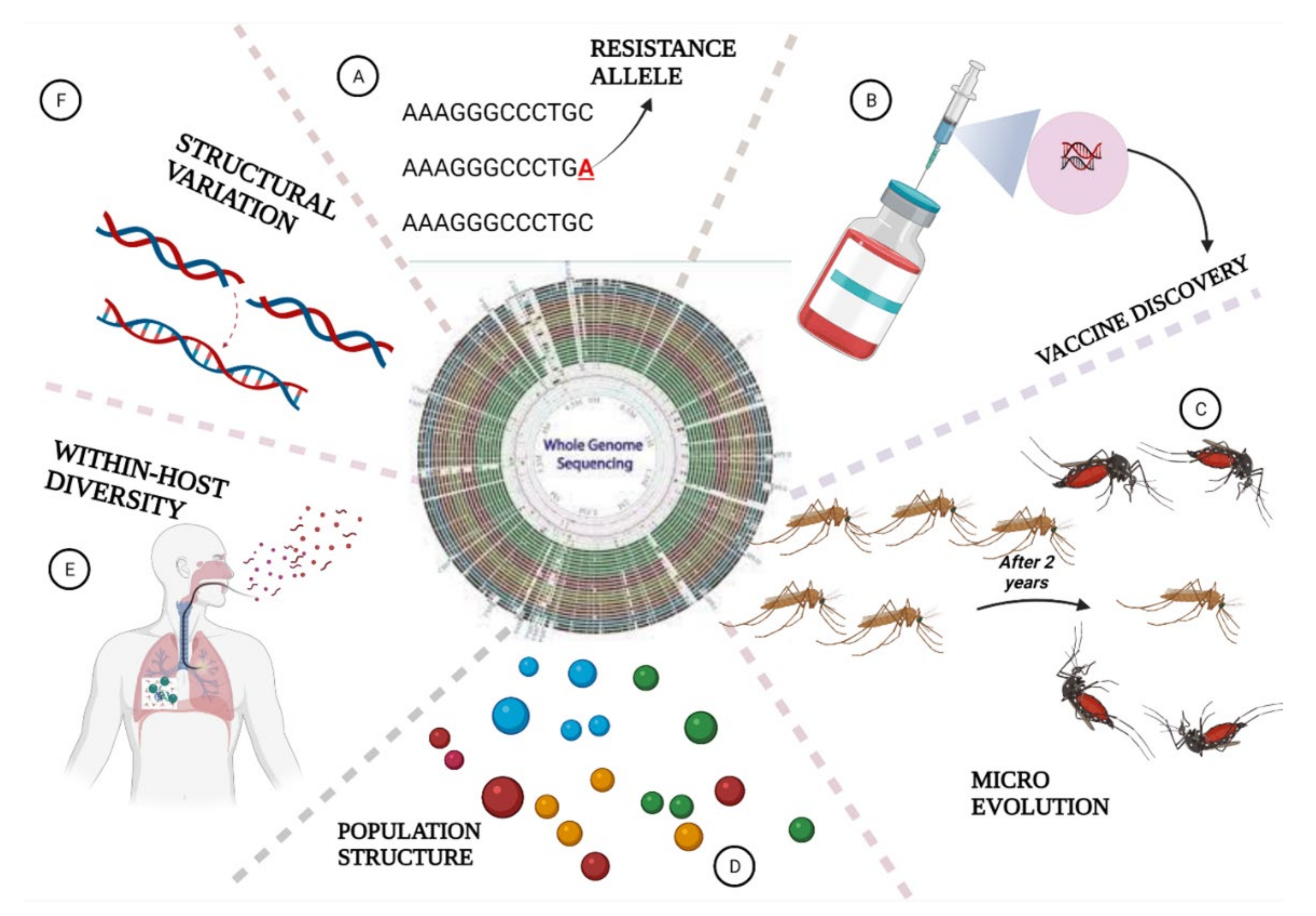
6. The Role of Whole Genome Sequencing in Malaria Elimination
6.1. Parasites Population Structure and Geographic Origin
6.2. Identification of Structural Variants in Plasmodium Species
6.3. Measuring Within-Host Genetic Diversity
6.4. Detection of Parasites Evolution
6.5. WGS Helps in Drug Resistance Surveillance
6.6. Application of WGS in Vaccine Discovery
6.7. WGS Deciphers Malaria Pathogens in Mosquitoes
7. Challenges of Plasmodium Whole Genome Sequence
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rénia, L.; Goh, Y.S. Malaria parasites: The great escape. Front. Immunol. 2016, 7, 463. [Google Scholar] [CrossRef] [PubMed]
- Cowman, A.F.; Healer, J.; Marapana, D.; Marsh, K. Malaria: Biology and disease. Cell 2016, 167, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Sato, S. Plasmodium—A brief introduction to the parasites causing human malaria and their basic biology. J. Physiol. Anthropol. 2021, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zareen, S.; Rehman, H.U.; Gul, N.; Zareen, H.; Hisham, M.; Rehman, M.U.; Bibi, S.; Bakht, A.; Khan, J.; Saeed, K.; et al. Malaria is still a life threatening disease review. J. Entomol. Zool. Stud. 2016, 105, 105–112. [Google Scholar]
- Ye, R.; Tian, Y.; Huang, Y.; Zhang, Y.; Wang, J.; Sun, X.; Zhou, H.; Zhang, D.; Pan, W. Genome-wide analysis of genetic diversity in plasmodium falciparum isolates from China–Myanmar border. Front. Genet. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Laporta, G.Z.; Linton, Y.M.; Wilkerson, R.C.; Bergo, E.S.; Nagaki, S.S.; Sant’Ana, D.C.; Sallum, M.A.M. Malaria vectors in South America: Current and future scenarios. Parasites Vectors 2015, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sanei-Dehkordi, A.; Soleimani-Ahmadi, M.; Jaberhashemi, S.A.; Zare, M. Species composition, seasonal abundance and distribution of potential anopheline vectors in a malaria endemic area of Iran: Field assessment for malaria elimination. Malar. J. 2019, 18, 1–9. [Google Scholar] [CrossRef]
- Guglielmi, G. Malaria cases are falling worldwide. Nature 2019, 1–4. [Google Scholar] [CrossRef]
- Nsanzabana, C. Strengthening surveillance systems for malaria elimination by integrating molecular and genomic data. Trop. Med. Infect. Dis. 2019, 4, 139. [Google Scholar] [CrossRef]
- Gatton, M.L.; Martin, L.B.; Cheng, Q. Evolution of resistance to sulfadoxine-pyrimethamine in plasmodium falciparum. Antimicrob. Agents Chemother. 2004, 48, 2116–2123. [Google Scholar] [CrossRef]
- Fagbemi, K.A.; Adebusuyi, S.A.; Nderu, D.; Adedokun, S.A.; Pallerla, S.R.; Amoo, A.O.J.; Thomas, B.N.; Velavan, T.P.; Ojurongbe, O. Analysis of sulphadoxine–Pyrimethamine resistance-associated mutations in plasmodium falciparum isolates obtained from asymptomatic pregnant women in Ogun State, Southwest Nigeria. Infect. Genet. Evol. 2020, 85, 104503. [Google Scholar] [CrossRef]
- Juge, N.; Moriyama, S.; Miyaji, T.; Kawakami, M.; Iwai, H.; Fukui, T.; Nelson, N.; Omote, H.; Moriyama, Y. Plasmodium falciparum chloroquine resistance transporter is a H+ -coupled polyspecific nutrient and drug exporter. Proc. Natl. Acad. Sci. USA 2015, 112, 3356–3361. [Google Scholar] [CrossRef]
- Roux, A.T.; Maharaj, L.; Oyegoke, O.; Akoniyon, O.P.; Adeleke, M.A.; Maharaj, R.; Okpeku, M. Chloroquine and sulfadoxine–pyrimethamine resistance in Sub-Saharan Africa—A review. Front. Genet. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Gimenez, A.M.; Marques, R.F.; Regiart, M.; Bargieri, D.Y. Diagnostic methods for non-falciparum malaria. Front. Cell. Infect. Microbiol. 2021, 11, 536. [Google Scholar] [CrossRef]
- Manning, L.; Laman, M.; Rosanas-Urgell, A.; Turlach, B.; Aipit, S.; Bona, C.; Warrell, J.; Siba, P.; Mueller, I.; Davis, T.M.E. Rapid antigen detection tests for malaria diagnosis in severely ill Papua New Guinean children: A comparative study using bayesian latent class models. PLoS ONE 2012, 7, e48701. [Google Scholar] [CrossRef]
- Berzosa, P.; De Lucio, A.; Romay-Barja, M.; Herrador, Z.; González, V.; García, L.; Fernández-Martínez, A.; Santana-Morales, M.; Ncogo, P.; Valladares, B.; et al. Comparison of three diagnostic methods (microscopy, RDT, and PCR) for the detection of malaria parasites in representative samples from Equatorial Guinea 11 medical and health sciences 1108 medical microbiology. Malar. J. 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Krishna, S.; Bharti, P.K.; Chandel, H.S.; Ahmad, A.; Kumar, R.; Singh, P.P.; Singh, M.P.; Singh, N. Detection of mixed infections with plasmodium spp. by PCR, India, 2014. Emerg. Infect. Dis. 2015, 21, 1853. [Google Scholar] [CrossRef]
- Wongsrichanalai, C.; Barcus, M.J.; Muth, S.; Sutamihardja, A.; Wernsdorfer, W.H. A review of malaria diagnostic tools: Microscopy and rapid diagnostic test (RDT). Am. J. Trop. Med. Hyg. 2007, 77, 119–127. [Google Scholar] [CrossRef]
- Tessema, S.K.; Raman, J.; Duffy, C.W.; Ishengoma, D.S.; Amambua-Ngwa, A.; Greenhouse, B. Applying next-generation sequencing to track falciparum malaria in Sub-Saharan Africa. Malar. J. 2019, 18, 1–9. [Google Scholar] [CrossRef]
- Neafsey, D.E.; Volkman, S.K. Malaria genomics in the era of eradication. Cold Spring Harb. Perspect. Med. 2017, 7, a025544. [Google Scholar] [CrossRef]
- Molina-Cruz, A.; Raytselis, N.; Withers, R.; Dwivedi, A.; Crompton, P.D.; Traore, B.; Carpi, G.; Silva, J.C.; Barillas-Mury, C. A genotyping assay to determine geographic origin and transmission potential of plasmodium falciparum malaria cases. Commun. Biol. 2021, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Usman-Yamman, H.; Omalu, C.J.I.; Abubakar, A.; Abolarinwa, S.O.; Eke, S.S.; Otuu, C.A. Genetic diversity of plasmodium falciparum isolates in Minna, North Central Nigeria inferred by PCR genotyping of merozoite surface protein 1 and 2. Infect. Genet. Evol. 2021, 96, 105143. [Google Scholar] [CrossRef] [PubMed]
- Akter, J.; Thriemer, K.; Khan, W.A.; Sullivan, D.J.; Noedl, H.; Haque, R. Genotyping of plasmodium falciparum using antigenic polymorphic markers and to study anti-malarial drug resistance markers in malaria endemic areas of Bangladesh. Malar. J. 2012, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Satta, G.; Lipman, M.; Smith, G.P.; Arnold, C.; Kon, O.M.; McHugh, T.D. Mycobacterium tuberculosis and whole-genome sequencing: How close are we to unleashing its full potential? Clin. Microbiol. Infect. 2018, 24, 604–609. [Google Scholar] [CrossRef]
- Mardis, E.R. New strategies and emerging technologies for massively parallel sequencing: Applications in medical research. Genome Med. 2009, 1, 1–4. [Google Scholar] [CrossRef]
- Winter, D.J.; Pacheco, M.A.; Vallejo, A.F.; Schwartz, R.S.; Arevalo-Herrera, M.; Herrera, S.; Cartwright, R.A.; Escalante, A.A. Whole genome sequencing of field isolates reveals extensive genetic diversity in plasmodium vivax from Colombia. PLoS Negl. Trop. Dis. 2015, 9, e0004252. [Google Scholar] [CrossRef]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-generation sequencing technologies. Cold Spring Harb. Perspect. Med. 2019, 9, a036798. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213–218. [Google Scholar] [CrossRef]
- Portmann, A.C.; Fournier, C.; Gimonet, J.; Ngom-Bru, C.; Barretto, C.; Baert, L. A validation approach of an end-to-end whole genome sequencing workflow for source tracking of listeria monocytogenes and salmonella enterica. Front. Microbiol. 2018, 9, 446. [Google Scholar] [CrossRef]
- Forrester, S.J.; Hall, N. The revolution of whole genome sequencing to study parasites. Mol. Biochem. Parasitol. 2014, 195, 77–81. [Google Scholar] [CrossRef]
- Leipzig, J. A review of bioinformatic pipeline frameworks. Brief. Bioinform. 2017, 18, 530–536. [Google Scholar] [CrossRef]
- Zanini, S.; Šečić, E.; Jelonek, L.; Kogel, K.H. A bioinformatics pipeline for the analysis and target prediction of rna effectors in bidirectional communication during plant–microbe interactions. Front. Plant. Sci. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Yin, R.; Kwoh, C.K.; Zheng, J. Whole genome sequencing analysis. Encycl. Bioinform. Comput. Biol. ABC Bioinform. 2018, 1–3, 176–183. [Google Scholar] [CrossRef]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: A joint recommendation of the association for molecular pathology and the college of American pathologists. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef]
- Flicek, P.; Birney, E. Sense from sequence reads: Methods for alignment and assembly. Nat. Methods 2009, 6, S6–S12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef]
- Schoumans, J.; Ruivenkamp, C.; Tools, B.; Gene, D.; Mooney, S.D.; Krishnan, V.G.; Evani, U.S. Chapter 12: Whole genome sequencing. Methods Mol. Biol. 2010, 628, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, M.; Chen, T.; Jiang, R. Whole genome sequencing and its applications in medical genetics. Quant. Biol. 2016, 4, 115–128. [Google Scholar] [CrossRef]
- Liu, X.; Han, S.; Wang, Z.; Gelernter, J.; Yang, B.Z. Variant callers for next-generation sequencing data: A comparison study. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Chen, S.B.; Cui, Y.B.; Xu, B.; Kassegne, K.; Abe, E.M.; Wang, Y.; Chen, J.H. Whole-genome sequencing and analysis of plasmodium falciparum isolates from China-Myanmar border area. Infect. Dis. Poverty 2018, 7, 118. [Google Scholar] [CrossRef]
- Kassegne, K.; Komi Koukoura, K.; Shen, H.-M.; Chen, S.-B.; Fu, H.-T.; Chen, Y.-Q.; Zhou, X.-N.; Chen, J.-H.; Cheng, Y. Genome-wide analysis of the malaria parasite plasmodium falciparum isolates from Togo reveals selective signals in immune selection-related antigen genes. Front. Immunol. 2020, 11, 2433. [Google Scholar] [CrossRef]
- Mobegi, V.A.; Duffy, C.W.; Amambua-Ngwa, A.; Loua, K.M.; Laman, E.; Nwakanma, D.C.; MacInnis, B.; Aspeling-Jones, H.; Murray, L.; Clark, T.G.; et al. Genome-wide analysis of selection on the malaria parasite plasmodium falciparum in west African populations of differing infection endemicity. Mol. Biol. Evol. 2014, 31, 1490–1499. [Google Scholar] [CrossRef]
- Carlton, J.M.; Escalante, A.A.; Neafsey, D.; Volkman, S.K. Comparative evolutionary genomics of human malaria parasites. Trends Parasitol. 2008, 24, 545–550. [Google Scholar] [CrossRef]
- Cornejo, O.E.; Fisher, D.; Escalante, A.A. Genome-wide patterns of genetic polymorphism and signatures of selection in plasmodium vivax. Genome Biol. Evol. 2014, 7, 106–119. [Google Scholar] [CrossRef]
- Ocholla, H.; Preston, M.D.; Mipando, M.; Jensen, A.T.R.; Campino, S.; Macinnis, B.; Alcock, D.; Terlouw, A.; Zongo, I.; Oudraogo, J.B.; et al. Whole-genome scans provide evidence of adaptive evolution in Malawian plasmodium falciparum isolates. J. Infect. Dis. 2014, 210, 1991–2000. [Google Scholar] [CrossRef]
- Loy, D.E.; Plenderleith, L.J.; Sundararaman, S.A.; Liu, W.; Gruszczyk, J.; Chen, Y.J.; Trimboli, S.; Learn, G.H.; MacLean, O.A.; Morgan, A.L.K.; et al. Evolutionary history of human plasmodium vivax revealed by genome-wide analyses of related ape parasites. Proc. Natl. Acad. Sci. USA 2018, 115, E8450–E8459. [Google Scholar] [CrossRef]
- Nair, S.; Miller, B.; Barends, M.; Jaidee, A.; Patel, J.; Mayxay, M.; Newton, P.; Nosten, F.; Ferdig, M.T.; Anderson, T.J.C. Adaptive copy number evolution in malaria parasites. PLoS Genet. 2008, 4, e1000243. [Google Scholar] [CrossRef]
- Kwong, J.C.; Mccallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef]
- Van El, C.G.; Cornel, M.C.; Borry, P.; Hastings, R.J.; Fellmann, F.; Hodgson, S.V.; Howard, H.C.; Cambon-Thomsen, A.; Knoppers, B.M.; Meijers-Heijboer, H.; et al. Whole-genome sequencing in health care. Eur. J. Hum. Genet. 2013, 21, 580–584. [Google Scholar] [CrossRef]
- Davis-Turak, J.; Courtney, S.M.; Hazard, E.S.; Glen, W.B.; da Silveira, W.A.; Wesselman, T.; Harbin, L.P.; Wolf, B.J.; Chung, D.; Hardiman, G. Genomics pipelines and data integration: Challenges and opportunities in the research setting. Expert Rev. Mol. Diagn. 2017, 17, 225–237. [Google Scholar] [CrossRef]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef]
- Stasiewicz, M.J.; Oliver, H.F.; Wiedmann, M.; Den Bakker, H.C. Whole-genome sequencing allows for improved identification of persistent listeria monocytogenes in food-associated environments. Appl. Environ. Microbiol. 2015, 81, 6024–6037. [Google Scholar] [CrossRef]
- Pightling, A.W.; Pettengill, J.B.; Luo, Y.; Baugher, J.D.; Rand, H.; Strain, E. Interpreting whole-genome sequence analyses of foodborne bacteria for regulatory applications and outbreak investigations. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Franz, E.; Delaquis, P.; Morabito, S.; Beutin, L.; Gobius, K.; Rasko, D.A.; Bono, J.; French, N.; Osek, J.; Lindstedt, B.A.; et al. Exploiting the explosion of information associated with whole genome sequencing to tackle shiga toxin-producing escherichia coli (STEC) in global food production systems. Int. J. Food Microbiol. 2014, 187, 57–72. [Google Scholar] [CrossRef]
- World Health Organization. Whole Genome Sequencing for Foodborne Disease Surveillance; WHO: Geneva, Switzerland, 2018; ISBN 9789241513869. [Google Scholar]
- Alghoribi, M.F.; Balkhy, H.H.; Woodford, N.; Ellington, M.J. The role of whole genome sequencing in monitoring antimicrobial resistance: A biosafety and public health priority in the Arabian peninsula. J. Infect. Public Health 2018, 11, 784–787. [Google Scholar] [CrossRef]
- Balloux, F.; Brønstad Brynildsrud, O.; Van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From theory to practice: Translating whole-genome sequencing (WGS) into the clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef]
- Manga, I.; Hasman, H.; Smidkova, J.; Medvecky, M.; Dolejska, M.; Cizek, A. Fecal carriage and whole-genome sequencing-assisted characterization of CMY-2 beta-lactamase-producing escherichia coli in calves at Czech dairy cow farm. Foodborne Pathog. Dis. 2019, 16, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Ronholm, J.; Nasheri, N.; Petronella, N.; Pagotto, F. Navigating microbiological food safety in the era of whole-genome sequencing. Clin. Microbiol. Rev. 2016, 29, 837–857. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Masim, M.A.L.; Gayeta, J.M.; Lagrada, M.L.; Macaranas, P.K.V.; Cohen, V.; Limas, M.T.; Espiritu, H.O.; Palarca, J.C.; Chilam, J.; et al. Integrating whole-genome sequencing within the national antimicrobial resistance surveillance program in the Philippines. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gygli, S.M.; Keller, P.M.; Ballif, M.; Reinhard, M.; Ritter, C.; Sander, P.; Borrell, S.; Loo, J.C.; Avihingsanon, A.; Gnokoro, J.; et al. Crossm prediction in mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, 1–13. [Google Scholar]
- Srivatsan, A.; Han, Y.; Peng, J.; Tehranchi, A.K.; Gibbs, R.; Wang, J.D.; Chen, R. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 2008, 4, e1000139. [Google Scholar] [CrossRef] [PubMed]
- Manara, S.; Pasolli, E.; Dolce, D.; Ravenni, N.; Campana, S.; Armanini, F.; Asnicar, F.; Mengoni, A.; Galli, L.; Montagnani, C.; et al. Whole-genome epidemiology, characterisation, and phylogenetic reconstruction of staphylococcus aureus strains in a paediatric hospital 11 medical and health sciences 1108 medical microbiology 06 biological sciences 0604 genetics 11 medical and health sciences 1103 clinical sciences. Genome Med. 2018, 10, 1–19. [Google Scholar] [CrossRef]
- Abdelbary, M.M.H.; Basset, P.; Blanc, D.S.; Feil, E.J. The Evolution and Dynamics of Methicillin-Resistant Staphylococcus Aureus; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780127999425. [Google Scholar]
- Price, J.; Gordon, N.C.; Crook, D.; Llewelyn, M.; Paul, J. The usefulness of whole genome sequencing in the management of staphylococcus aureus infections. Clin. Microbiol. Infect. 2013, 19, 784–789. [Google Scholar] [CrossRef]
- Greig, D.R.; Schaefer, U.; Octavia, S.; Hunter, E.; Chattaway, M.A.; Dallman, T.J.; Jenkins, C. Crossm and typing of vibrio cholerae. J. Clin. Microbiol. 2018, 56, 1–8. [Google Scholar]
- Reuter, S.; Ellington, M.J.; Cartwright, E.J.P.; Köser, C.U.; Török, M.E.; Gouliouris, T.; Harris, S.R.; Brown, N.M.; Holden, M.T.G.; Quail, M.; et al. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern. Med. 2013, 173, 1397–1404. [Google Scholar] [CrossRef]
- Shen, Y.; Sarin, S.; Liu, Y.; Hobert, O.; Pe’er, I. Comparing platforms for c. elegans mutant identification using high-throughput whole-genome sequencing. PLoS ONE 2008, 3, 2–7. [Google Scholar] [CrossRef][Green Version]
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole genome sequencing of mycobacterium tuberculosis: Current standards and open issues. Nat. Rev. Microbiol. 2019, 17, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, U.; Regier, A.; Tan, A.; Desany, B.A.; Collins, B.; Tan, J.C.; Emrich, S.J.; Ferdig, M.T. High-throughput 454 resequencing for allele discovery and recombination mapping in plasmodium falciparum. BMC Genom. 2011, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhong, D.; Cao, J.; Zhou, H.; Li, J.; Liu, Y.; Bai, L.; Xu, S.; Wang, M.H.; Zhou, G.; et al. Transcriptome profiling of pyrethroid resistant and susceptible mosquitoes in the malaria vector, anopheles sinensis. BMC Genom. 2014, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ning, B.; Fang, H.; Hong, H.; Perkins, R.; Tong, W.; Shi, L. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev. Mol. Diagn. 2011, 11, 333–343. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Rask, T.S.; Petersen, B.; Chen, D.S.; Day, K.P.; Pedersen, A.G. Using expected sequence features to improve basecalling accuracy of amplicon pyrosequencing data. BMC Bioinform. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Van Vliet, A.H.M. Next generation sequencing of microbial transcriptomes: Challenges and opportunities. FEMS Microbiol. Lett. 2010, 302, 1–7. [Google Scholar] [CrossRef]
- Zavodna, M.; Bagshaw, A.; Brauning, R.; Gemmell, N.J. The accuracy, feasibility and challenges of sequencing short tandem repeats using next-generation sequencing platforms. PLoS ONE 2014, 9, e113862. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Ding, L.; Mardis, E.R.; Wilson, R.K. Challenges of sequencing human genomes. Brief. Bioinform. 2010, 11, 484–498. [Google Scholar] [CrossRef]
- Gregory, R.; Darby, A.C.; Irving, H.; Coulibaly, M.B.; Hughes, M.; Koekemoer, L.L.; Coetzee, M.; Ranson, H.; Hemingway, J.; Hall, N.; et al. A de novo expression profiling of anopheles funestus, malaria vector in Africa, using 454 pyrosequencing. PLoS ONE 2011, 6, e17418. [Google Scholar] [CrossRef]
- Tachibana, S.I.; Sullivan, S.A.; Kawai, S.; Nakamura, S.; Kim, H.R.; Goto, N.; Arisue, N.; Palacpac, N.M.Q.; Honma, H.; Yagi, M.; et al. Plasmodium cynomolgi genome sequences provide insight into plasmodium vivax and the monkey malaria clade. Nat. Genet. 2012, 44, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Barnetche, J.; Gómez-Barreto, R.E.; Ovilla-Muñoz, M.; Téllez-Sosa, J.; López, D.E.G.; Dinglasan, R.R.; Mohien, C.U.; MacCallum, R.M.; Redmond, S.N.; Gibbons, J.G.; et al. Transcriptome of the adult female malaria mosquito vector anopheles albimanus. BMC Genom. 2012, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lalremruata, A.; Jeyaraj, S.; Engleitner, T.; Joanny, F.; Lang, A.; Bélard, S.; Mombo-Ngoma, G.; Ramharter, M.; Kremsner, P.G.; Mordmüller, B.; et al. Species and genotype diversity of plasmodium in malaria patients from Gabon analysed by next generation sequencing. Malar. J. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pringle, J.C.; Wesolowski, A.; Berube, S.; Kobayashi, T.; Gebhardt, M.E.; Mulenga, M.; Chaponda, M.; Bobanga, T.; Juliano, J.J.; Meshnick, S.; et al. High plasmodium falciparum genetic diversity and temporal stability despite control efforts in high transmission settings along the international border between Zambia and the Democratic Republic of the Congo. Malar. J. 2019, 18, 1–13. [Google Scholar] [CrossRef]
- Le Roch, K.G.; Chung, D.W.D.; Ponts, N. Genomics and integrated systems biology in plasmodium falciparum: A path to malaria control and eradication. Parasite Immunol. 2012, 34, 50–60. [Google Scholar] [CrossRef]
- Xuan, J.; Yu, Y.; Qing, T.; Guo, L.; Shi, L. Next-generation sequencing in the clinic: Promises and challenges. Cancer Lett. 2013, 340, 284–295. [Google Scholar] [CrossRef]
- Neafsey, D.E.; Galinsky, K.; Jiang, R.H.Y.; Young, L.; Sykes, S.M.; Saif, S.; Gujja, S.; Goldberg, J.M.; Young, S.; Zeng, Q.; et al. The malaria parasite plasmodium vivax exhibits greater genetic diversity than plasmodium falciparum. Nat. Genet. 2012, 44, 1046–1050. [Google Scholar] [CrossRef]
- Manske, M.; Miotto, O.; Campino, S.; Auburn, S.; Almagro-Garcia, J.; Maslen, G.; O’Brien, J.; Djimde, A.; Doumbo, O.; Zongo, I.; et al. Analysis of plasmodium falciparum diversity in natural infections by deep sequencing. Nature 2012, 487, 375–379. [Google Scholar] [CrossRef]
- Campino, S.; Auburn, S.; Kivinen, K.; Zongo, I.; Ouedraogo, J.B.; Mangano, V.; Djimde, A.; Doumbo, O.K.; Kiara, S.M.; Nzila, A.; et al. Population genetic analysis of plasmodium falciparum parasites using a customized illumina goldengate genotyping assay. PLoS ONE 2011, 6, e20251. [Google Scholar] [CrossRef]
- Dharia, N.V.; Bright, A.T.; Westenberger, S.J.; Barnes, S.W.; Batalov, S.; Kuhen, K.; Borboa, R.; Federe, G.C.; McClean, C.M.; Vinetz, J.M.; et al. Whole-genome sequencing and microarray analysis of ex vivo plasmodium vivax reveal selective pressure on putative drug resistance genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20045–20050. [Google Scholar] [CrossRef]
- Ghansah, A.; Kamau, E.; Amambua-Ngwa, A.; Ishengoma, D.S.; Maiga-Ascofare, O.; Amenga-Etego, L.; Deme, A.; Yavo, W.; Randrianarivelojosia, M.; Ochola-Oyier, L.I.; et al. Targeted next generation sequencing for malaria research in Africa: Current status and outlook. Malar. J. 2019, 18, 324. [Google Scholar] [CrossRef]
- Noviyanti, R.; Miotto, O.; Barry, A.; Marfurt, J.; Siegel, S.; Thuy-Nhien, N.; Quang, H.H.; Anggraeni, N.D.; Laihad, F.; Liu, Y.; et al. Implementing parasite genotyping into national surveillance frameworks: Feedback from control programmes and researchers in the Asia-Pacific region. Malar. J. 2020, 19, 1–20. [Google Scholar] [CrossRef]
- Buyon, L.E.; Santamaria, A.M.; Early, A.M.; Quijada, M.; Barahona, I.; Lasso, J.; Avila, M.; Volkman, S.K.; Marti, M.; Neafsey, D.E.; et al. Population genomics of plasmodium vivax in Panama to assess the risk of case importation on malaria elimination. PLoS Negl. Trop. Dis. 2020, 14, e0008962. [Google Scholar] [CrossRef]
- Nag, S.; Dalgaard, M.D.; Kofoed, P.E.; Ursing, J.; Crespo, M.; Andersen, L.O.B.; Aarestrup, F.M.; Lund, O.; Alifrangis, M. High throughput resistance profiling of plasmodium falciparum infections based on custom dual indexing and illumina next generation sequencing-technology. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Ranade, S.S.; Chung, C.B.; Zon, G.; Boyd, V.L. Preparation of genome-wide dna fragment libraries using bisulfite in polyacrylamide gel electrophoresis slices with formamide denaturation and quality control for massively parallel sequencing by oligonucleotide ligation and detection. Anal. Biochem. 2009, 390, 126–135. [Google Scholar] [CrossRef]
- Huang, Y.F.; Chen, S.C.; Chiang, Y.S.; Chen, T.H.; Chiu, K.P. Palindromic sequence impedes sequencing-by-ligation mechanism. BMC Syst. Biol. 2012, 6, 1–7. [Google Scholar] [CrossRef]
- Nardella, F.; Halby, L.; Hammam, E.; Erdmann, D.; Cadet-Daniel, V.; Peronet, R.; Ménard, D.; Witkowski, B.; Mecheri, S.; Scherf, A.; et al. DNA methylation bisubstrate inhibitors are fast-acting drugs active against artemisinin-resistant plasmodium falciparum parasites. ACS Cent. Sci. 2020, 6, 16–21. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Senabouth, A.; Andersen, S.; Shi, Q.; Shi, L.; Jiang, F.; Zhang, W.; Wing, K.; Daniszewski, M.; Lukowski, S.W.; Hung, S.S.C.; et al. Comparative performance of the BGI and illumina sequencing technology for single-cell RNA-sequencing. NAR Genom. Bioinform. 2020, 2, lqaa034. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, Z.; Tang, C.; Tang, Y.; Cai, Y.; Zhong, H.; Wang, X.; Zhang, W.; Xu, C.; Wang, J.; et al. A new massively parallel nanoball sequencing platform for whole exome research. BMC Bioinform. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Garrido-Cardenas, J.A.; Garcia-Maroto, F.; Alvarez-Bermejo, J.A.; Manzano-Agugliaro, F. DNA sequencing sensors: An overview. Sensors 2017, 17, 588. [Google Scholar] [CrossRef]
- Levitt, B.; Obala, A.; Langdon, S.; Corcoran, D.; O’Meara, W.P.; Taylor, S.M. Overlap extension barcoding for the next generation sequencing and genotyping of plasmodium falciparum in individual patients in Western Kenya. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Miller, R.H.; Hathaway, N.J.; Kharabora, O.; Mwandagalirwa, K.; Tshefu, A.; Meshnick, S.R.; Taylor, S.M.; Juliano, J.J.; Stewart, V.A.; Bailey, J.A. A deep sequencing approach to estimate plasmodium falciparum complexity of infection (COI) and explore apical membrane antigen 1 diversity. Malar. J. 2017, 16, 1–15. [Google Scholar] [CrossRef]
- Lin, J.T.; Hathaway, N.J.; Saunders, D.L.; Lon, C.; Balasubramanian, S.; Kharabora, O.; Gosi, P.; Sriwichai, S.; Kartchner, L.; Chuor, C.M.; et al. Using amplicon deep sequencing to detect genetic signatures of plasmodium vivax relapse. J. Infect. Dis. 2015, 212, 999–1008. [Google Scholar] [CrossRef]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A Window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 1–4. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Vembar, S.S.; Seetin, M.; Lambert, C.; Nattestad, M.; Schatz, M.C.; Baybayan, P.; Scherf, A.; Smith, M.L. Complete telomere-to-telomere de novo assembly of the plasmodium falciparum genome through long-read (>11 kb), single molecule, real-time sequencing. DNA Res. 2016, 23, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Dara, A.; Travassos, M.A.; Adams, M.; Schaffer Deroo, S.; Drábek, E.F.; Agrawal, S.; Laufer, M.K.; Plowe, C.V.; Silva, J.C. A new method for sequencing the hypervariable plasmodium falciparum gene var2csa from clinical samples. Malar. J. 2017, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.M.; Baumgarten, S.; Lorthiois, A.; Scheidig-Benatar, C.; Claës, A.; Scherf, A. De novo genome assembly of a plasmodium falciparum NF54 clone using single-molecule real-time sequencing. Genome Announc. 2018, 6, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Steinmann, K.E. Single molecule sequencing with a heliscope genetic analysis system. Curr. Protoc. Mol. Biol. 2010, 92, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.D.; Buzby, P.R.; Babcock, H.; Beer, E.; Bowers, J.; Braslavsky, I.; Causey, M.; Colonell, J.; DiMeo, J.; Efcavitch, J.W.; et al. Single-molecule DNA sequencing of a viral genome. Science 2008, 320, 106–109. [Google Scholar] [CrossRef]
- Deamer, D.W.; Akeson, M. Nanopores and nucleic acids: Prospects for ultrarapid sequencing. Trends Biotechnol. 2000, 18, 147–151. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Runtuwene, L.R.; Tuda, J.S.B.; Mongan, A.E.; Makalowski, W.; Frith, M.C.; Imwong, M.; Srisutham, S.; Nguyen Thi, L.A.; Tuan, N.N.; Eshita, Y.; et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, plasmodium falciparum. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef]
- Imai, K.; Tarumoto, N.; Runtuwene, L.R.; Sakai, J.; Hayashida, K.; Eshita, Y.; Maeda, R.; Tuda, J.; Ohno, H.; Murakami, T.; et al. An innovative diagnostic technology for the codon mutation C580Y in Kelch13 of plasmodium falciparum with MinION manopore sequencer. Malar. J. 2018, 17, 1–11. [Google Scholar] [CrossRef]
- Imai, K.; Tarumoto, N.; Misawa, K.; Runtuwene, L.R.; Sakai, J.; Hayashida, K.; Eshita, Y.; Maeda, R.; Tuda, J.; Murakami, T.; et al. A novel diagnostic method for malaria using loop-mediated isothermal amplification (LAMP) and MinIONTM nanopore sequencer. BMC Infect. Dis. 2017, 17, 1–9. [Google Scholar] [CrossRef]
- Zamyatin, A.; Avdeyev, P.; Liang, J.; Sharma, A.; Chen, C.; Lukyanchikova, V.; Alexeev, N.; Tu, Z.; Alekseyev, M.A.; Sharakhov, I.V. Chromosome-level genome assemblies of the malaria vectors anopheles coluzzii and anopheles arabiensis. GigaScience 2021, 10, giab017. [Google Scholar] [CrossRef]
- Chen, J.H.; Fen, J.; Zhou, X.N. From 30 million to zero malaria cases in China: Lessons learned for China–Africa collaboration in malaria elimination. Infect. Dis. Poverty 2021, 10, 1–4. [Google Scholar] [CrossRef]
- Karunaweera, N.D.; Galappaththy, G.N.; Wirth, D.F. On the road to eliminate malaria in Sri Lanka: Lessons from history, challenges, gaps in knowledge and research needs. Malar. J. 2014, 13, 1–10. [Google Scholar] [CrossRef]
- Gunawardena, S.; Ferreira, M.U.; Kapilananda, G.M.G.; Wirth, D.F.; Karunaweera, N.D. The Sri Lankan paradox: High genetic diversity in plasmodium vivax populations despite decreasing levels of malaria transmission. Parasitology 2014, 141, 880–890. [Google Scholar] [CrossRef]
- Yin, R.; Kwoh, C.K.; Zheng, J. Whole genome sequencing analysis. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 176–183. ISBN 9780128114322. [Google Scholar]
- Revez, J.; Espinosa, L.; Albiger, B.; Leitmeyer, K.C.; Struelens, M.J.; ECDC National Microbiology Focal Points and Experts Group. Survey on the use of whole-genome sequencing for infectious diseases surveillance: Rapid expansion of European national capacities, 2015–2016. Front. Public Health 2017, 5, 347. [Google Scholar] [CrossRef]
- Teyssier, N.B.; Chen, A.; Duarte, E.M.; Sit, R.; Greenhouse, B.; Tessema, S.K. Optimization of whole-genome sequencing of plasmodium falciparum from low-density dried blood spot samples. Malar. J. 2021, 20, 116. [Google Scholar] [CrossRef]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Llinás, M.; Bozdech, Z.; Wong, E.D.; Adai, A.T.; DeRisi, J.L. Comparative whole genome transcriptome analysis of three plasmodium falciparum strains. Nucleic Acids Res. 2006, 34, 1166–1173. [Google Scholar] [CrossRef]
- Lucas, E.R.; Miles, A.; Harding, N.J.; Clarkson, C.S.; Lawniczak, M.K.N.; Kwiatkowski, D.P.; Weetman, D.; Donnelly, M.J.; Anopheles Gambiae Genomes Consortium. Whole-genome sequencing reveals high complexity of copy number variation at insecticide resistance loci in malaria mosquitoes. Genome Res. 2019, 29, 1250–1261. [Google Scholar] [CrossRef]
- Bright, A.T.; Tewhey, R.; Abeles, S.; Chuquiyauri, R.; Llanos-Cuentas, A.; Ferreira, M.U.; Schork, N.J.; Vinetz, J.M.; Winzeler, E.A. Whole genome sequencing analysis of plasmodium vivax using whole genome capture. BMC Genom. 2012, 13, 262. [Google Scholar] [CrossRef]
- Da Veiga Leal, S.; Ward, D.; Campino, S.; Benavente, E.D.; Ibrahim, A.; Claret, T.; Isaias, V.; Monteiro, D.; Clark, T.G.; Goncalves, L.; et al. Drug resistance profile and clonality of plasmodium falciparum parasites in Cape Verde: The 2017 malaria outbreak. Malar. J. 2021, 20, 172. [Google Scholar] [CrossRef]
- Bright, A.T.; Alenazi, T.; Shokoples, S.; Tarning, J.; Paganotti, G.M.; White, N.J.; Houston, S.; Winzeler, E.A.; Yanow, S.K. Genetic analysis of primaquine tolerance in a patient with relapsing vivax malaria. Emerg Infect. Dis. 2013, 19, 802–805. [Google Scholar] [CrossRef]
- Fola, A.A.; Kattenberg, E.; Razook, Z.; Lautu-Gumal, D.; Lee, S.; Mehra, S.; Bahlo, M.; Kazura, J.; Robinson, L.J.; Laman, M.; et al. SNP barcodes provide higher resolution than microsatellite markers to measure plasmodium vivax population genetics. Malar. J. 2020, 19, 1–15. [Google Scholar] [CrossRef]
- Auburn, S.; Barry, A.E. Dissecting malaria biology and epidemiology using population genetics and genomics. Int. J. Parasitol. 2017, 47, 77–85. [Google Scholar] [CrossRef]
- Shen, H.M.; Chen, S.B.; Wang, Y.; Xu, B.; Abe, E.M.; Chen, J.H. Genome-wide scans for the identification of plasmodium vivax genes under positive selection. Malar. J. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Park, C.; Qian, W.; Zhang, J. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 2012, 13, 1123–1129. [Google Scholar] [CrossRef]
- Hupalo, D.N.; Luo, Z.; Melnikov, A.; Sutton, P.L.; Rogov, P.; Escalante, A.; Vallejo, A.F.; Herrera, S.; Arévalo-Herrera, M.; Fan, Q.; et al. Population genomics studies identify signatures of global dispersal and drug resistance in plasmodium vivax. Nat. Genet. 2016, 48, 953–958. [Google Scholar] [CrossRef]
- Chen, S.B.; Wang, Y.; Kassegne, K.; Xu, B.; Shen, H.M.; Chen, J.H. Whole-genome sequencing of a plasmodium vivax clinical isolate exhibits geographical characteristics and high genetic variation in China-Myanmar border area. BMC Genom. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Shi, S.M.; Shi, T.Q.; Chen, S.B.; Cui, Y.B.; Kassegne, K.; Okpeku, M.; Chen, J.H.; Shen, H.M. Genome-wide scans for Ghanaian plasmodium falciparum genes under selection from local and Chinese host populations. Front. Cell. Infect. Microbiol. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Mu, J.; Myers, R.A.; Jiang, H.; Liu, S.; Ricklefs, S.; Waisberg, M.; Chotivanich, K.; Wilairatana, P.; Krudsood, S.; White, N.J.; et al. Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat. Genet. 2010, 42, 268–271. [Google Scholar] [CrossRef]
- Ibrahim, A.; Diez Benavente, E.; Nolder, D.; Proux, S.; Higgins, M.; Muwanguzi, J.; Gomez Gonzalez, P.J.; Fuehrer, H.P.; Roper, C.; Nosten, F.; et al. Selective whole genome amplification of plasmodium malariae DNA from clinical samples reveals insights into population structure. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Chan, E.R.; Menard, D.; David, P.H.; Ratsimbasoa, A.; Kim, S.; Chim, P.; Do, C.; Witkowski, B.; Mercereau-Puijalon, O.; Zimmerman, P.A.; et al. Whole genome sequencing of field isolates provides robust characterization of genetic diversity in plasmodium vivax. PLoS Negl. Trop. Dis. 2012, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Obaldia, N.; Baro, N.K.; Calzada, J.E.; Santamaria, A.M.; Daniels, R.; Wong, W.; Chang, H.H.; Hamilton, E.J.; Arevalo-Herrera, M.; Herrera, S.; et al. Clonal outbreak of plasmodium falciparum infection in Eastern Panama. J. Infect. Dis. 2015, 211, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Urban, A.E.; Affourtit, J.P.; Godwin, B.; Grubert, F.; Simons, J.F.; Kim, P.M.; Palejev, D.; Carriero, N.J.; Du, L.; et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 2007, 318, 420–426. [Google Scholar] [CrossRef]
- Huckaby, A.C.; Granum, C.S.; Carey, M.A.; Szlachta, K.; Al-Barghouthi, B.; Wang, Y.H.; Guler, J.L. Complex DNA structures trigger copy number variation across the plasmodium falciparum genome. Nucleic Acids Res. 2019, 47, 1615–1627. [Google Scholar] [CrossRef]
- Simam, J.; Rono, M.; Ngoi, J.; Nyonda, M.; Mok, S.; Marsh, K.; Bozdech, Z.; Mackinnon, M. Gene copy number variation in natural populations of plasmodium falciparum in Eastern Africa. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, J.; Guo, Y.; Yang, Y.; Teng, T.; Yu, Q.; Wang, T.; Zhou, M.; Zhu, Q.; Wang, W.; et al. Genome-wide detection of CNVs and association with body weight in sheep based on 600k SNP arrays. Front. Genet. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Beghain, J.; Langlois, A.C.; Legrand, E.; Grange, L.; Khim, N.; Witkowski, B.; Duru, V.; Ma, L.; Bouchier, C.; Ménard, D.; et al. Plasmodium copy number variation scan: Gene copy numbers evaluation in haploid genomes. Malar. J. 2016, 15, 1–6. [Google Scholar] [CrossRef]
- Eastman, R.T.; Dharia, N.V.; Winzeler, E.A.; Fidock, D.A. Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured plasmodium falciparum parasites. Antimicrob. Agents Chemother. 2011, 55, 3908–3916. [Google Scholar] [CrossRef]
- Ravenhall, M.; Benavente, E.D.; Sutherland, C.J.; Baker, D.A.; Campino, S.; Clark, T.G. An Analysis of large structural variation in global plasmodium falciparum isolates identifies a novel duplication of the chloroquine resistance associated gene. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Auburn, S.; Campino, S.; Miotto, O.; Djimde, A.A.; Zongo, I.; Manske, M.; Maslen, G.; Mangano, V.; Alcock, D.; MacInnis, B.; et al. Characterization of within-host plasmodium falciparum diversity using next-generation sequence data. PLoS ONE 2012, 7, e32891. [Google Scholar] [CrossRef]
- Murray, L.; Mobegi, V.A.; Duffy, C.W.; Assefa, S.A.; Kwiatkowski, D.P.; Laman, E.; Loua, K.M.; Conway, D.J. Microsatellite genotyping and genome-wide single nucleotide polymorphism-based indices of plasmodium falciparum diversity within clinical infections. Malar. J. 2016, 15, 1–6. [Google Scholar] [CrossRef]
- De Roode, J.C.; Helinski, M.E.H.; Anwar, M.A.; Read, A.F. Dynamics of multiple infection and within-host competition in genetically diverse malaria infections. Am. Nat. 2005, 166, 531–542. [Google Scholar] [CrossRef]
- Nkhoma, S.C.; Trevino, S.G.; Gorena, K.M.; Nair, S.; Khoswe, S.; Jett, C.; Garcia, R.; Daniel, B.; Dia, A.; Terlouw, D.J.; et al. Co-transmission of related malaria parasite lineages shapes within-host parasite diversity. Cell Host Microbe 2020, 27, 93–103. [Google Scholar] [CrossRef]
- Guiguemde, T.; Zampa, O.; Kazienga, A.; Derra, K.; Valea, I.; Lefevre, T.; Ouedraogo, J.; Diallo-Nakanabo, S.; Sondo, P.; Tinto, H.; et al. Genetically diverse plasmodium falciparum infections, within-host competition and symptomatic malaria in humans. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Bushman, M.; Antia, R.; Udhayakumar, V.; de Roode, J.C. Within-host competition can delay evolution of drug resistance in malaria. PLoS Biol. 2018, 16, 1–25. [Google Scholar] [CrossRef]
- Nkhoma, S.C.; Trevino, S.G.; Gorena, K.M.; Nair, S.; Khoswe, S.; Jett, C.; Garcia, R.; Daniel, B.; Dia, A.; Terlouw, D.J.; et al. Resolving within-host malaria parasite diversity using single-cell sequencing. BioRxiv 2018, 10, 391268. [Google Scholar] [CrossRef]
- Early, A.M.; Lievens, M.; MacInnis, B.L.; Ockenhouse, C.F.; Volkman, S.K.; Adjei, S.; Agbenyega, T.; Ansong, D.; Gondi, S.; Greenwood, B.; et al. Host-mediated selection impacts the diversity of plasmodium falciparum antigens within infections. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Gwarinda, H.B.; Tessema, S.K.; Raman, J.; Greenhouse, B.; Birkholtz, L.M. Parasite genetic diversity reflects continued residual malaria transmission in vhembe district, a hotspot in the limpopo province of South Africa. Malar. J. 2021, 20, 1–13. [Google Scholar] [CrossRef]
- Trevino, S.G.; Nkhoma, S.C.; Nair, S.; Daniel, B.J.; Moncada, K.; Khoswe, S.; Banda, R.L.; Nosten, F.; Cheeseman, I.H. High-resolution single-cell sequencing of malaria parasites. Genome Biol. Evol. 2017, 9, 3373–3383. [Google Scholar] [CrossRef]
- Su, X.Z.; Lane, K.D.; Xia, L.; Sá, J.M.; Wellems, T.E. Plasmodium genomics and genetics: New insights into malaria pathogenesis, drug resistance, epidemiology, and evolution. Clin. Microbiol. Rev. 2019, 32, 1–29. [Google Scholar] [CrossRef]
- World Health Organization. Status report on artemisinin resistance and ACT efficacy. World Health Organ. 2018, 10, 1–6. [Google Scholar]
- Hall, N.; Carlton, J. Comparative genomics of malaria parasites. Curr. Opin. Genet. Dev. 2005, 15, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Rayner, J.C.; Böhme, U.; Pain, A.; Spottiswoode, N.; Sanders, M.; Quail, M.; Ollomo, B.; Renaud, F.; Thomas, A.W.; et al. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cowell, A.N.; Istvan, E.S.; Lukens, A.K.; Gomez-Lorenzo, M.G.; Vanaerschot, M.; Sakata-Kato, T.; Flannery, E.L.; Magistrado, P.; Owen, E.; Abraham, M.; et al. Mapping the malaria parasite druggable genome by using in vitroevolution and chemogenomics. Science 2018, 359, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Slater, H.C.; Griffin, J.T.; Ghani, A.C.; Okell, L.C. Assessing the potential impact of artemisinin and partner drug resistance in Sub-Saharan Africa. Malar. J. 2016, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tacoli, C.; Gai, P.P.; Bayingana, C.; Sifft, K.; Geus, D.; Ndoli, J.; Sendegeya, A.; Gahutu, J.B.; Mockenhaupt, F.P. Artemisinin resistance-associated K13 polymorphisms of plasmodium falciparum in Southern Rwanda, 2010–2015. Am. J. Trop. Med. Hyg. 2016, 95, 1090–1093. [Google Scholar] [CrossRef]
- Raobela, O.; Andriantsoanirina, V.; Rajaonera, D.G.; Rakotomanga, T.A.; Rabearimanana, S.; Ralinoro, F.; Ménard, D.; Ratsimbasoa, A. Efficacy of artesunate-amodiaquine in the treatment of falciparum uncomplicated malaria in Madagascar. Malar. J. 2018, 17, 5–11. [Google Scholar] [CrossRef]
- Byakika-Kibwika, P.; Nyakato, P.; Lamorde, M.; Kiragga, A.N. Assessment of parasite clearance following treatment of severe malaria with intravenous artesunate in Ugandan Children enrolled in a randomized controlled clinical trial PACTR201110000321348 PACTR. Malar. J. 2018, 17, 4–9. [Google Scholar] [CrossRef]
- Park, D.J.; Lukens, A.K.; Neafsey, D.E.; Schaffner, S.F.; Chang, H.H.; Valim, C.; Ribacke, U.; Van Tyne, D.; Galinsky, K.; Galligan, M.; et al. Sequence-based association and selection scans identify drug resistance loci in the plasmodium falciparum malaria parasite. Proc. Natl. Acad. Sci. USA 2012, 109, 13052–13057. [Google Scholar] [CrossRef]
- Diakité, S.A.S.; Traoré, K.; Sanogo, I.; Clark, T.G.; Campino, S.; Sangaré, M.; Dabitao, D.; Dara, A.; Konaté, D.S.; Doucouré, F.; et al. A comprehensive analysis of drug resistance molecular markers and plasmodium falciparum genetic diversity in two malaria endemic sites in Mali. Malar. J. 2019, 18, 1–9. [Google Scholar] [CrossRef]
- Apinjoh, T.; Ouattara, A.; Titanji, V.; Djimde, A.; Amambua-Ngwa, A. Genetic diversity and drug resistance surveillance of plasmodium falciparum for malaria elimination: Is there an ideal tool for resource-limited Sub-Saharan Africa? Malar. J. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Cravo, P.; Napolitano, H.; Culleton, R. How genomics is contributing to the fight against artemisinin-resistant malaria parasites. Acta Trop. 2015, 148, 1–7. [Google Scholar] [CrossRef]
- Flannery, E.L.; Wang, T.; Akbari, A.; Corey, V.C.; Gunawan, F.; Bright, A.T.; Abraham, M.; Sanchez, J.F.; Santolalla, M.L.; Baldeviano, G.C.; et al. Next-generation sequencing of plasmodium vivax patient samples shows evidence of direct evolution in drug-resistance genes. ACS Infect. Dis. 2016, 1, 367–379. [Google Scholar] [CrossRef]
- Nsanzabana, C.; Ariey, F.; Beck, H.P.; Ding, X.C.; Kamau, E.; Krishna, S.; Legrand, E.; Lucchi, N.; Miotto, O.; Nag, S.; et al. Molecular assays for antimalarial drug resistance surveillance: A target product profile. PLoS ONE 2018, 13, e0204347. [Google Scholar] [CrossRef]
- Price, R.N.; Auburn, S.; Marfurt, J.; Cheng, Q. Phenotypic and genotypic characterisation of drug-resistant plasmodium vivax. Trends Parasitol. 2012, 28, 522–529. [Google Scholar] [CrossRef]
- Vegyari, C.; Underwood, A.; Kekre, M.; Argimon, S.; Muddyman, D.; Abrudan, M.; Carlos, C.; Donado-Godoy, P.; Okeke, I.N.; Ravikumar, K.L.; et al. Whole-genome sequencing as part of national and international surveillance programmes for antimicrobial resistance: A roadmap. BMJ Glob. Health 2020, 5, e002244. [Google Scholar]
- Bambini, S.; Rappuoli, R. The use of genomics in microbial vaccine development. Drug Discov. Today 2009, 14, 252–260. [Google Scholar] [CrossRef]
- Conway, D.J. Paths to a malaria vaccine illuminated by parasite genomics. Trends Genet. 2015, 31, 97–107. [Google Scholar] [CrossRef]
- Fowkes, F.J.I.; Richards, J.S.; Simpson, J.A.; Beeson, J.G. The relationship between anti-merozoite antibodies and incidence of plasmodium falciparum malaria: A systematic review and meta-analysis. PLoS Med. 2010, 7, e1000218. [Google Scholar] [CrossRef]
- Cowman, A.F.; Berry, D.; Baum, J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J. Cell Biol. 2012, 198, 961–971. [Google Scholar] [CrossRef]
- Kassegne, K.; Abe, E.M.; Chen, J.H.; Zhou, X.N. Immunomic approaches for antigen discovery of human parasites. Expert Rev. Proteom. 2016, 13, 1091–1101. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, D.; Ding, G.; Shi, L.; Hou, Q.; Ye, Y.; Xu, Y.; Zhou, H.; Xiong, C.; Li, S.; et al. Genome sequence of anopheles sinensis provides insight into genetics basis of mosquito competence for malaria parasites. BMC Genom. 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Sharma, A.; Jangid, K.; Shouche, Y. Microbes, mosquitoes and malaria. Curr. Sci. 2012, 103, 254. [Google Scholar]
- Boissière, A.; Tchioffo, M.T.; Bachar, D.; Abate, L.; Marie, A.; Nsango, S.E.; Shahbazkia, H.R.; Awono-Ambene, P.H.; Levashina, E.A.; Christen, R.; et al. Midgut microbiota of the malaria mosquito vector anopheles gambiae and interactions with plasmodium falciparum infection. PLoS Pathog. 2012, 8, 1–12. [Google Scholar] [CrossRef]
- World Health Organisation. World Malaria Report. Available online: https://www.who.int/malaria/publications/world-repot-malaria-report-2019/en (accessed on 10 September 2020).
- Zhang, S.-S.; Zhou, S.-S.; Zhou, Z.-B.; Chen, T.-M.; Wang, X.-Z.; Shi, W.-Q.; Jiang, W.-K.; Li, J.-L.; Zhou, X.-N.; Frutos, R.; et al. Monitoring of malaria vectors at the China-Myanmar border while approaching malaria elimination. Parasites Vectors 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Karunasena, V.M.; Marasinghe, M.; Koo, C.; Amarasinghe, S.; Senaratne, A.S.; Hasantha, R.; Hewavitharana, M.; Hapuarachchi, H.C.; Herath, H.D.B.; Wickremasinghe, R.; et al. The first introduced malaria case reported from Sri Lanka after elimination: Implications for preventing the re-introduction of malaria in recently eliminated countries. Malar. J. 2019, 18, 1–10. [Google Scholar] [CrossRef]
- Premaratne, R.; Wickremasinghe, R.; Ranaweera, D.; Gunasekera, W.M.; Hevawitharana, M.; Pieris, L.; Fernando, D.; Mendis, K. Technical and operational underpinnings of malaria elimination from Sri Lanka. Malar. J. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Chang, X.; Zhong, D.; Wang, X.; Bonizzoni, M.; Li, Y.; Zhou, G.; Cui, L.; Wei, X.; Yan, G. Genomic variant analyses in pyrethroid resistant and susceptible malaria vector, anopheles sinensis. G3 Genes Genomes Genet. 2020, 10, 2185–2193. [Google Scholar] [CrossRef]
- Muhammad, A.; Ibrahim, S.S.; Mukhtar, M.M.; Irving, H.; Abajue, M.C.; Edith, N.M.A.; Dau, S.S.; Paine, M.J.I.; Wondji, C.S. High pyrethroid/DDT resistance in major malaria vector anopheles coluzzii from Niger-delta of Nigeria is probably driven by metabolic resistance mechanisms. PLoS ONE 2021, 16, e0247944. [Google Scholar] [CrossRef]
- Awolola, T.S.; Adeogun, A.; Olakiigbe, A.K.; Oyeniyi, T.; Olukosi, Y.A.; Okoh, H.; Arowolo, T.; Akila, J.; Oduola, A.; Amajoh, C.N. Pyrethroids resistance intensity and resistance mechanisms in anopheles Gambiae from malaria vector surveillance sites in Nigeria. PLoS ONE 2018, 13, e0205230. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.H.; Brooke, B.D.; Pillay, C.; Koekemoer, L.L.; Coetzee, M. Laboratory selection for and characteristics of pyrethroid resistance in the malaria vector anopheles funestus. Med. Vet. Entomol. 2005, 19, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Tomlinson, S.; Kleinschmidt, I.; Donnelly, M.J.; Akogbeto, M.; Adechoubou, A.; Massougbodji, A.; Okê-Sopoh, M.; Corbel, V.; Cornelie, S.; et al. Implications of insecticide resistance for malaria vector control with long-lasting insecticidal nets: Trends in pyrethroid resistance during a WHO—Coordinated multi-country prospective study 11 medical and health sciences 1117 public health and health services. Parasites Vectors 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Mint Mohamed Lemine, A.; Ould Lemrabott, M.A.; Niang, E.H.A.; Basco, L.K.; Bogreau, H.; Faye, O.; Ould Mohamed Salem Boukhary, A. Pyrethroid resistance in the major malaria vector anopheles arabiensis in Nouakchott, Mauritania. Parasites Vectors 2018, 11, 344. [Google Scholar] [CrossRef] [PubMed]
- Akuamoah-Boateng, Y.; Brenyah, R.C.; Kwarteng, S.A.; Obuam, P.; Owusu-Frimpong, I.; Agyapong, A.K.; Badu, K. Malaria transmission, vector diversity, and insecticide resistance at a peri-urban site in the forest zone of Ghana. Front. Trop. Dis. 2021, 2, 739771. [Google Scholar] [CrossRef]
- Rinker, D.C.; Pitts, R.J.; Zwiebel, L.J. Disease vectors in the era of next generation sequencing. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef]
- Rohani, A.; Ahmad Fakhriy, H.; Suzilah, I.; Zurainee, M.N.; Wan Najdah, W.M.A.; Mohd Ariffin, M.; Mohamad Shakirudin, N.; Mohd Afiq, M.S.; Jenarun, J.; Tanrang, Y.; et al. Indoor and outdoor residual spraying of a novel formulation of deltamethrin K-Othrine® (Polyzone) for the control of simian malaria in Sabah, Malaysia. PLoS ONE 2020, 15, e0230860. [Google Scholar] [CrossRef]
- Scudellari, M. Self-destructing mosquitoes and sterilized rodents: The promise of gene drives. Nature 2019, 571, 160–163. [Google Scholar] [CrossRef]
- Carballar-Lejarazú, R.; Ogaugwu, C.; Tushar, T.; Kelsey, A.; Pham, T.B.; Murphy, J.; Schmidt, H.; Lee, Y.; Lanzaro, G.C.; James, A.A. Next-generation gene drive for population modification of the malaria vector mosquito, anopheles gambiae. Proc. Natl. Acad. Sci. USA 2020, 117, 22805–22814. [Google Scholar] [CrossRef]
- Jiang, X.; Peery, A.; Hall, A.B.; Sharma, A.; Chen, X.G.; Waterhouse, R.M.; Komissarov, A.; Riehle, M.M.; Shouche, Y.; Sharakhova, M.V.; et al. Genome analysis of a major urban malaria vector mosquito, anopheles stephensi. Genome Biol. 2014, 15, 459. [Google Scholar] [CrossRef]
- Ghurye, J.; Koren, S.; Small, S.T.; Redmond, S.; Howell, P.; Phillippy, A.M.; Besansky, N.J. A Chromosome-scale assembly of the major african malaria vector anopheles funestus. Gigascience 2019, 8, giz063. [Google Scholar] [CrossRef]
- Nag, S.; Kofoed, P.-E.; Ursing, J.; Lemvigh, C.K.; Allesøe, R.L.; Rodrigues, A.; Svendsen, C.A.; Jensen, J.D.; Alifrangis, M.; Lund, O.; et al. Direct whole-genome sequencing of plasmodium falciparum specimens from dried erythrocyte spots. Malar. J. 2018, 17, 91. [Google Scholar] [CrossRef]
- Auburn, S.; Marfurt, J.; Maslen, G.; Campino, S.; Ruano Rubio, V.; Manske, M.; MacHunter, B.; Kenangalem, E.; Noviyanti, R.; Trianty, L.; et al. Effective preparation of plasmodium vivax field isolates for high-throughput whole genome sequencing. PLoS ONE 2013, 8, e53160. [Google Scholar] [CrossRef]
- Auburn, S.; Campino, S.; Clark, T.G.; Djimde, A.A.; Zongo, I.; Pinches, R.; Manske, M.; Mangano, V.; Alcock, D.; Anastasi, E.; et al. An effective method to purify plasmodium falciparum DNA directly from clinical blood samples for whole genome high-throughput sequencing. PLoS ONE 2011, 6, e22213. [Google Scholar] [CrossRef]
- Taylor, B.J.; Martin, K.A.; Arango, E.; Agudelo, O.M.; Maestre, A.; Yanow, S.K. Real-time PCR detection of plasmodium directly from whole blood and filter paper samples. Malar. J. 2011, 10, 1–8. [Google Scholar] [CrossRef]
- Amambua-Ngwa, A.; Amenga-Etego, L.; Kamau, E.; Amato, R.; Ghansah, A.; Golassa, L.; Randrianarivelojosia, M.; Ishengoma, D.; Apinjoh, T.; Maïga-Ascofaré, O.; et al. Major subpopulations of plasmodium falciparum in Sub-Saharan Africa. Science 2019, 365, 813–816. [Google Scholar] [CrossRef]
- Ishengoma, D.S.; Saidi, Q.; Sibley, C.H.; Roper, C.; Alifrangis, M. Deployment and utilization of next-generation sequencing of plasmodium falciparum to guide anti-malarial drug policy decisions in Sub-Saharan Africa: Opportunities and challenges. Malar. J. 2019, 18, 1–10. [Google Scholar] [CrossRef]
- Oakeson, K.F.; Wagner, J.M.; Mendenhall, M.; Rohrwasser, A.; Atkinson-Dunn, R. Bioinformatic analyses of whole-genome sequence data in a public health laboratory. Emerg. Infect. Dis. 2017, 23, 1441. [Google Scholar] [CrossRef]
- Miles, A.; Iqbal, Z.; Vauterin, P.; Pearson, R.; Campino, S.; Theron, M.; Gould, K.; Mead, D.; Drury, E.; O’Brien, J.; et al. Indels, structural variation, and recombination drive genomic diversity in plasmodium falciparum. Genome Res. 2016, 26, 1288–1299. [Google Scholar] [CrossRef]
- Cowell, A.N.; Valdivia, H.O.; Bishop, D.K.; Winzeler, E.A. Exploration of plasmodium vivax transmission dynamics and recurrent infections in the Peruvian Amazon using whole genome sequencing. Genome Med. 2018, 10, 1–12. [Google Scholar] [CrossRef]
- Oyola, S.O.; Ariani, C.V.; Hamilton, W.L.; Kekre, M.; Amenga-Etego, L.N.; Ghansah, A.; Rutledge, G.G.; Redmond, S.; Manske, M.; Jyothi, D.; et al. Whole genome sequencing of plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar. J. 2016, 15, 597. [Google Scholar] [CrossRef]
- Cowell, A.N.; Loy, D.E.; Sundararaman, S.A.; Valdivia, H.; Fisch, K.; Lescano, A.G.; Baldeviano, G.C.; Durand, S.; Gerbasi, V.; Sutherland, C.J.; et al. Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of plasmodium vivax from unprocessed clinical samples. MBio 2017, 8, e02257-16. [Google Scholar] [CrossRef]
- Li, H.; Yang, B.; Hu, H.; Wang, W.; Wang, W.; Li, X.; Li, C.; Huang, G. Observation on parasite density in the early stage of vivax malaria. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 1994, 12, 119–121. [Google Scholar]
- Iskandar, K.; Molinier, L.; Hallit, S.; Sartelli, M.; Hardcastle, T.C.; Haque, M.; Lugova, H.; Dhingra, S.; Sharma, P.; Islam, S.; et al. Surveillance of antimicrobial resistance in low- and middle-income countries: A scattered picture. Antimicrob. Resist. Infect. Control 2021, 10, 1–19. [Google Scholar] [CrossRef]
- Farhat, M.R.; Shapiro, B.J.; Kieser, K.J.; Sultana, R.; Jacobson, K.R.; Victor, T.C.; Warren, R.M.; Streicher, E.M.; Calver, A.; Sloutsky, A.; et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant mycobacterium tuberculosis. Nat. Genet. 2013, 45, 1183–1189. [Google Scholar] [CrossRef]
- Chewapreecha, C.; Marttinen, P.; Croucher, N.J.; Salter, S.J.; Harris, S.R.; Mather, A.E.; Hanage, W.P.; Goldblatt, D.; Nosten, F.H.; Turner, C.; et al. Comprehensive identification of single nucleotide polymorphisms associated with beta-lactam resistance within pneumococcal mosaic genes. PLoS Genet. 2014, 10, e1004547. [Google Scholar] [CrossRef]
- Peters, J.; Cresswell, F.; Amor, L.; Cole, K.; Dean, G.; Didelot, X.; De Silva, D.; Eyre, D.W.; Paul, J. Whole genome sequencing of neisseria gonorrhoeae reveals transmission clusters involving patients of mixed HIV serostatus. Sex. Transm. Infect. 2018, 94, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Penedos, A.R.; Myers, R.; Hadef, B.; Aladin, F.; Brown, K.E. Assessment of the utility of whole genome sequencing of measles virus in the characterisation of outbreaks. PLoS ONE 2015, 10, e0143081. [Google Scholar] [CrossRef]
- Probert, W.S.; Glenn-Finer, R.; Espinosa, A.; Yen, C.; Stockman, L.; Harriman, K.; Hacker, J.K. Molecular epidemiology of measles in California, United States—2019. J. Infect. Dis. 2021, 224, 1015–1023. [Google Scholar] [CrossRef]
- Cherukuri, P.; Vilboux, T.; Kothiyal, P.; Black, A.; Eley, G.; Huddleston, K.; Iyer, R.; Solomon, B.; Vockley, J.; Niederhuber, J. P-B23 prevalence of ebola viral entry resistance in a diverse population. JAIDS J. Acquir. Immune Defic. Syndr. 2016, 71, 83. [Google Scholar] [CrossRef]
- Bwire, G.; Sack, D.A.; Almeida, M.; Li, S.; Voeglein, J.B.; Debes, A.K.; Kagirita, A.; Buyinza, A.W.; Orach, C.G.; Stine, O.C. Molecular characterization of vibrio cholerae responsible for cholera epidemics in Uganda by PCR, MLVA and WGS. PLoS Negl. Trop. Dis. 2018, 12, e0006492. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, Z.; Chen, W.; Chen, X.; Hosseini, M.; Yang, Z.; Li, J.; Ho, D.; Turay, D.; Gheorghe, C.P.; et al. A benchmarking study of SARS-CoV-2 whole-genome sequencing protocols using COVID-19 patient samples. IScience 2021, 24, 102892. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Power, B.J.; Waters, A.P. Recent advances in malaria genomics and epigenomics. Genome Med. 2016, 8, 1–17. [Google Scholar] [CrossRef]
- Adebamowo, S.N.; Francis, V.; Tambo, E.; Diallo, S.H.; Landouré, G.; Nembaware, V.; Dareng, E.; Muhamed, B.; Odutola, M.; Akeredolu, T.; et al. Implementation of genomics research in Africa: Challenges and recommendations. Glob. Health Action 2018, 11, 1419033. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| WGS Platforms | Pros | Cons | Reads |
|---|---|---|---|
| Solexa/Illumina | High precision Analysis with a high throughput The most widely available technology | Read length is rather short. Machines are expensive Could be time consuming | 3 G |
| ABI SOLiD | High accuracy High-throughput analysis | Short read length High cost of machine Time consuming | 1.2–1.4 G |
| Roche 454 | Long read length High accuracy Short run time | Expensive machine Moderately low throughput analysis | 1 M |
| Ion Torrent Technologies | Moderate to long read length High throughput analysis High accuracy Relatively cheap when compared to others Shorter run time | No appropriate products | 6 × 107 |
| Oxford Nanopore | High-throughput analysis Short processing time | Less accuracy when compared to others Short read length | 6 × 104 |
| BGI Retrovolocity | Long reads Short time | Prone to error | 1 × 109 |
| Heliscope/Helicos | 7 × 109 | ||
| Pac Bio SMRT | High-throughput analysis Long read length | Prone to error High cost of machine | 1 × 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akoniyon, O.P.; Adewumi, T.S.; Maharaj, L.; Oyegoke, O.O.; Roux, A.; Adeleke, M.A.; Maharaj, R.; Okpeku, M. Whole Genome Sequencing Contributions and Challenges in Disease Reduction Focused on Malaria. Biology 2022, 11, 587. https://doi.org/10.3390/biology11040587
Akoniyon OP, Adewumi TS, Maharaj L, Oyegoke OO, Roux A, Adeleke MA, Maharaj R, Okpeku M. Whole Genome Sequencing Contributions and Challenges in Disease Reduction Focused on Malaria. Biology. 2022; 11(4):587. https://doi.org/10.3390/biology11040587
Chicago/Turabian StyleAkoniyon, Olusegun Philip, Taiye Samson Adewumi, Leah Maharaj, Olukunle Olugbenle Oyegoke, Alexandra Roux, Matthew A. Adeleke, Rajendra Maharaj, and Moses Okpeku. 2022. "Whole Genome Sequencing Contributions and Challenges in Disease Reduction Focused on Malaria" Biology 11, no. 4: 587. https://doi.org/10.3390/biology11040587
APA StyleAkoniyon, O. P., Adewumi, T. S., Maharaj, L., Oyegoke, O. O., Roux, A., Adeleke, M. A., Maharaj, R., & Okpeku, M. (2022). Whole Genome Sequencing Contributions and Challenges in Disease Reduction Focused on Malaria. Biology, 11(4), 587. https://doi.org/10.3390/biology11040587