
A Boolean Model of the Proliferative Role of the lncRNA XIST in Non-Small Cell Lung Cancer Cells


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
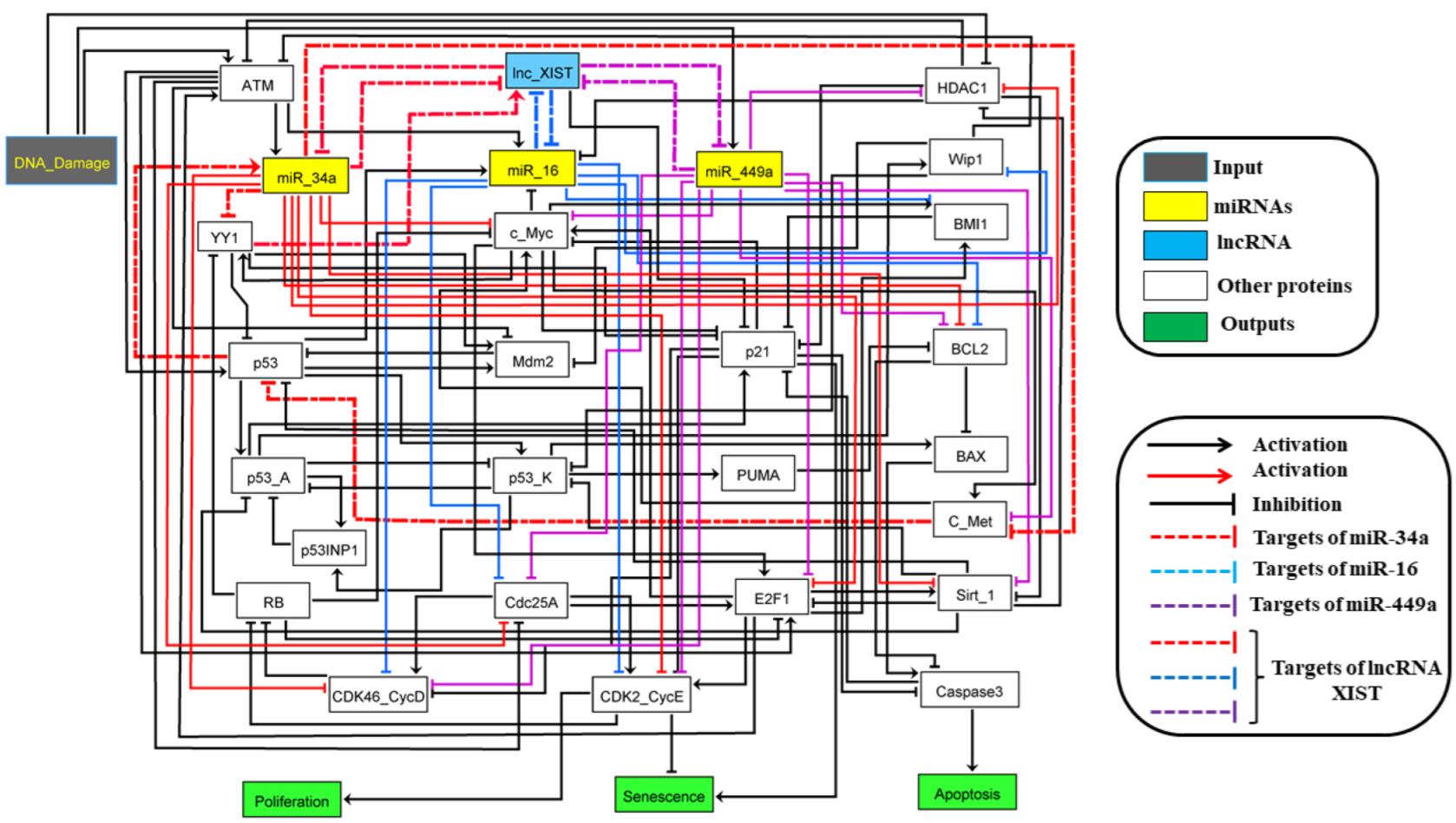
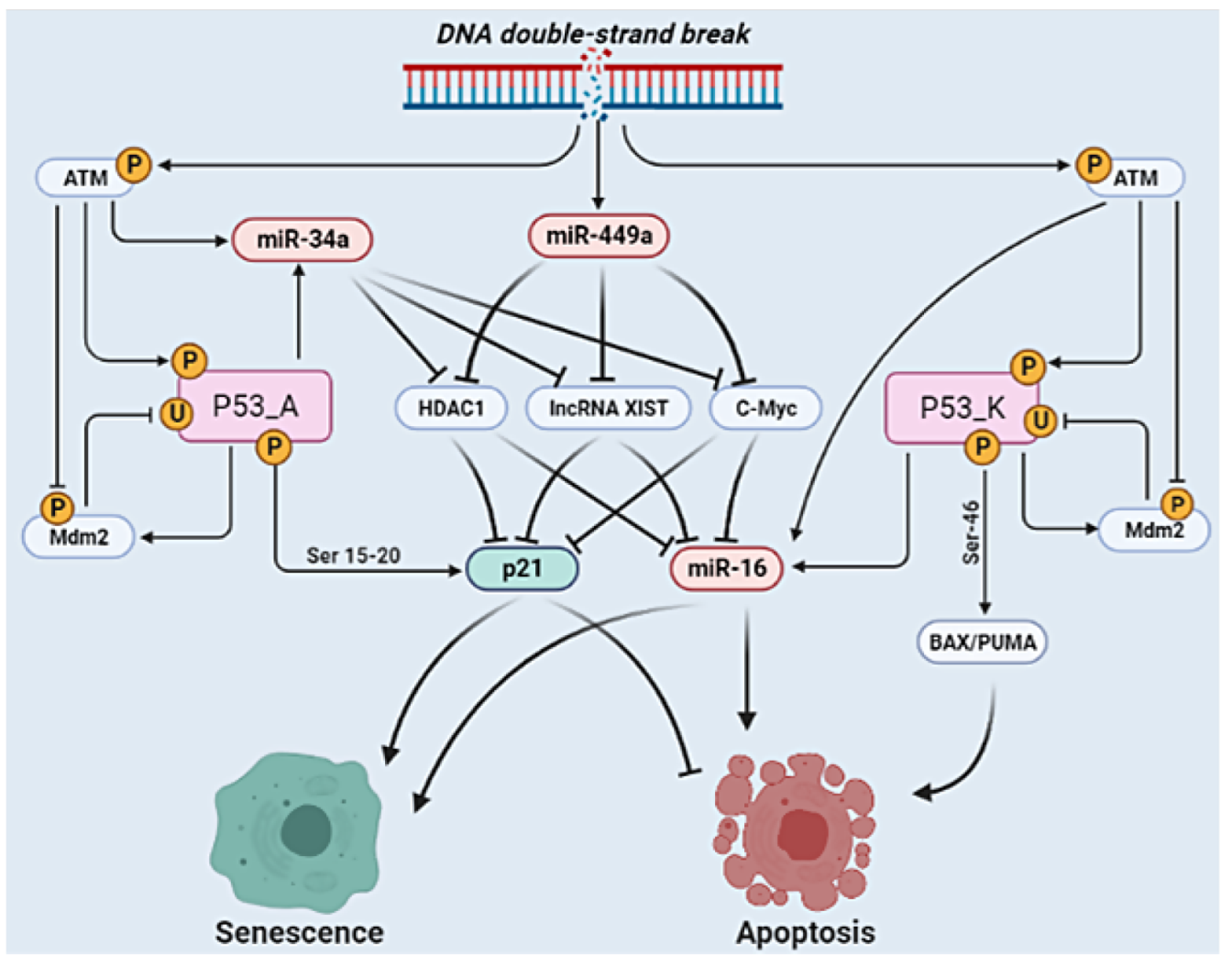
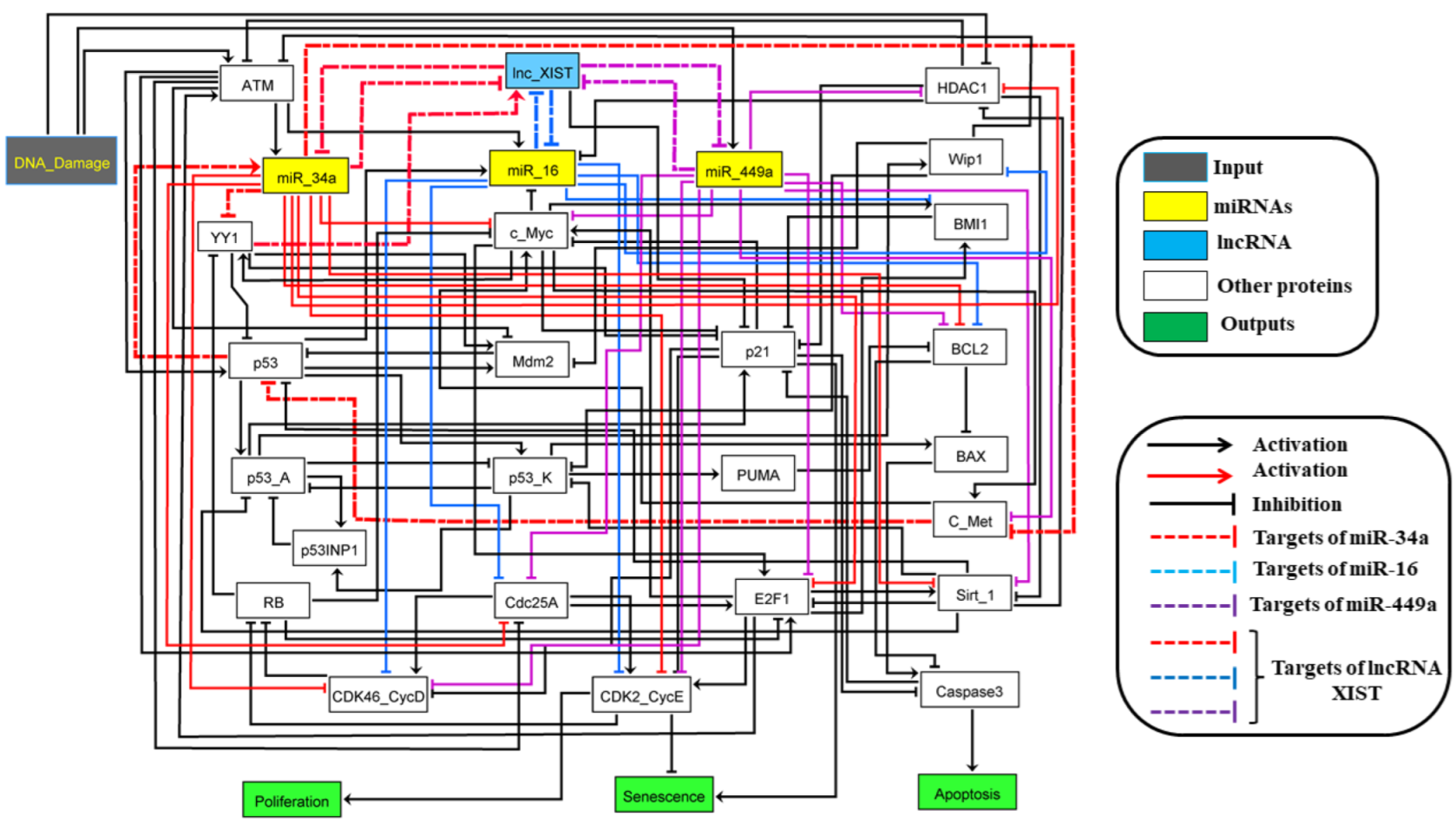
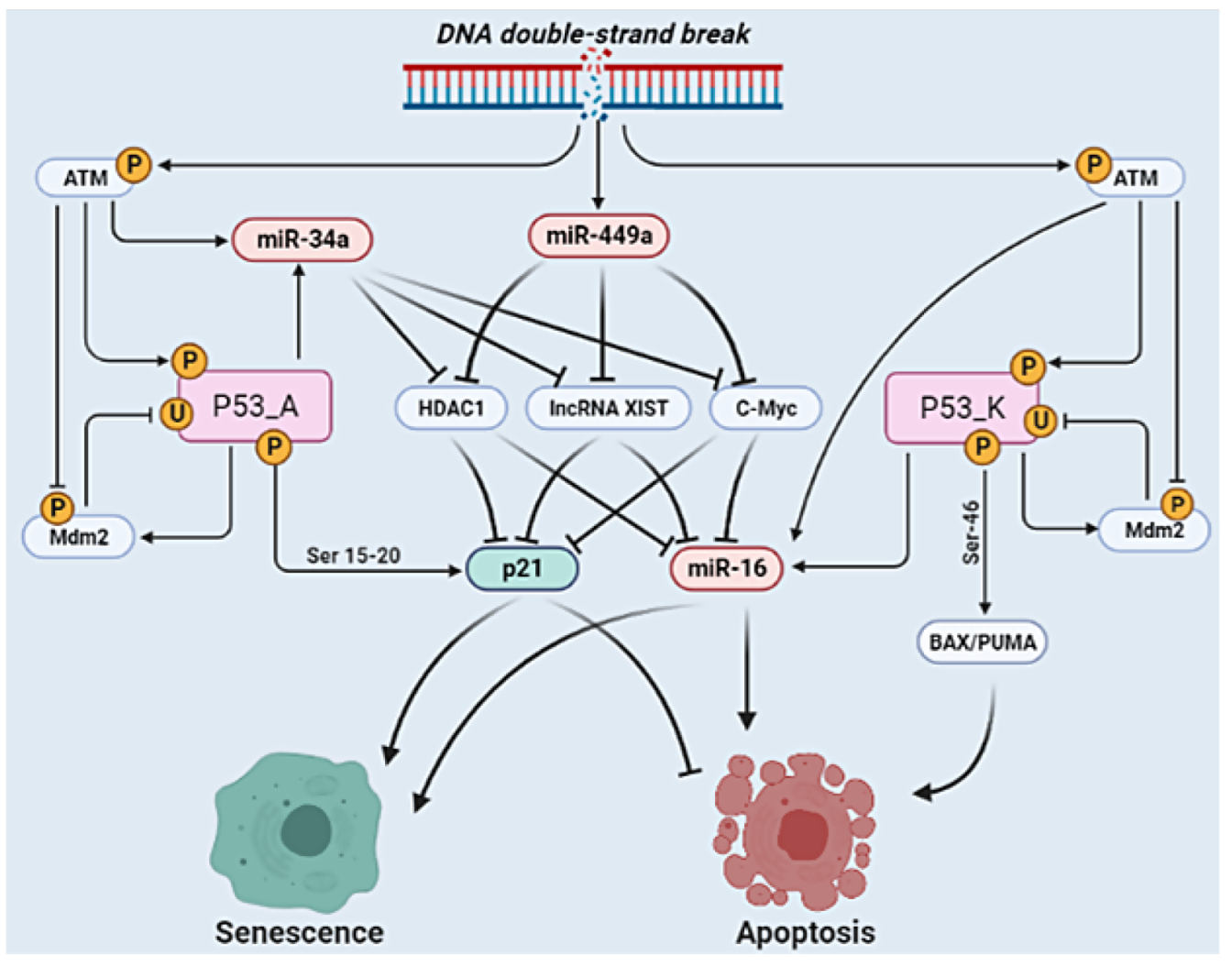
2.1. The Molecular Mechanisms of the G1/S Checkpoint in NSCLC
2.2. Boolean Methods
2.3. Database and Tools
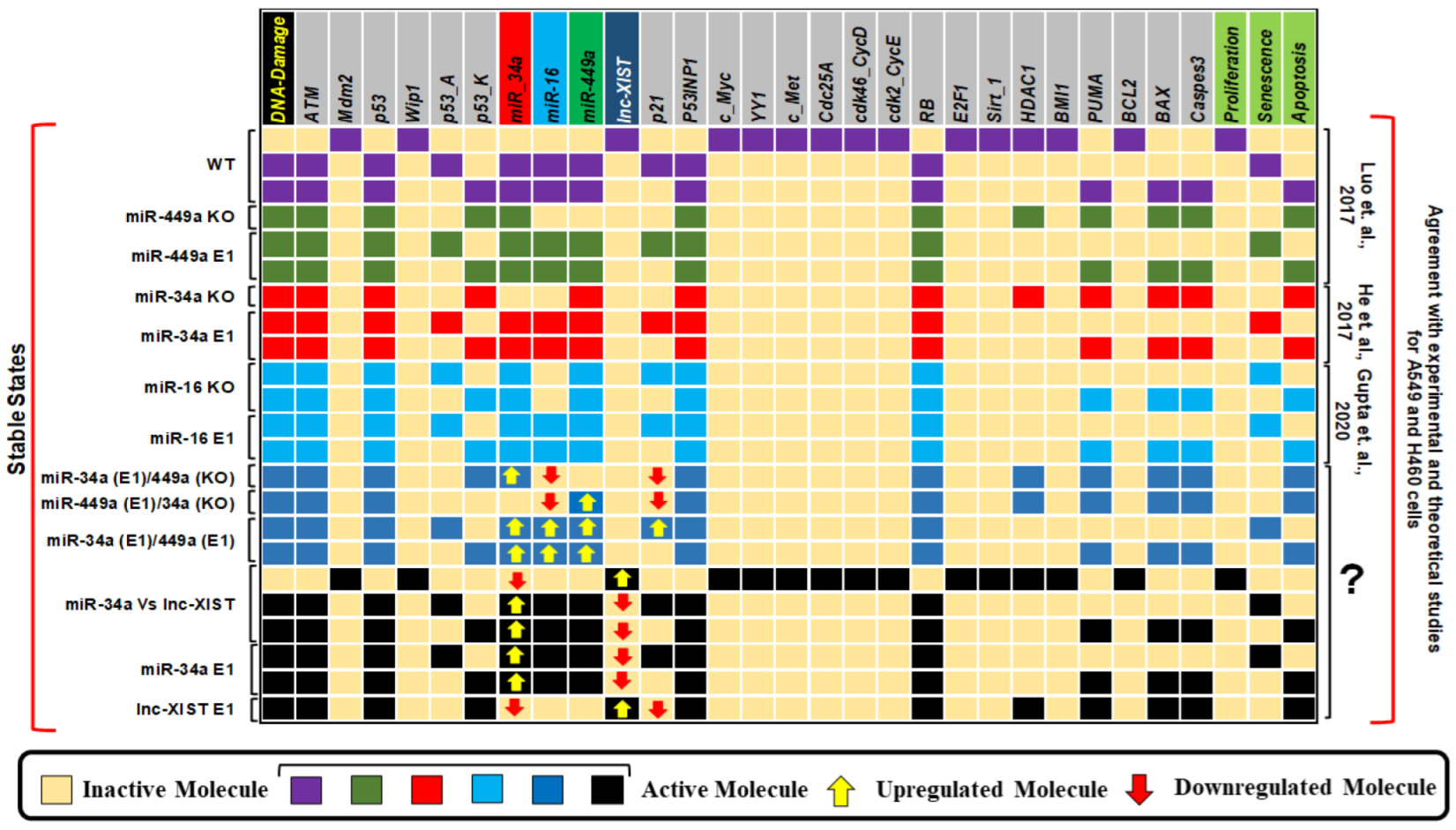
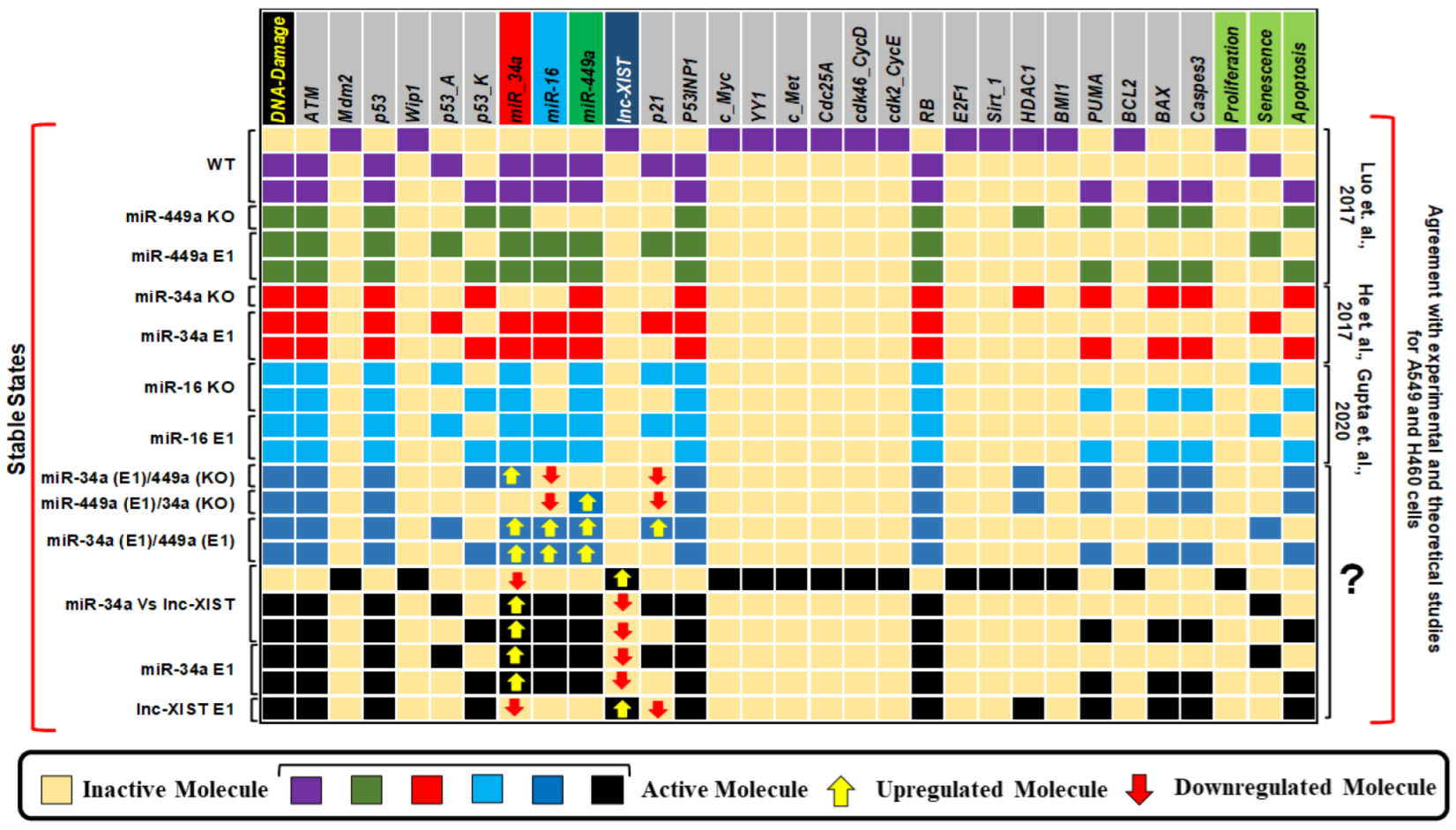
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [Green Version]
- do Valle, Í.F.; Menichetti, G.; Simonetti, G.; Bruno, S.; Zironi, I.; Durso, D.F.; Mombach, J.C.M.; Martinelli, G.; Castellani, G.; Remondini, D. Network integration of multi-tumour omics data suggests novel targeting strategies. Nat. Commun. 2018, 9, 4514. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Ju, H.; Lu, Y.; Chen, L.; Zeng, Z.; Zhang, D.; Luo, H.; Wang, F.; Qiu, M.; Wang, D.; et al. Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J. Exp. Clin. Cancer Res. 2016, 35, 142. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Li, Z.; Ma, K.; Li, X.; Tian, N.; Duan, J.; Xiao, X.; Wang, Y. Long non-coding RNA XIST promotes glioma tumorigenicity and angiogenesis by acting as a molecular sponge of miR-429. J. Cancer 2017, 8, 4106. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, X.; Chen, Z.; Gao, C.; Zhao, L.; Cui, Y. Silencing of lncRNA XIST inhibits non-small cell lung cancer growth and promotes chemosensitivity to cisplatin. Aging (Albany NY) 2020, 12, 4711. [Google Scholar] [CrossRef]
- Tian, L.J.; Wu, Y.P.; Wang, D.; Zhou, Z.H.; Xue, S.B.; Zhang, D.Y.; Wei, Y.G.; Liu, W. Upregulation of long noncoding RNA (lncRNA) X-inactive specific transcript (XIST) is associated with cisplatin resistance in non-small cell lung cancer (NSCLC) by downregulating microRNA-144-3p. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 8095. [Google Scholar] [CrossRef]
- Marshall, E.A.; Stewart, G.L.; Sage, A.P.; Lam, W.L.; Brown, C.J. Beyond sequence homology: Cellular biology limits the potential of XIST to act as a miRNA sponge. PLoS ONE 2019, 14, e0221371. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wan, L.; Liu, Z.; Xu, G.; Wang, S.; Su, Z.; Zhang, Y.; Zhang, C.; Liu, X.; Lei, Z.; et al. Long non-coding RNA XIST promotes TGF-β-induced epithelial-mesenchymal transition by regulating miR-367/141-ZEB2 axis in non-small-cell lung cancer. Cancer Lett. 2018, 418, 185–195. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, G.; Cheng, Z.; Dai, L.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA-XIST suppresses proliferation and TGF-β1-induced EMT in NSCLC through the Notch-1 pathway by regulation of miR-137. Genet. Test. Mol. Biomark. 2018, 22, 333–342. [Google Scholar] [CrossRef]
- Wang, J.; Cai, H.; Dai, Z.; Wang, G. Down-regulation of lncRNA XIST inhibits cell proliferation via regulating miR-744/RING1 axis in non-small cell lung cancer. Clin. Sci. 2019, 133, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Modeling the role of microRNA-449a in the regulation of the G2/M cell cycle checkpoint in prostate LNCaP cells under ionizing radiation. PLoS ONE 2018, 13, e0200768. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Towards DNA-damage induced autophagy: A Boolean model of p53-induced cell fate mechanisms. DNA Repair 2020, 96, 102971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Hou, Y.; Fang, N.; You, J.; Zhou, Q. The lncRNA XIST exhibits oncogenic properties via regulation of miR-449a and Bcl-2 in human non-small cell lung cancer. Acta Pharmacol. Sin. 2017, 38, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Xu, X.; Gao, C.; Cui, Y. XIST promote the proliferation and migration of non-small cell lung cancer cells via sponging miR-16 and regulating CDK8 expression. Am. J. Transl. Res. 2019, 11, 6196–6206. [Google Scholar]
- Liu, H.; Deng, H.; Zhao, Y.; Li, C.; Liang, Y. LncRNA XIST/miR-34a axis modulates the cell proliferation and tumor growth of thyroid cancer through MET-PI3K-AKT signaling. J. Exp. Clin. Cancer Res. 2018, 37, 279. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Barbé-Tuana, F.M.; Mombach, J.C.M. Integrative data modeling from lung and lymphatic cancer predicts functional roles for miR-34a and miR-16 in cell fate regulation. Sci. Rep. 2020, 10, 2511. [Google Scholar] [CrossRef] [Green Version]
- Lizé, M.; Klimke, A.; Dobbelstein, M. MicroRNA-449 in cell fate determination. Cell Cycle 2011, 10, 2874–2882. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Zhang, Z.; Pan, L.; Zhang, Y. MicroRNA-34/449 family and viral infections. Virus Res. 2019, 260, 1–6. [Google Scholar] [CrossRef]
- Aqeilan, R.; Calin, G.; Croce, C. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2010, 17, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Mao, A.; Zhao, Q.; Zhou, X.; Sun, C.; Si, J.; Zhou, R.; Gan, L.; Zhang, H. MicroRNA-449a enhances radiosensitivity by downregulation of c-Myc in prostate cancer cells. Sci. Rep. 2016, 6, 27346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhlouf, M.; Ouimette, J.F.; Oldfield, A.; Navarro, P.; Neuillet, D.; Rougeulle, C. A prominent and conserved role for YY1 in Xist transcriptional activation. Nat. Commun. 2014, 5, 4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, J.; Ge, X.; Geng, Y.; Cao, H.; Zhu, W.; Jiao, Y.; Wu, J.; Zhou, J.; Cao, J. miR-34a inhibits the migration and invasion of esophageal squamous cell carcinoma by targeting Yin Yang-1. Oncol. Rep. 2015, 34, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Jiang, C.; Zhu, J.; Xu, K.; Ren, X.; Xu, L.; Hu, P.; Wang, B.; Yuan, Q.; Guo, Y.; et al. miR-449a inhibits cell proliferation, migration, and inflammation by regulating high-mobility group box protein 1 and forms a mutual inhibition loop with Yin Yang 1 in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Res. Ther. 2019, 21, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grönroos, E.; Terentiev, A.A.; Punga, T.; Ericsson, J. YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 2004, 101, 12165–12170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.Y.; Joo, H.J.; Park, J.K.; Kim, Y.H. The blocking of c-Met signaling induces apoptosis through the increase of p53 protein in lung cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2012, 44, 251. [Google Scholar] [CrossRef] [PubMed]
- Imani, S.; Wu, R.C.; Fu, J. MicroRNA-34 family in breast cancer: From research to therapeutic potential. J. Cancer 2018, 9, 3765. [Google Scholar] [CrossRef] [Green Version]
- Abou-Jaoudé, W.; Traynard, P.; Monteiro, P.T.; Saez-Rodriguez, J.; Helikar, T.; Thieffry, D.; Chaouiya, C. Logical modeling and dynamical analysis of cellular networks. Front. Genet. 2016, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Naldi, A.; Hernandez, C.; Abou-Jaoudé, W.; Monteiro, P.T.; Chaouiya, C.; Thieffry, D. Logical modeling and analysis of cellular regulatory networks with ginsim 3.0. Front. Physiol. 2018, 9, 646. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Hamberg, M.; Backes, C.; Fehlmann, T.; Hart, M.; Meder, B.; Meese, E.; Keller, A. MiRTargetLink—miRNAs, genes and interaction networks. Int. J. Mol. Sci. 2016, 17, 564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. ATM/miR-34a-5p axis regulates a p21-dependent senescence-apoptosis switch in non-small cell lung cancer: A Boolean model of G1/S checkpoint regulation. FEBS Lett. 2020, 594, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Silveira, D.A.; Gupta, S.; Mombach, J.C.M. p53/E2F1/miR-25 axis regulates apoptosis induction in glioblastoma cells: A qualitative model. J. Phys. Complex. 2020, 1, 035001. [Google Scholar] [CrossRef]
- He, X.; Yang, A.; McDonald, D.G.; Riemer, E.C.; Vanek, K.N.; Schulte, B.A.; Wang, G.Y. MiR-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget 2017, 8, 69797. [Google Scholar] [CrossRef]
- Bandi, N.; Zbinden, S.; Gugger, M.; Arnold, M.; Kocher, V.; Hasan, L.; Kappeler, A.; Brunner, T.; Vassella, E. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non–small cell lung cancer. Cancer Res. 2009, 69, 5553–5559. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Huang, B.; Li, Z.; Li, H.; Sun, L.; Zhang, Q.; Qiu, X.; Wang, E. MicroRNA-449a is downregulated in non-small cell lung cancer and inhibits migration and invasion by targeting c-Met. PLoS ONE 2013, 8, e64759. [Google Scholar] [CrossRef] [Green Version]
- Bandi, N.; Vassella, E. miR-34a and miR-15a/16 are co-regulated in non-small cell lung cancer and control cell cycle progression in a synergistic and Rb-dependent manner. Mol. Cancer 2011, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, B.; Adamiok, A.; Mercey, O.; Revinski, D.R.; Zaragosi, L.E.; Pasini, A.; Kodjabachian, L.; Barbry, P.; Marcet, B. miR-34/449 control apical actin network formation during multiciliogenesis through small GTPase pathways. Nat. Commun. 2015, 6, 8386. [Google Scholar] [CrossRef] [Green Version]
- Coller, H.A.; Forman, J.J.; Legesse-Miller, A. “Myc’ed messages”: Myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genet. 2007, 3, e146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fujita, N.; Tsuruo, T. Caspase-mediated cleavage of p21 Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 1999, 18, 1131–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.R.; Yu, L.R.; Tsang, P.; Wei, J.S.; Song, Y.K.; Cheuk, A.; Chung, J.Y.; Hewitt, S.M.; Veenstra, T.D.; Khan, J. Systematic proteome analysis identifies transcription factor YY1 as a direct target of miR-34a. J. Proteome Res. 2011, 10, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844. [Google Scholar]
- Pomerening, J.R. Positive-feedback loops in cell cycle progression. FEBS Lett. 2009, 583, 3388–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänicke, R.U.; Sohn, D.; Essmann, F.; Schulze-Osthoff, K. The multiple battles fought by anti-apoptotic p21. Cell Cycle 2007, 6, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Bar-Or, R.L.; Maya, R.; Segel, L.A.; Alon, U.; Levine, A.J.; Oren, M. Generation of oscillations by the p53-Mdm2 feedback loop: A theoretical and experimental study. Proc. Natl. Acad. Sci. USA 2000, 97, 11250–11255. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Valdecanas, D.; Niknam, S.; Peltier, H.J.; Diao, L.; Giri, U.; Komaki, R.; Calin, G.A.; Gomez, D.R.; Chang, J.Y.; et al. In vivo delivery of miR-34a sensitizes lung tumors to radiation through RAD51 regulation. Mol. Ther.-Nucleic Acids 2015, 4, e270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.H.; Chai, K.; Tashiro, S.I.; Onodera, S.; Ikejima, T. Inhibition of c-M et promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A 549 lung cancer cells. J. Pharm. Pharmacol. 2013, 65, 1622–1642. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Yu, J.; Artandi, S.; Calame, K. YY1 and c-Myc associate in vivo in a manner that depends on c-Myc levels. Proc. Natl. Acad. Sci. USA 1996, 93, 10638–10641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Day, M.L. RB activation and repression of C-MYC transcription precede apoptosis of human prostate epithelial cells. Urology 2001, 57, 860–865. [Google Scholar] [CrossRef]
- Williams, M.; Cheng, Y.Y.; Kirschner, M.B.; Sarun, K.H.; Schelch, K.; Winata, P.; McCaughan, B.; Kao, S.; Van Zandwijk, N.; Reid, G. Transcriptional suppression of the miR-15/16 family by c-Myc in malignant pleural mesothelioma. Oncotarget 2019, 10, 4125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fu, H.; Sun, F.; Zhang, H.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008, 36, 5391. [Google Scholar] [CrossRef]
- Vigneron, A.; Cherier, J.; Barré, B.; Gamelin, E.; Coqueret, O. The cell cycle inhibitor p21waf1 binds to the myc and cdc25A promoters upon DNA damage and induces transcriptional repression. J. Biol. Chem. 2006, 281, 34742–34750. [Google Scholar] [CrossRef] [Green Version]
- Shamloo, B.; Usluer, S. p21 in cancer research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Mikami, K.; Medova, M.; Nisa, L.; Francica, P.; Glück, A.A.; Tschan, M.; Blaukat, A.; Bladt, F.; Aebersold, D.; Zimmer, Y. Impact of p53 Status on Radiosensitization of Tumor Cells by MET Inhibition—Associated Checkpoint Abrogation. Mol. Cancer Res. 2015, 13, 1544–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Hashimoto, R.F. Dynamical Analysis of a Boolean Network Model of the Oncogene Role of lncRNA ANRIL and lncRNA UFC1 in Non-Small Cell Lung Cancer. Biomolecules 2022, 12, 420. [Google Scholar] [CrossRef]
- Silveira, D.A.; Gupta, S.; Mombach, J.C.M. Systems biology approach suggests new miRNAs as phenotypic stability factors in the epithelial–mesenchymal transition. J. R. Soc. Interface 2020, 17, 20200693. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Salzman, D.W.; Nakamura, K.; Nallur, S.; Dookwah, M.T.; Metheetrairut, C.; Slack, F.J.; Weidhaas, J.B. miR-34 activity is modulated through 5’-end phosphorylation in response to DNA damage. Nat. Commun. 2016, 7, 10954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wan, G.; Berger, F.G.; He, X.; Lu, X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol. Cell 2011, 41, 371–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063. [Google Scholar] [CrossRef]
- Xu, T.; Jiang, W.; Fan, L.; Gao, Q.; Li, G. Upregulation of long noncoding RNA Xist promotes proliferation of osteosarcoma by epigenetic silencing of P21. Oncotarget 2017, 8, 101406. [Google Scholar] [CrossRef]
- Jeong, J.H.; Kang, S.S.; Park, K.K.; Chang, H.W.; Magae, J.; Chang, Y.C. p53-independent induction of G1 arrest and p21WAF1/CIP1 expression by ascofuranone, an isoprenoid antibiotic, through downregulation of c-Myc. Mol. Cancer Ther. 2010, 9, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Sampath, D.; Liu, C.; Vasan, K.; Sulda, M.; Puduvalli, V.K.; Wierda, W.G.; Keating, M.J. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood J. Am. Soc. Hematol. 2012, 119, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.; Dalton, W.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noonan, E.; Place, R.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Lammers, P.; Torrance, C.J.; Bader, A.G. TP53-independent Function of miR-34a via HDAC1 and p21CIP1/WAF1. Mol. Ther. 2013, 21, 1678–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Stimulus/Perturbations | Model Response/Phenotype | Cell Lines | References |
|---|---|---|---|
| miR-34a in the absence of DNA Damage | Proliferation | A549, H460 | [36] |
| miR-34a in the presence of DNA Damage | Triggers senescence and apoptosis | A549, H460 | [36] |
| miR-34a Vs c-Myc in response to DNA Damage | Negative correlation | A549, H460 | [36] |
| miR-34a E1 | Induces Senescence and Apoptosis | A549, H460 | [36] |
| c-Myc KO | Activating Senescence and Apoptosis | A549, H460 | [36] |
| miR-16 E1 and RB E1 | Induces Senescence and Apoptosis | A549 | [37] |
| miR-16 E1 and RB KO | Induces only Apoptosis | A549 | [37] |
| miR-16 E1 | Activating Senescence and Apoptosis | A549 | [37] |
| miR-449a Vs C-Met in response to DNA Damage | Negative correlation | A549 | [38] |
| miR-449a KO | Proliferation | A549 | [38] |
| miR-449a E1 | Induces Senescence and Apoptosis | A549 | [38] |
| C-Met KO | Activating Senescence and Apoptosis | A549 | [38] |
| miR-34a E1 and miR-16 E1 | Activating Senescence and Apoptosis | A549 | [39] |
| miR-34a E1 and RB E1 | Induces Senescence and Apoptosis | A549 | [39] |
| miR-34a KO and miR-16 KO | Induces only Apoptosis | A549 | [39] |
| RB E1 | Induces Senescence and Apoptosis | A549 | [39] |
| miR-34a KO and miR-449a KO | Proliferation | A549 | [40] |
| miR-34a E1 and miR-449a E1 | Induces Senescence and Apoptosis | A549 | [40] |
| miR-449a Vs lncRNA XIST in DNA Damage Response | Negative correlation | A549, H460 | [14] |
| miR-449a E1 | Activating Senescence and Apoptosis | A549, H460 | [14] |
| lncRNA XIST KO | Induces Senescence and Apoptosis | A549, H460 | [14] |
| miR-16 Vs lncRNA XIST in DNA Damage Response | Negative correlation | A549 | [15] |
| miR-16 E1 | Triggers senescence and apoptosis | A549 | [15] |
| lncRNA XIST KO | Induces Senescence and Apoptosis | A549 | [15] |
| Number | Circuits | Reference |
|---|---|---|
| Positive | ||
| 1 | c-Myc/E2F1 | [41] |
| 2 | p21/Caspase3 | [42] |
| 3 | ATM/miR-34a/HDAC1 | [34] |
| 4 | miR-449a/lncRNA XIST | ? |
| 5 | miR-16/lncRNA XIST | ? |
| 6 | miR-34a/lncRNA XIST | ? |
| 7 | p53/miR-34a/C-Met | ? |
| 8 | miR-34a/YY1/lncRNA XIST | ? |
| 9 | YY1/p53/miR-34a/c-Myc | ? |
| 10 | YY1/lncRNA XIST/miR-34a/c-Myc | ? |
| 11 | CDK4-6-CycD/RB/c-Myc/miR-16 | ? |
| 12 | CDK2-CycE/RB/c-Myc/miR-16 | ? |
| 13 | p21/c-Myc/YY1/p53 | ? |
| 14 | p21/c-Myc/c-Met/p53 | ? |
| 15 | miR-449a/c-Myc/YY1/lncRNA XIST | ? |
| 16 | p53-K/p53-A | [33] |
| 17 | p53/miR-34a/YY1 | [43] |
| 18 | p53/miR-34a/Sirt1 | [44] |
| 19 | E2F1/ATM | [45] |
| 20 | E2F1/CDK2-CycE/RB | [46] |
| 21 | p21/c-Myc | [47] |
| Negative | ||
| 22 | p53/Mdm2 | [48] |
| 23 | p53INP1/p53-A | [33] |
| 24 | ATM/miR-34a/E2F1 | [34] |
| 25 | E2F1/Sirt1 | [49] |
| Positive Circuits | Circuit Molecules | Target | Direct/indirect Interaction | References |
|---|---|---|---|---|
| miR-449a/lncRNA XIST | miR-449a | lncRNA XIST | Direct inhibition | [14] |
| lncRNA XIST | miR-449a | Direct inhibition | [14] | |
| miR-16/lncRNA XIST | miR-16 | lncRNA XIST | Direct inhibition | [15] |
| lncRNA XIST | miR-16 | Direct inhibition | [15] | |
| miR-34a/lncRNA XIST | miR-34a | lncRNA XIST | Direct inhibition | [16] |
| lncRNA XIST | miR-34a | Direct inhibition | [16] | |
| p53/miR-34a/C-Met | p53 | miR-34a | Direct activation | [50] |
| miR-34a | C-Met | Direct inhibition | [51] | |
| C-Met | p53 | Direct inhibition | [52] | |
| miR-34a/YY1/lncRNA XIST | miR-34a | YY1 | Direct inhibition | [43] |
| YY1 | lncRNA XIST | Direct activation | [22] | |
| lncRNA XIST | miR-34a | Direct inhibition | [16] | |
| YY1/p53/miR-34a/c-Myc | YY1 | p53 | Direct inhibition | [25] |
| p53 | miR-34a | Direct activation | [50] | |
| miR-34a | c-Myc | Direct inhibition | [36] | |
| c-Myc | YY1 | Direct activation | [53] | |
| YY1/lncRNA XIST/miR-34a/c-Myc | YY1 | lncRNA XIST | Direct activation | [22] |
| lncRNA XIST | miR-34a | Direct inhibition | [16] | |
| miR-34a | c-Myc | Direct inhibition | [36] | |
| c-Myc | YY1 | Direct activation | [53] | |
| CDK4-6-CycD/RB/c-Myc/miR-16 | CDK4-6-CycD | RB | Direct inhibition | [54] |
| RB | c-Myc | Direct inhibition | [55] | |
| c-Myc | miR-16 | Direct inhibition | [56] | |
| miR-16 | CDK4-6-CycD | Direct inhibition | [57] | |
| CDK2-CycE/RB/c-Myc/miR-16 | CDK2-cycE | RB | Direct inhibition | [54] |
| RB | c-Myc | Direct inhibition | [55] | |
| c-Myc | miR-16 | Direct inhibition | [56] | |
| miR-16 | CDK2-CycE | Direct inhibition | [57] | |
| p21/c-Myc/YY1/p53 | p21 | c-Myc | Direct inhibition | [58] |
| c-Myc | YY1 | Direct activation | [53] | |
| YY1 | p53 | Direct inhibition | [25] | |
| p53 | p21 | Direct activation | [59] | |
| p21/c-Myc/c-Met/p53 | p21 | c-Myc | Direct inhibition | [58] |
| c-Myc | C-Met | Direct activation | [60] | |
| C-Met | p53 | Direct inhibition | [61] | |
| p53 | p21 | Direct activation | [59] | |
| miR-449a/c-Myc/YY1/lncRNA XIST | miR-449a | c-Myc | Direct inhibition | [21] |
| c-Myc | YY1 | Direct activation | [53] | |
| YY1 | lncRNA XIST | Direct activation | [22] | |
| lncRNA XIST | miR-449a | Direct inhibition | [14] |
| Positive Circuits | Removed Interactions | Phenotype Observation |
|---|---|---|
| miR-449a/lncRNA XIST | miR-449a/lncRNA XIST | Senescence and Apoptosis |
| lncRNA XIST/miR-449a | Senescence and Apoptosis | |
| miR-16/lncRNA XIST | miR-16/lncRNA XIST | Senescence and Apoptosis |
| lncRNA XIST/miR-16 | Senescence and Apoptosis | |
| miR-34a/lncRNA XIST | miR-34a/lncRNA XIST | Senescence and Apoptosis |
| lncRNA XIST/miR-34a | Senescence and Apoptosis | |
| p53/miR-34a/C-Met | p53/miR-34a | Senescence and Apoptosis |
| miR-34a/C-Met | Senescence and Apoptosis | |
| C-Met/p53 | Senescence and Apoptosis | |
| miR-34a/YY1/lncRNA XIST | miR-34a/YY1 | Senescence and Apoptosis |
| YY1/lncRNA XIST | Two states for Apoptosis and one state for senescence | |
| lncRNA XIST/miR-34a | Senescence and Apoptosis | |
| YY1/p53/miR-34a/c-Myc | YY1/p53 | Senescence and Apoptosis |
| p53/miR-34a | Senescence and Apoptosis | |
| miR-34a/c-Myc | Senescence and Apoptosis | |
| c-Myc/YY1 | Senescence and Apoptosis | |
| YY1/lncRNA XIST/miR-34a/c-Myc | YY1/lncRNA XIST | Two states for Apoptosis and one state for senescence |
| lncRNA XIST/miR-34a | Senescence and Apoptosis | |
| miR-34a/c-Myc | Senescence and Apoptosis | |
| c-Myc/YY1 | Senescence and Apoptosis | |
| CDK4-6-CycD/RB/c-Myc/miR-16 | CDK4-6-CycD/RB | Senescence and Apoptosis |
| RB/c-Myc | 4 States (2 apoptotic and 1 senescence and other 1) | |
| c-Myc/miR-16 | Senescence and Apoptosis | |
| miR-16/CDK4-6-CycD | Senescence and Apoptosis | |
| CDK2-CycE/RB/c-Myc/miR-16 | CDK2-CycE/RB | Senescence and Apoptosis |
| RB/c-Myc | 4 States (2 apoptotic and 1 senescence and other 1) | |
| c-Myc/miR-16 | Senescence and Apoptosis | |
| miR-16/CDK2-CycE | Senescence and Apoptosis | |
| p21/c-Myc/YY1/p53 | p21/c-Myc | Senescence and Apoptosis |
| c-Myc/YY1 | Senescence and Apoptosis | |
| YY1/p53 | Senescence and Apoptosis | |
| p53/p21 | Senescence and Apoptosis | |
| p21/c-Myc/c-Met/p53 | p21/c-Myc | Senescence and Apoptosis |
| c-Myc/C-Met | Senescence and Apoptosis | |
| C-Met/p53 | Senescence and Apoptosis | |
| p53/p21 | Senescence and Apoptosis | |
| miR-449a/c-Myc/YY1/lncRNA XIST | miR-449a/c-Myc | Senescence and Apoptosis |
| c-Myc/YY1 | Senescence and Apoptosis | |
| YY1/lncRNA XIST | Two states for Apoptosis and one state for senescence | |
| lncRNA XIST/miR-449a | Senescence and Apoptosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Silveira, D.A.; Hashimoto, R.F.; Mombach, J.C.M. A Boolean Model of the Proliferative Role of the lncRNA XIST in Non-Small Cell Lung Cancer Cells. Biology 2022, 11, 480. https://doi.org/10.3390/biology11040480
Gupta S, Silveira DA, Hashimoto RF, Mombach JCM. A Boolean Model of the Proliferative Role of the lncRNA XIST in Non-Small Cell Lung Cancer Cells. Biology. 2022; 11(4):480. https://doi.org/10.3390/biology11040480
Chicago/Turabian StyleGupta, Shantanu, Daner A. Silveira, Ronaldo F. Hashimoto, and Jose Carlos M. Mombach. 2022. "A Boolean Model of the Proliferative Role of the lncRNA XIST in Non-Small Cell Lung Cancer Cells" Biology 11, no. 4: 480. https://doi.org/10.3390/biology11040480
APA StyleGupta, S., Silveira, D. A., Hashimoto, R. F., & Mombach, J. C. M. (2022). A Boolean Model of the Proliferative Role of the lncRNA XIST in Non-Small Cell Lung Cancer Cells. Biology, 11(4), 480. https://doi.org/10.3390/biology11040480