
Spatial Metagenomic Analysis in Understanding the Microbial Diversity of Thar Desert



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sampling
2.2. Physicochemical Analyses
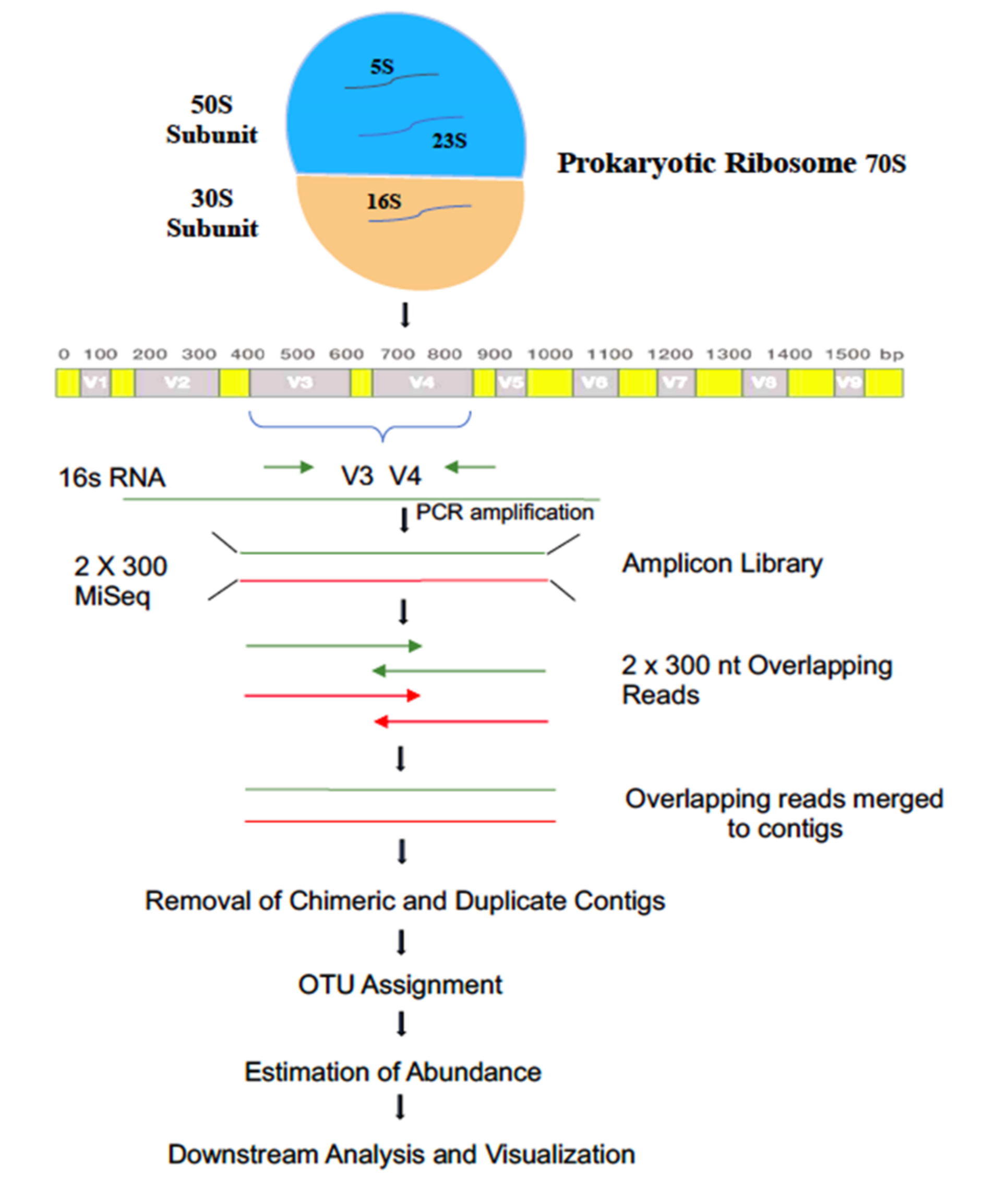
2.3. DNA Extraction, PCR Amplification, and Illumina MiSeq Sequencing
2.4. Technical Validation
2.5. Phylotype Analysis
2.6. Sequence Data Quality Control
2.7. NGS Analyses
3. Results and Discussions
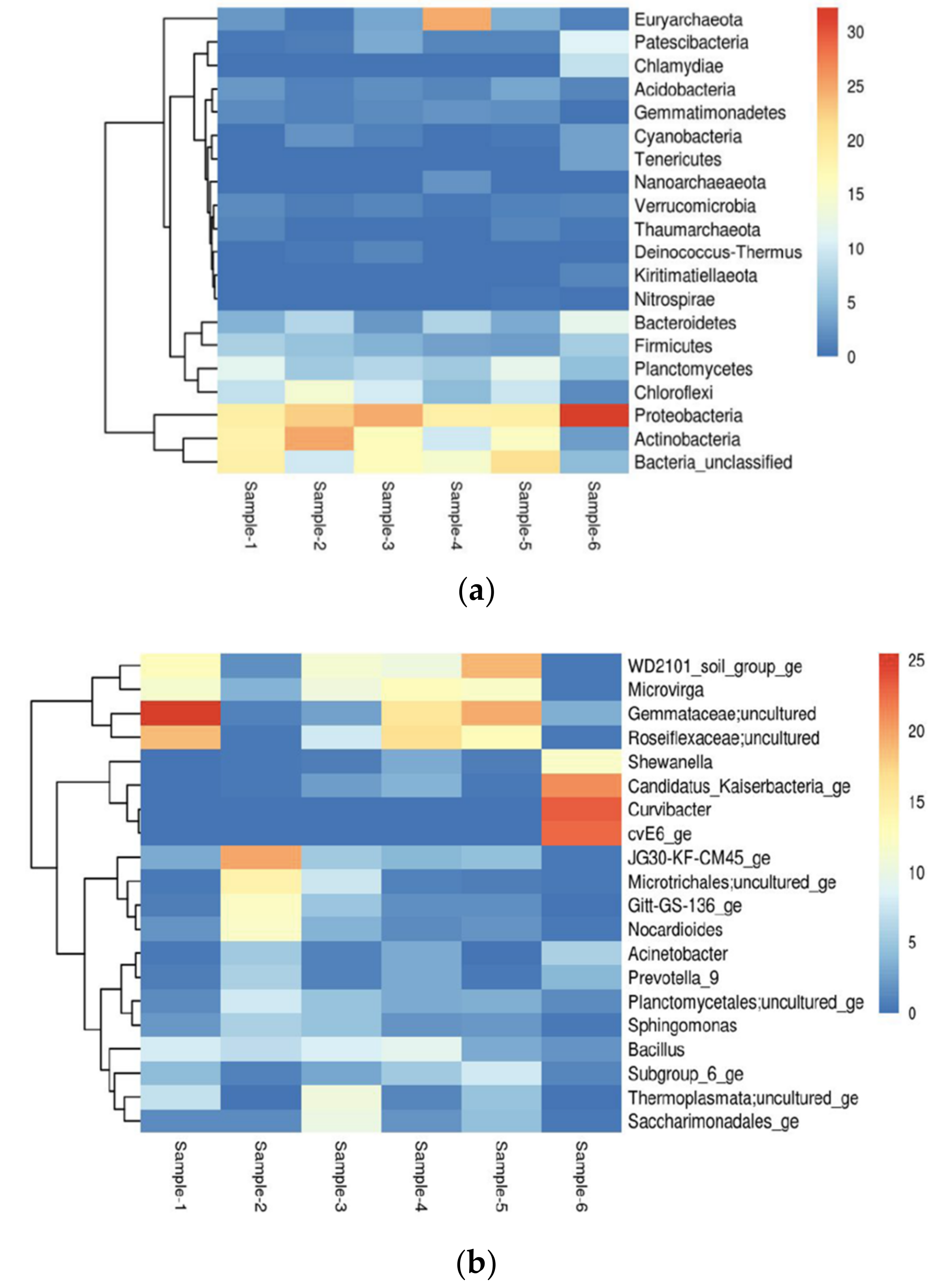
3.1. Microbial Structure, Diversity and Richness
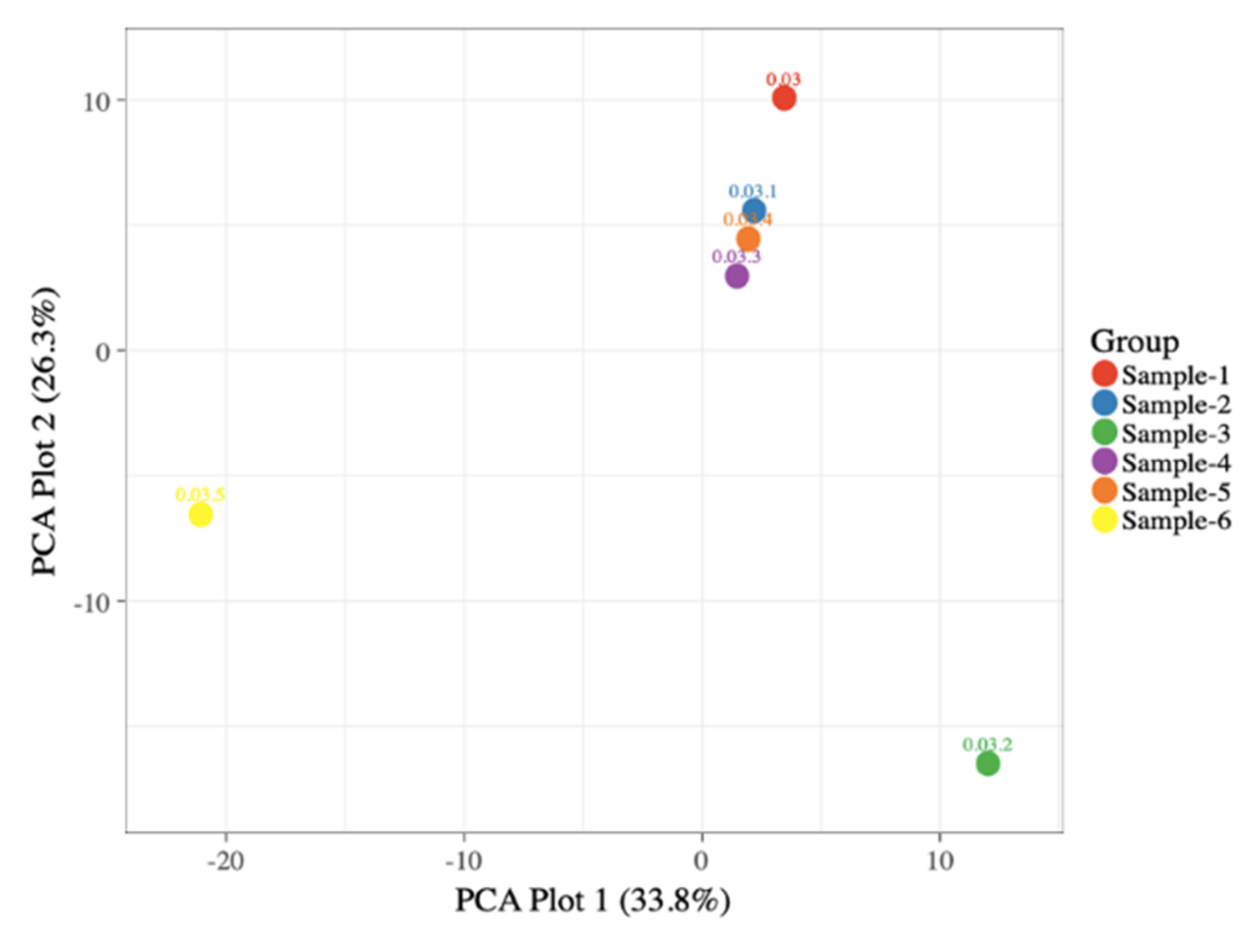
3.2. Alpha Diversity and Beta Diversity Analysis
3.3. Comparative Analysis from Different Deserts

4. Conclusions and Future Aspects
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, S.; Suyal, D.C.; Yadav, A.; Shouche, Y.; Goel, R. Microbial diversity and soil physiochemical characteristic of higher altitude. PLoS ONE 2019, 14, e0213844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghadikolaei, K.K.; Gharechahi, J.; Haghbeen, K.; Akbari, N.K.; Hosseini, S.G.; Shahbani, Z.H. A cold-adapted endoglucanase from camel rumen with high catalytic activity at moderate and low temperatures: An anomaly of truly cold-adapted evolution in a mesophilic environment. Extremophiles 2018, 22, 315–326. [Google Scholar] [CrossRef]
- Escuder-Rodríguez, J.-J.; DeCastro, M.E.; Cerdán, M.E.; Rodríguez-Belmonte, E.; Becerra, M.; González-Siso, M.I. Cellulases from thermophiles found by metagenomics. Microorganisms 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagaria, A.; Parihar, J. The Extremes of Life and Extremozymes: Diversity and Perspectives. Acta Sci. Microbiol. 2019, 3, 13. [Google Scholar]
- Le, P.T.; Makhalanyane, T.P.; Guerrero, L.D.; Vikram, S.; Van de Peer, Y.; Cowan, D.A. Comparative metagenomic analysis reveals mechanisms for stress response in hypoliths from extreme hyperarid deserts. Genome Biol. Evol. 2016, 8, 2737–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parihar, J.; Bagaria, A. Isolation, Identification and Characterization of a Thermo-halotolerant Aeribacillus Pallidus SHJP4, from the Arid Region of Rajasthan. IOP Conf. Ser. Earth Environ. Sci. 2021, 796, 012050. [Google Scholar] [CrossRef]
- Koo, H.; Hakim, J.A.; Morrow, C.D.; Crowley, M.R.; Andersen, D.T.; Bej, A.K. Metagenomic analysis of microbial community compositions and cold-responsive stress genes in selected Antarctic lacustrine and soil ecosystems. Life 2018, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Cowan, D.A.; Ramond, J.B.; Makhalanyane, T.P.; De Maayer, P. Metagenomics of extreme environments. Curr. Opin. Microbiol. 2015, 25, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Delattre, H.; Quéméner, D.L.; Duquennoi, C.; Filali, A.; Bouchez, T. Consistent microbial dynamics and functional community patterns derived from first principles. ISME J. 2019, 13, 263–276. [Google Scholar] [CrossRef]
- Jagdish, P.; Ashok, K.; Kamakhya, P.M.; Ashima, B. Extracellular Synthesis of Zinc Oxide Nanoparticles Using Thermo-Halotolerant Aeribacillus pallidus strain SJP 27: Characterization and Antibacterial Potential. J. Nano Electron. Phys. 2021, 13, 02007. [Google Scholar]
- Batson, J.; Noe, G.B.; Hupp, C.R.; Krauss, K.W.; Rybicki, N.B.; Schenk, E.R. Soil greenhouse gas emissions and carbon budgeting in a short-hydroperiod floodplain wetland. J. Geophys. Res. Biogeosci. 2015, 120, 77–95. [Google Scholar] [CrossRef]
- Li, C.; Fultz, L.M.; Moore-Kucera, J.; Acosta-Martínez, V.; Kakarla, M.; Weindorf, D.C. Soil microbial community restoration in Conservation Reserve Program semi-arid grasslands. Soil Biol. Biochem. 2018, 118, 166–177. [Google Scholar] [CrossRef]
- Delmont, T.O.; Robe, P.; Cecillon, S.; Clark, I.M.; Constancias, F.; Simonet, P.; Hirsch, P.R.; Vogel, T.M. Accessing microbial diversity for soil metagenomic studies. Appl. Environ. Microbiol. 2011, 77, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocali, S.; Benedetti, A. Exploring research frontiers in microbiology: The challenge of metagenomics in soil microbiology. Res. Microbiol. 2010, 161, 497–505. [Google Scholar] [CrossRef]
- Rondon, M.R.; August, P.R.; Bettermann, A.D.; Brady, S.F.; Grossman, T.H.; Liles, M.R.; Loiacono, K.A.; Lynch, B.A.; MacNeil, I.A.; Minor, C.; et al. Cloning the soil metagenome: A strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl. Environ. Microbiol. 2000, 66, 2541–2547. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The impact of next-generation sequencing on genomics. J. Genet. Genom. 2011, 38, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Torsvik, V.; Øvreås, L. Microbial diversity and function in soil: From genes to ecosystems. Curr. Opin. Microbiol. 2002, 5, 240–245. [Google Scholar] [CrossRef]
- Wang, Y.; Osman, J.R.; DuBow, M.S. Bacterial communities on the surface of the mineral sandy soil from the Desert of Maine (USA). Curr. Microbiol. 2020, 77, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hansen, M.A.; Hansen, L.H.; Jacquiod, S.; Sørensen, S.J. Bioinformatic approaches reveal metagenomic characterization of soil microbial community. PLoS ONE 2014, 9, e93445. [Google Scholar]
- McHugh, T.A.; McHugh, T.A.; Compson, Z.; van Gestel, N.; Hayer, M.; Ballard, L.; Haverty, M.; Hines, J.; Irvine, N.; Krassner, D.; et al. Climate controls prokaryotic community composition in desert soils of the southwestern United States. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Buse, H.Y.; Lu, J.; Lu, X.; Mou, X.; Ashbolt, N.J. Microbial diversities (16S and 18S rRNA gene pyrosequencing) and environmental pathogens within drinking water biofilms grown on the common premise plumbing materials unplasticized polyvinylchloride and copper. FEMS Microbiol. Ecol. 2014, 88, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef]
- Wu, S.; Xiong, J.; Yu, Y. Taxonomic resolutions based on 18S rRNA genes: A case study of subclass Copepoda. PLoS ONE 2015, 10, e0131498. [Google Scholar] [CrossRef]
- Balvočiūtė, M.; Huson, D.H. OTT: How do these taxonomies compare. BMC Genom. 2017, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Wang, Y.; Qian, P.-Y. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform. 2016, 17, 135. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Dhakan, D.B.; Mittal, P.; Waiker, P.; Chowdhury, A.; Ghatak, A.; Sharma, V.K. Metagenomic analysis of hot springs in Central India reveals hydrocarbon degrading thermophiles and pathways essential for survival in extreme environments. Front. Microbiol. 2017, 7, 2123. [Google Scholar] [CrossRef]
- Coller, E.; Cestaro, A.; Zanzotti, R.; Bertoldi, D.; Pindo, M.; Larger, S.; Albanese, D.; Mescalchin, E.; Donati, C. Microbiome of vineyard soils is shaped by geography and management. Microbiome 2019, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Orellana, L.H.; Chee-Sanford, J.C.; Sanford, R.A.; Löffler, F.E.; Konstantinidis, K.T. Year-round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl. Environ. Microbiol. 2018, 84, e01646-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsharif, W.; Saad, M.M.; Hirt, H. Desert microbes for boosting sustainable agriculture in extreme environments. Front. Microbiol. 2020, 11, 1666. [Google Scholar] [CrossRef] [PubMed]
- Kambura, A.K.; Mwirichia, R.K.; Kasili, R.W.; Karanja, E.N.; Makonde, H.M.; Boga, H.I. Bacteria and Archaea diversity within the hot springs of Lake Magadi and Little Magadi in Kenya. BMC Microbiol. 2016, 16, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-López, O.; Cerdan, M.E.; Siso, M.I.G. New extremophilic lipases and esterases from metagenomics. Curr. Protein Pept. Sci. 2014, 15, 445–455. [Google Scholar] [CrossRef]
- Cota-Ruiz, K.; Hernández-Viezcas, J.A.; Varela-Ramírez, A.; Valdés, C.; Núñez-Gastélum, J.A.; Martínez-Martínez, A.; Delgado-Rios, M.; Peralta-Videa, J.R.; Gardea-Torresdey, J.L. Toxicity of copper hydroxide nanoparticles, bulk copper hydroxide, and ionic copper to alfalfa plants: A spectroscopic and gene expression study. Environ. Pollut. 2018, 243, 703–712. [Google Scholar] [CrossRef]
- Bandyopadhyay, K.K.; Aggarwal, P.; Chakraborty, D.; Pradhan, S.; Garg, R.N.; Bhattacharyya, R.; Pramanik, P.; Singh, R. Practical Manual on Soil and Water Erosion; Division of Agricultural Physics, Indian Agricultural Research Institute: New Delhi, India, 2013; 46p. [Google Scholar]
- Lindsay, W.L.; Norvell, W. Development of a DTPA soil test for zinc, iron, manganese, and copper 1. Soil Sci. Soc. Am. J. 1978, 42, 421–428. [Google Scholar] [CrossRef]
- Sivakala, K.K.; Jose, P.A.; Anandham, R.; Thinesh, T.; Jebakumar, S.R.D.; Samaddar, S.; Chatterjee, P.; Sivakumar, N.; Sa, T. Spatial physicochemical and metagenomic analysis of desert environment. J. Microbiol. Biotechnol. 2018, 28, 1517–1526. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1. 0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 January 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioinformatics, B. Trim Galore! 2017. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 1 January 2022).
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Hadizadeh, F.; Walter, S.; Belheouane, M.; Bonfiglio, F.; Heinsen, F.A.; Andreasson, A.; Agreus, L.; Engstrand, L.; Baines, J.F.; Rafter, J.; et al. Stool frequency is associated with gut microbiota composition. Gut 2017, 66, 559–560. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Kutovaya, O.V.; Lebedeva, M.P.; Tkhakakhova, A.K.; Ivanova, E.A.; Andronov, E.E. Metagenomic characterization of biodiversity in the extremely arid desert soils of Kazakhstan. Eurasian Soil Sci. 2015, 48, 493–500. [Google Scholar] [CrossRef]
- Vikram, S.; Guerrero, L.D.; Makhalanyane, T.P.; Le, P.T.; Seely, M.; Cowan, D.A. Metagenomic analysis provides insights into functional capacity in a hyperarid desert soil niche community. Environ. Microbiol. 2016, 18, 1875–1888. [Google Scholar] [CrossRef]
- Bull, A.T.; Asenjo, J.A.; Goodfellow, M.; Gomez-Silva, B. The Atacama Desert: Technical resources and the growing importance of novel microbial diversity. Annu. Rev. Microbiol. 2016, 70, 215–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, D.R.; Fitak, R.R.; Munguia-Vega, A.; Racolta, A.; Martinson, V.G.; Dontsova, K. Abiotic factors shape microbial diversity in Sonoran Desert soils. Appl. Environ. Microbiol. 2012, 78, 7527–7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stomeo, F.; Valverde, A.; Pointing, S.B.; McKay, C.P.; Warren-Rhodes, K.A.; Tuffin, M.I.; Seely, M.; Cowan, D.A. Hypolithic and soil microbial community assembly along an aridity gradient in the Namib Desert. Extremophiles 2013, 17, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Chan, Y.; Bugler-Lacap, D.C.; Bhatnagar, A.; Bhatnagar, M.; Pointing, S.B. Microbial diversity in soil, sand dune and rock substrates of the Thar Monsoon Desert, India. Indian J. Microbiol. 2016, 56, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodfellow, M.; Nouioui, I.; Sanderson, R.; Xie, F.; Bull, A.T. Rare taxa and dark microbial matter: Novel bioactive actinobacteria abound in Atacama Desert soils. Antonie Van Leeuwenhoek 2018, 111, 1315–1332. [Google Scholar] [CrossRef]
- Mhete, M.; Eze, P.N.; Rahube, T.O.; Akinyemi, F.O. Soil properties influence bacterial abundance and diversity under different land-use regimes in semi-arid environments. Sci. Afr. 2020, 7, e00246. [Google Scholar] [CrossRef]
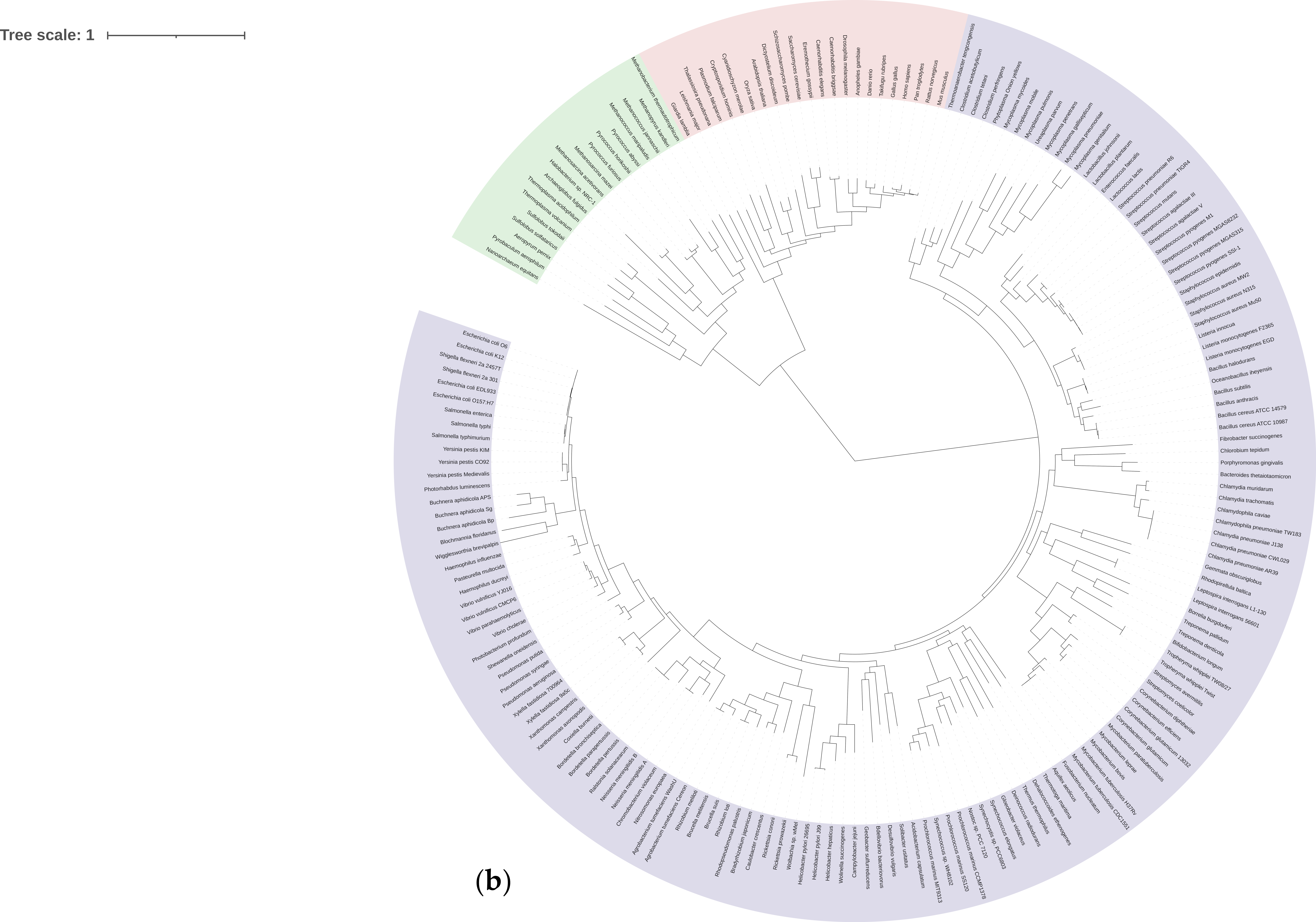
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parihar, J.; Parihar, S.P.; Suravajhala, P.; Bagaria, A. Spatial Metagenomic Analysis in Understanding the Microbial Diversity of Thar Desert. Biology 2022, 11, 461. https://doi.org/10.3390/biology11030461
Parihar J, Parihar SP, Suravajhala P, Bagaria A. Spatial Metagenomic Analysis in Understanding the Microbial Diversity of Thar Desert. Biology. 2022; 11(3):461. https://doi.org/10.3390/biology11030461
Chicago/Turabian StyleParihar, Jagdish, Suraj P. Parihar, Prashanth Suravajhala, and Ashima Bagaria. 2022. "Spatial Metagenomic Analysis in Understanding the Microbial Diversity of Thar Desert" Biology 11, no. 3: 461. https://doi.org/10.3390/biology11030461
APA StyleParihar, J., Parihar, S. P., Suravajhala, P., & Bagaria, A. (2022). Spatial Metagenomic Analysis in Understanding the Microbial Diversity of Thar Desert. Biology, 11(3), 461. https://doi.org/10.3390/biology11030461