
AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium


 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Rats and Diet Protocol
2.2. Mice and Diet Protocol
2.3. Echocardiography
2.4. RNA Extraction
2.5. Real-Time PCR
2.6. Protein Extraction and Western Blotting
2.7. Respirometric Measurements
2.8. Measurement of LV ATP Content
2.9. Plasma Analyses
2.10. Statistical Analysis
3. Results
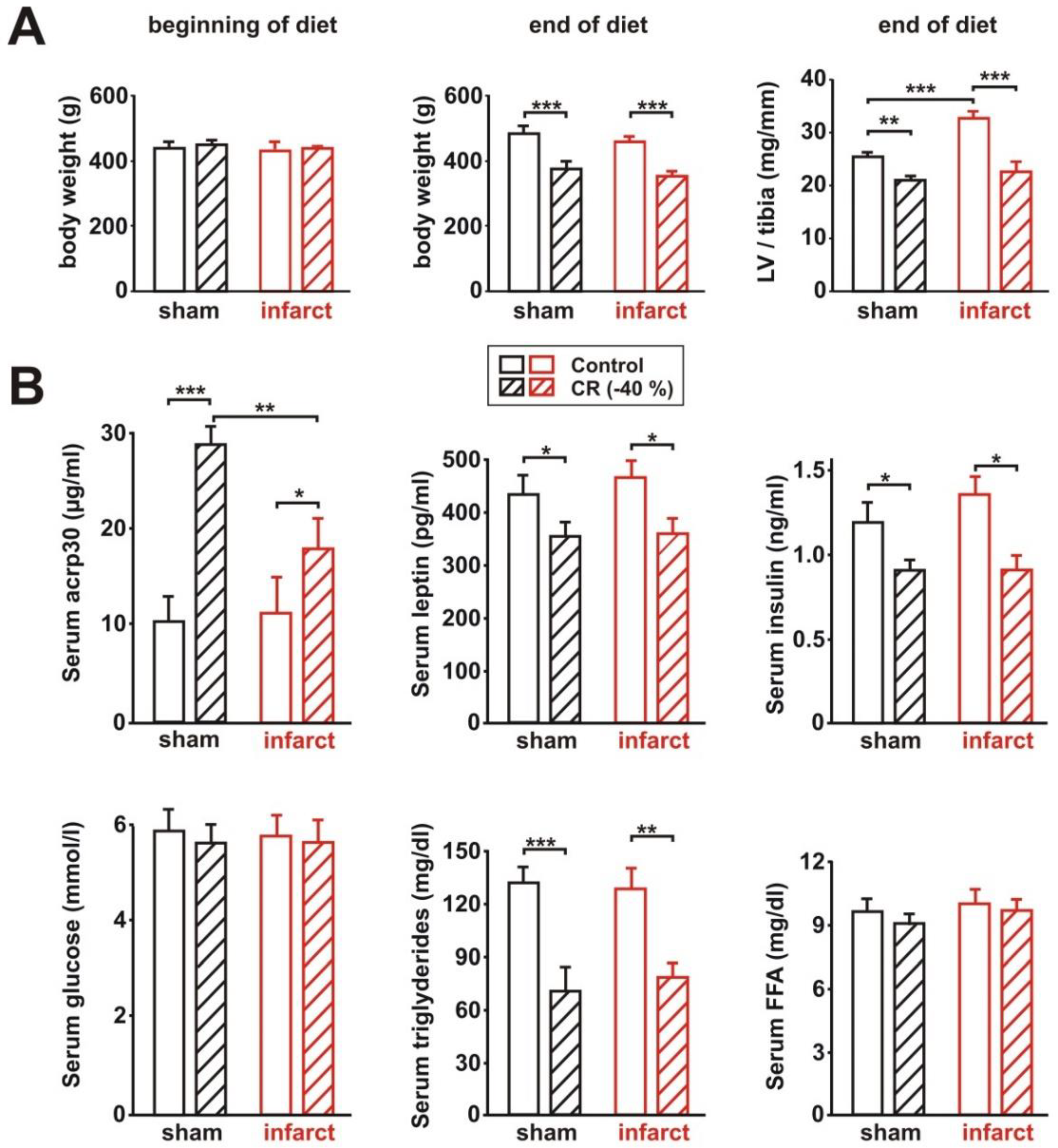
3.1. General Characteristics of the Rat Model
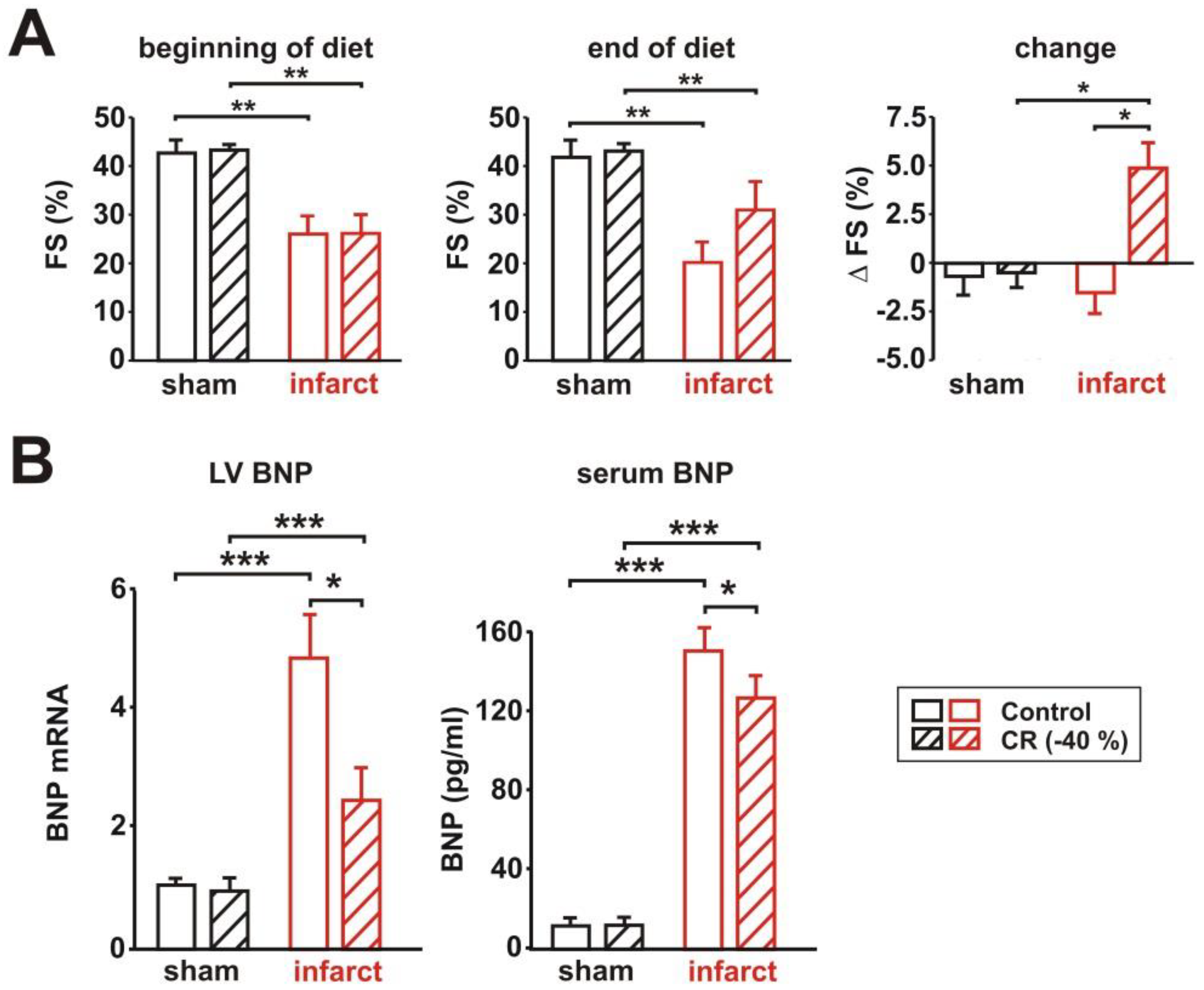
3.2. Characterization of Cardiac Function
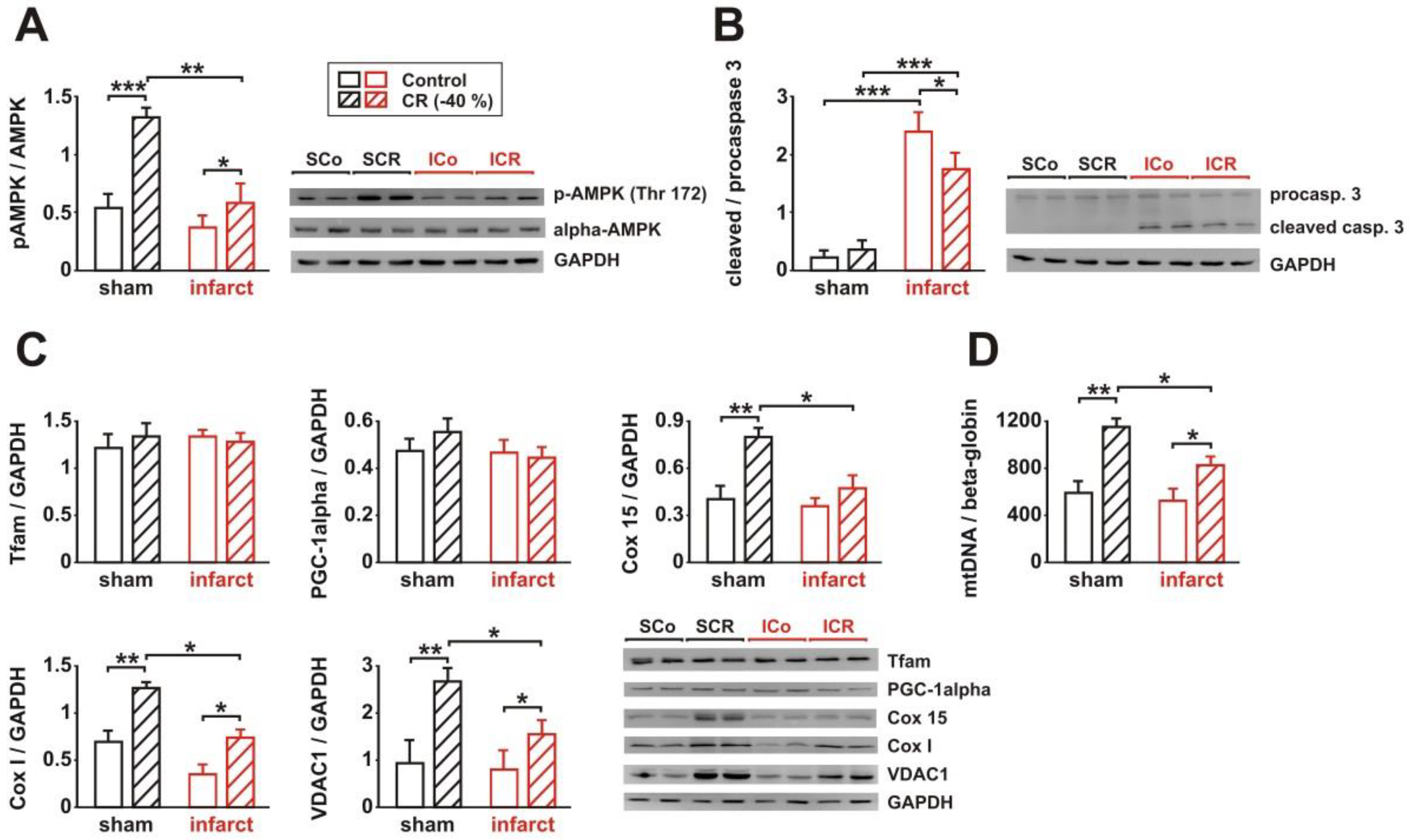
3.3. Signaling Pathway Activation and Mitochondrial Biogenesis
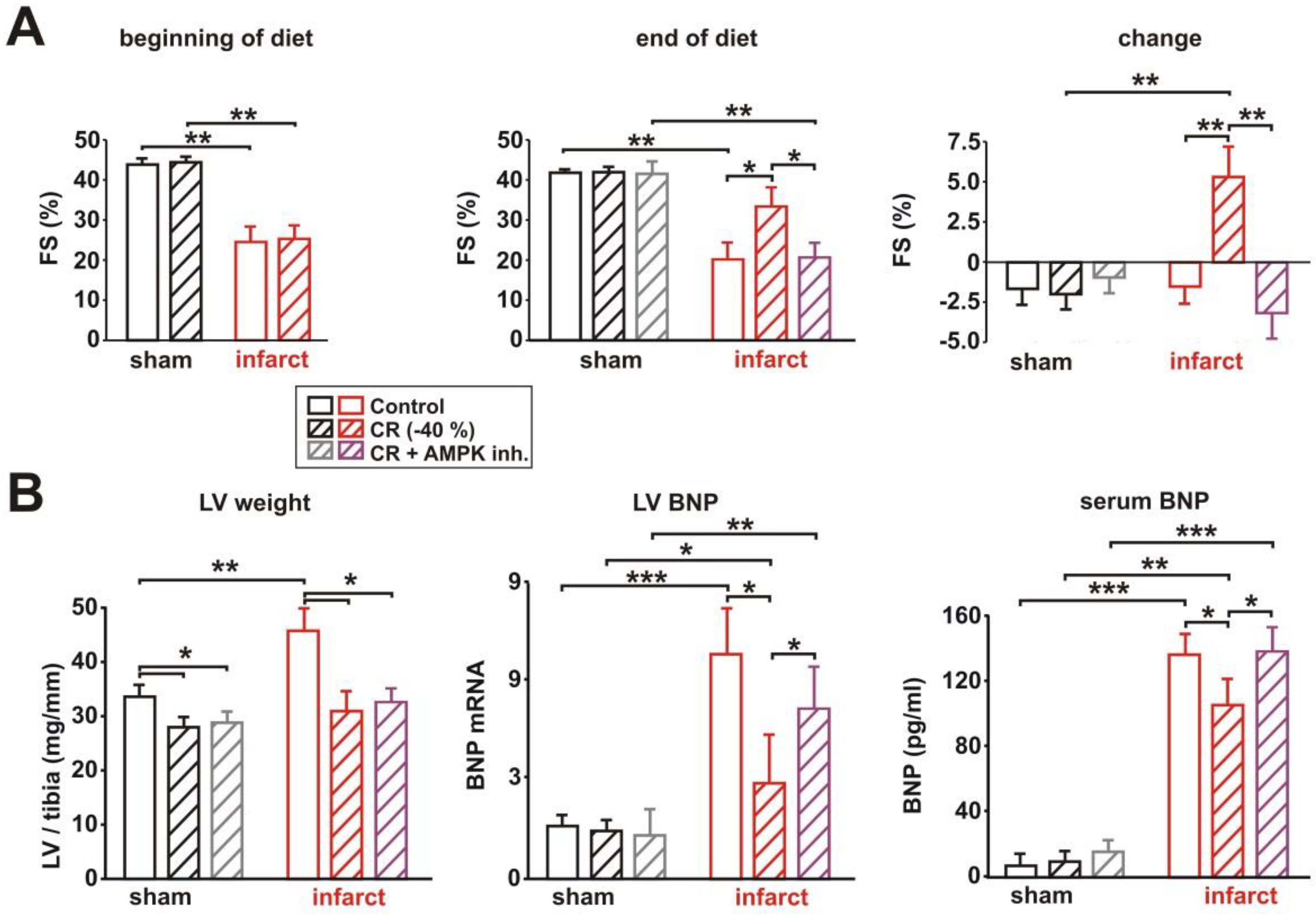
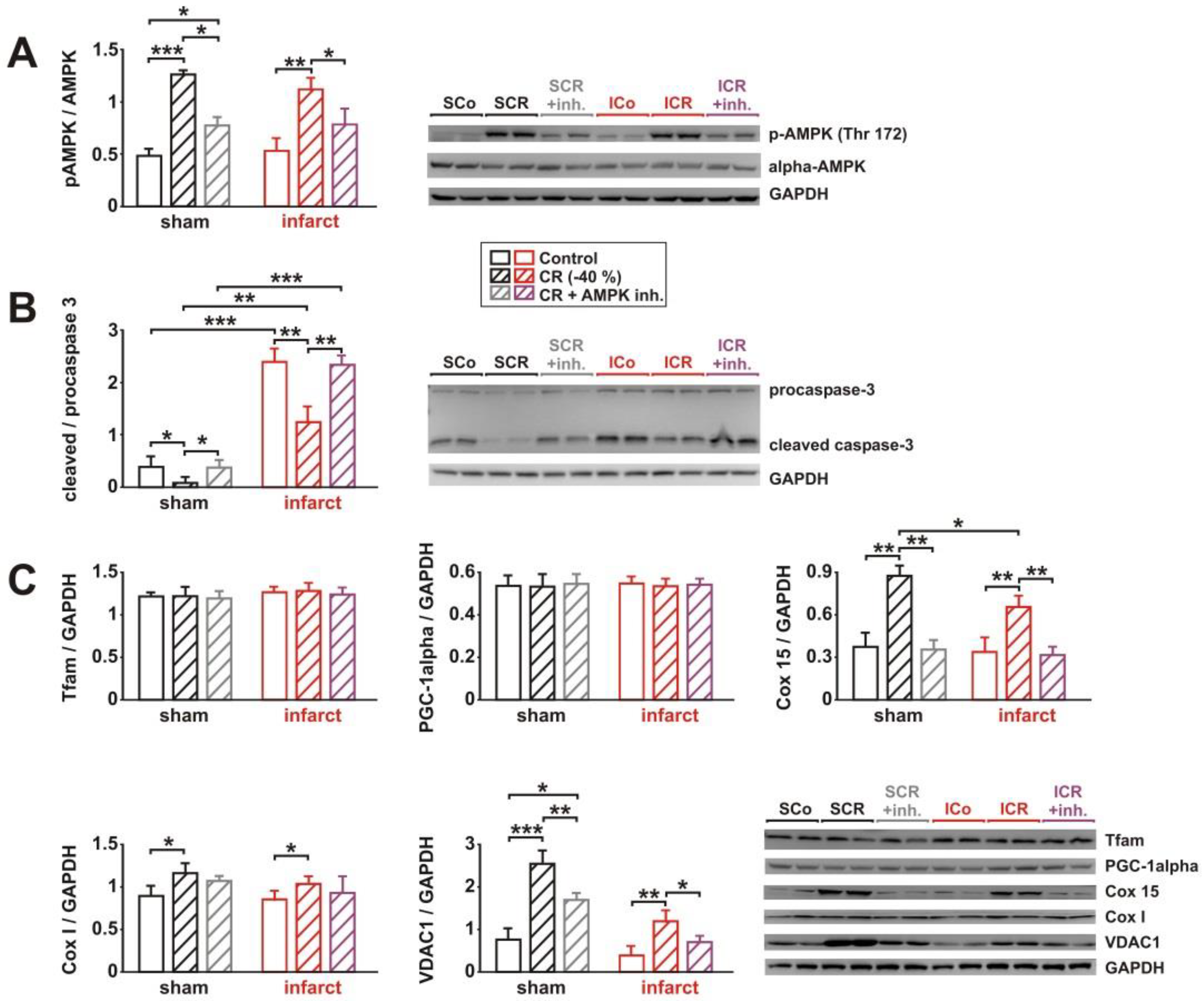
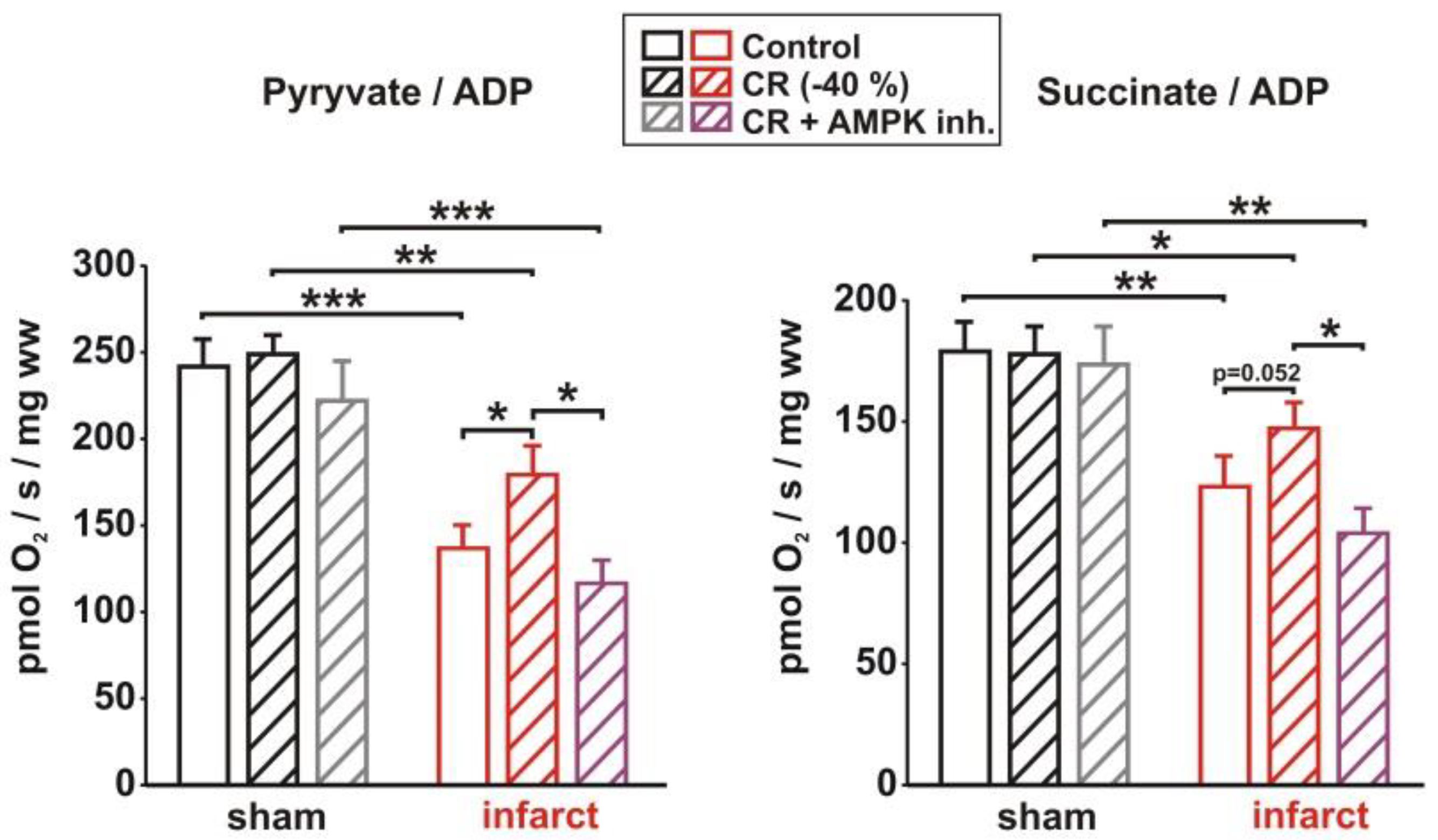
3.4. Impact of AMPK Inhibition on CR-Induced Cardioprotection
3.5. Impact of AMPK Inhibition on CR-Induced Effects on Signaling Pathway Activation, Mitochondrial Biogenesis and Respiration
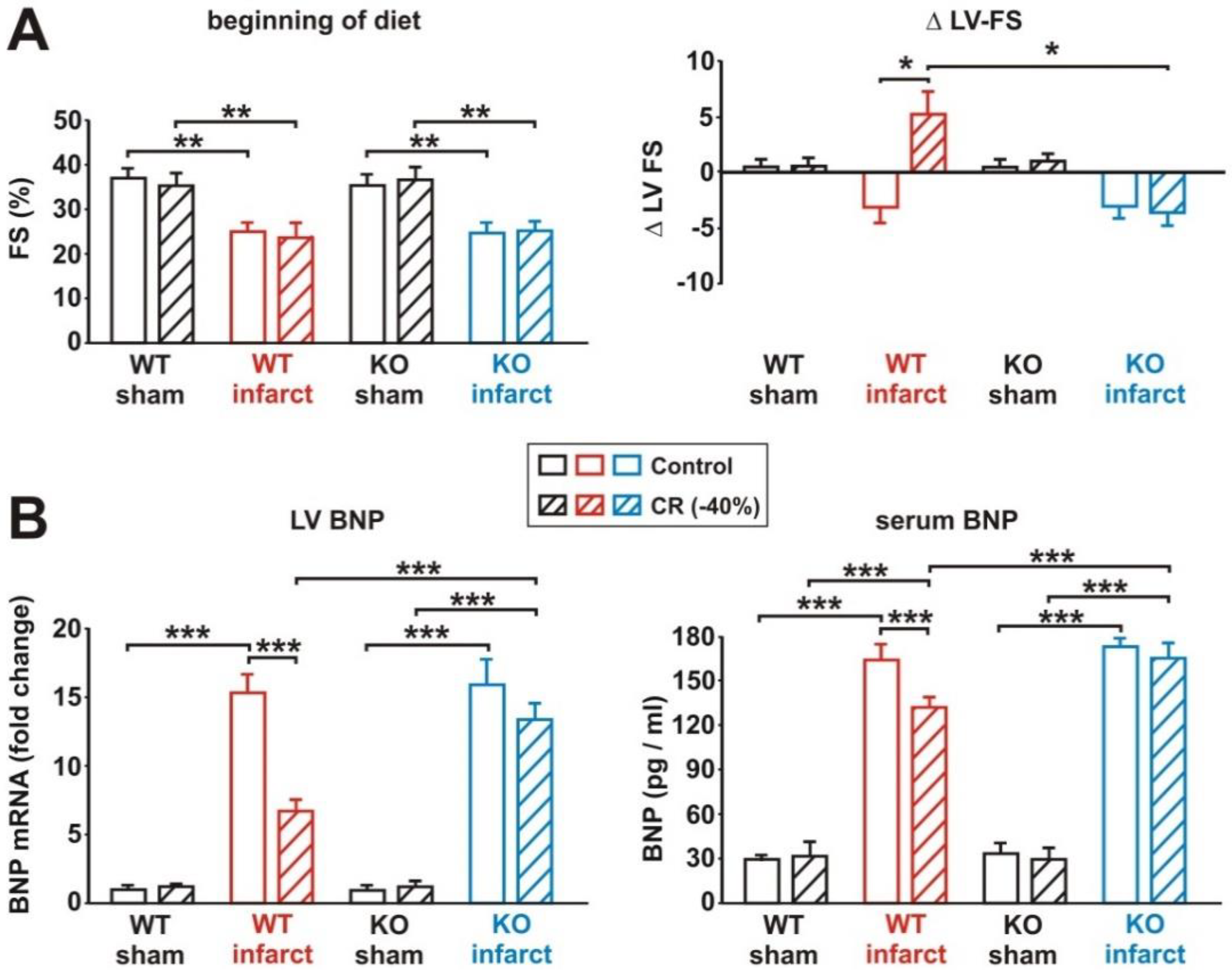
3.6. Impact of Loss of Alpha 2 AMPK on CR-Induced Cardioprotection
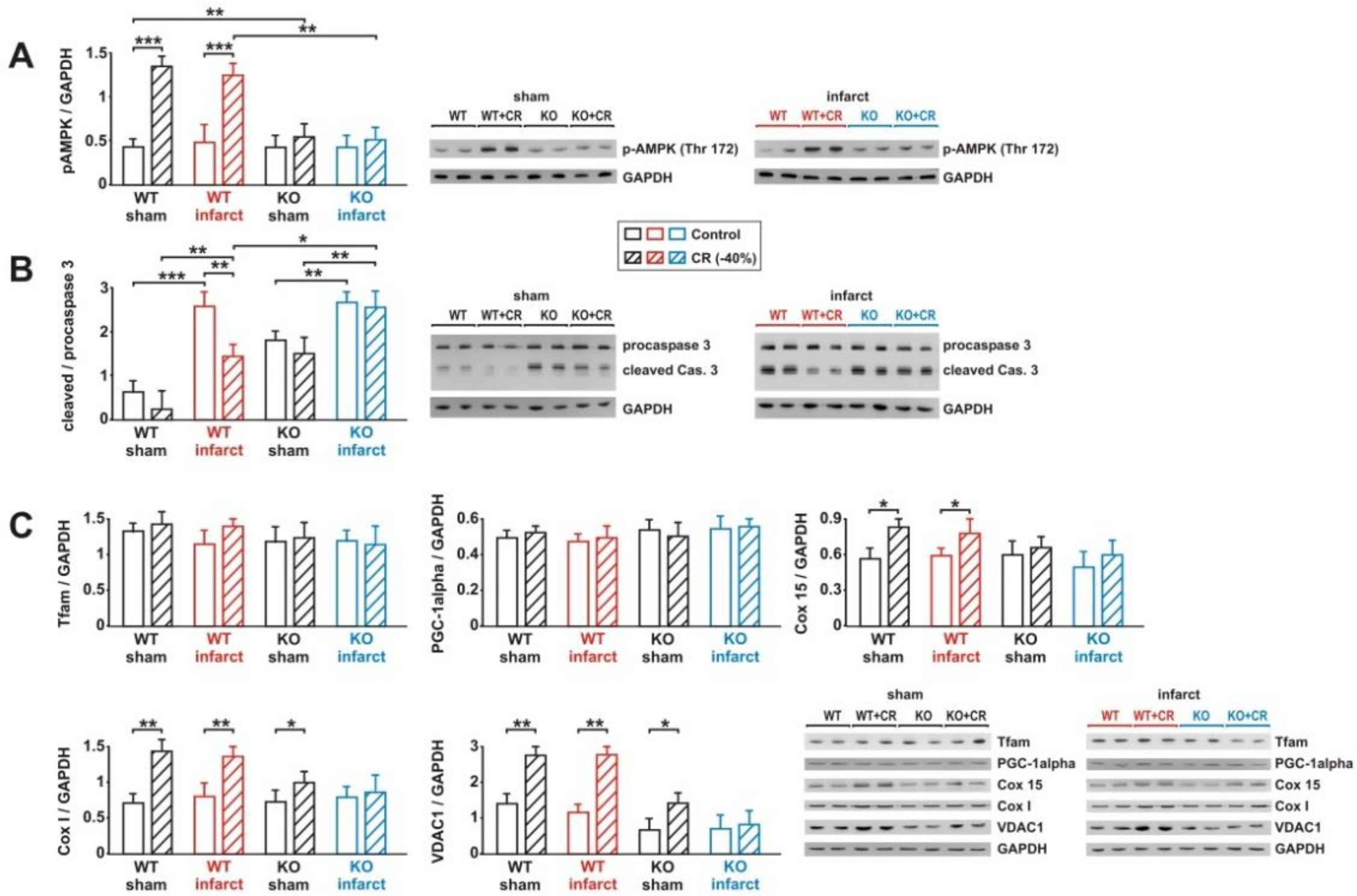
3.7. Impact of Loss of Alpha 2 AMPK on CR-Induced Effects on Signaling Pathway Activation and Mitochondrial Biogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weindruch, R. The retardation of aging by caloric restriction: Studies in rodents and primates. Toxicol. Pathol. 1996, 24, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Willcox, D.C. Caloric restriction, caloric restriction mimetics, and healthy aging in Okinawa: Controversies and clinical implications. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6659–6663. [Google Scholar] [CrossRef] [PubMed]
- Dorling, J.L.; van Vliet, S.; Huffman, K.M.; Kraus, W.E.; Bhapkar, M.; Pieper, C.F.; Stewart, T.; Das, S.K.; Racette, S.B.; Roberts, S.B.; et al. Effects of caloric restriction on human physiological, psychological, and behavioral outcomes: Highlights from CALERIE phase 2. Nutr. Rev. 2021, 79, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Kirkham, A.A.; Beka, V.; Prado, C.M. The effect of caloric restriction on blood pressure and cardiovascular function: A systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2021, 40, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. The future of obesity: New drugs versus lifestyle interventions. Expert Opin. Investig. Drugs 2008, 17, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Lv, S.; Huang, M.; Lv, Y.; Yu, J.; Liu, J.; Tang, T.; Qi, H.; Di, W.; Ding, G. Opposing effects on cardiac function by calorie restriction in different-aged mice. Aging Cell 2017, 16, 1155–1167. [Google Scholar] [CrossRef]
- Shinmura, K.; Tamaki, K.; Sano, M.; Murata, M.; Yamakawa, H.; Ishida, H.; Fukuda, K. Impact of long-term caloric restriction on cardiac senescence: Caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J. Mol. Cell. Cardiol. 2011, 50, 117–127. [Google Scholar] [CrossRef]
- Taffet, G.E.; Pham, T.T.; Hartley, C.J. The age-associated alterations in late diastolic function in mice are improved by caloric restriction. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1997, 52, B285–B290. [Google Scholar] [CrossRef]
- Seymour, E.M.; Parikh, R.V.; Singer, A.A.; Bolling, S.F. Moderate calorie restriction improves cardiac remodeling and diastolic dysfunction in the Dahl-SS rat. J. Mol. Cell. Cardiol. 2006, 41, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Silber, R.E.; Rohrbach, S. Age-specific effects of short- and long-term caloric restriction on the expression of adiponectin and adiponectin receptors: Influence of intensity of food restriction. Exp. Gerontol. 2008, 43, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Chen, Y.; Issa, H.; Silber, R.E.; Rohrbach, S. Caloric restriction delays cardiac ageing in rats: Role of mitochondria. Cardiovasc. Res. 2010, 88, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Li, L.; Simm, A.; Molenda, N.; Kockskamper, J.; Boening, A.; Rohrbach, S. Caloric restriction reduces sympathetic activity similar to beta-blockers but conveys additional mitochondrio-protective effects in aged myocardium. Sci. Rep. 2021, 11, 1931. [Google Scholar] [CrossRef] [PubMed]
- Abete, P.; Ferrara, N.; Cioppa, A.; Ferrara, P.; Bianco, S.; Calabrese, C.; Cacciatore, F.; Longobardi, G.; Rengo, F. Preconditioning does not prevent postischemic dysfunction in aging heart. J. Am. Coll. Cardiol. 1996, 27, 1777–1786. [Google Scholar] [CrossRef]
- Boengler, K.; Buechert, A.; Heinen, Y.; Roeskes, C.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ. Res. 2008, 102, 131–135. [Google Scholar] [CrossRef]
- Shinmura, K.; Tamaki, K.; Bolli, R. Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. J. Mol. Cell. Cardiol. 2005, 39, 285–296. [Google Scholar] [CrossRef]
- Shinmura, K.; Tamaki, K.; Bolli, R. Impact of 6-mo caloric restriction on myocardial ischemic tolerance: Possible involvement of nitric oxide-dependent increase in nuclear Sirt1. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2348–H2355. [Google Scholar] [CrossRef]
- Edwards, A.G.; Donato, A.J.; Lesniewski, L.A.; Gioscia, R.A.; Seals, D.R.; Moore, R.L. Life-long caloric restriction elicits pronounced protection of the aged myocardium: A role for AMPK. Mech. Ageing Dev. 2010, 131, 739–742. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kobayashi, S.; Xu, X.; Viollet, B.; Liang, Q. AMP activated protein kinase is indispensable for myocardial adaptation to caloric restriction in mice. PLoS ONE 2013, 8, e59682. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Yu, H.; Wang, J.; Gao, J.; Li, J. Chronic caloric restriction and exercise improve metabolic conditions of dietary-induced obese mice in autophagy correlated manner without involving AMPK. J. Diabetes Res. 2013, 2013, 852754. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.A.; Kumar, R.; Mulligan, J.D.; Davis, A.J.; Weindruch, R.; Saupe, K.W. Metabolic adaptations to fasting and chronic caloric restriction in heart, muscle, and liver do not include changes in AMPK activity. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E1032–E1037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Viollet, B.; Andreelli, F.; Jorgensen, S.B.; Perrin, C.; Geloen, A.; Flamez, D.; Mu, J.; Lenzner, C.; Baud, O.; Bennoun, M.; et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Investig. 2003, 111, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Robbins, J.; Sugden, P.H. Phenotyping hypertrophy: Eschew obfuscation. Circ. Res. 2003, 92, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Mu, N.; Yin, Y.; Yu, L.; Ma, H. Targeting AMP-Activated Protein Kinase in Aging-Related Cardiovascular Diseases. Aging Dis. 2020, 11, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Rios, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; Costoya, J.A.; Senaris, R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013, 73, 2628–2638. [Google Scholar] [CrossRef]
- Kim, E.K.; Miller, I.; Aja, S.; Landree, L.E.; Pinn, M.; McFadden, J.; Kuhajda, F.P.; Moran, T.H.; Ronnett, G.V. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J. Biol. Chem. 2004, 279, 19970–19976. [Google Scholar] [CrossRef]
- Dyck, J.R.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Seibel, W. Compound C/Dorsomorphin: Its Use and Misuse as an AMPK Inhibitor. Methods Mol. Biol. 2018, 1732, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Zarrinpashneh, E.; Budas, G.R.; Pouleur, A.C.; Dutta, A.; Prescott, A.R.; Vanoverschelde, J.L.; Ashworth, A.; Jovanovic, A.; Alessi, D.R.; et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E780–E788. [Google Scholar] [CrossRef]
- Lee-Young, R.S.; Griffee, S.R.; Lynes, S.E.; Bracy, D.P.; Ayala, J.E.; McGuinness, O.P.; Wasserman, D.H. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J. Biol. Chem. 2009, 284, 23925–23934. [Google Scholar] [CrossRef] [PubMed]
- Melo, D.S.; Costa-Pereira, L.V.; Santos, C.S.; Mendes, B.F.; Costa, K.B.; Santos, C.F.; Rocha-Vieira, E.; Magalhaes, F.C.; Esteves, E.A.; Ferreira, A.J.; et al. Severe Calorie Restriction Reduces Cardiometabolic Risk Factors and Protects Rat Hearts from Ischemia/Reperfusion Injury. Front. Physiol. 2016, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Tamaki, K.; Ito, K.; Yan, X.; Yamamoto, T.; Katsumata, Y.; Matsuhashi, T.; Sano, M.; Fukuda, K.; Suematsu, M.; et al. Indispensable role of endothelial nitric oxide synthase in caloric restriction-induced cardioprotection against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H894–H903. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, S.; Aslam, M.; Niemann, B.; Schulz, R. Impact of caloric restriction on myocardial ischaemia/reperfusion injury and new therapeutic options to mimic its effects. Br. J. Pharmacol. 2014, 171, 2964–2992. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; See Hoe, L.; Pepe, S.; Johnson, P.; Headrick, J.P. Opposing effects of age and calorie restriction on molecular determinants of myocardial ischemic tolerance. Rejuvenation Res. 2012, 15, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Verweij, M.; Brand, K.; van de Ven, M.; Goemaere, N.; van den Engel, S.; Chu, T.; Forrer, F.; Muller, C.; de Jong, M.; et al. Short-term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell 2010, 9, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Katare, R.G.; Kakinuma, Y.; Arikawa, M.; Yamasaki, F.; Sato, T. Chronic intermittent fasting improves the survival following large myocardial ischemia by activation of BDNF/VEGF/PI3K signaling pathway. J. Mol. Cell. Cardiol. 2009, 46, 405–412. [Google Scholar] [CrossRef] [PubMed]
- de Lucia, C.; Gambino, G.; Petraglia, L.; Elia, A.; Komici, K.; Femminella, G.D.; D’Amico, M.L.; Formisano, R.; Borghetti, G.; Liccardo, D.; et al. Long-Term Caloric Restriction Improves Cardiac Function, Remodeling, Adrenergic Responsiveness, and Sympathetic Innervation in a Model of Postischemic Heart Failure. Circ. Heart Fail. 2018, 11, e004153. [Google Scholar] [CrossRef] [PubMed]
- Okoshi, K.; Cezar, M.D.M.; Polin, M.A.M.; Paladino, J.R., Jr.; Martinez, P.F.; Oliveira, S.A., Jr.; Lima, A.R.R.; Damatto, R.L.; Paiva, S.A.R.; Zornoff, L.A.M.; et al. Influence of intermittent fasting on myocardial infarction-induced cardiac remodeling. BMC Cardiovasc. Disord. 2019, 19, 126. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Muller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, X.; Zhang, J.; Zhang, Y. Omentin-1: Protective impact on ischemic stroke via ameliorating atherosclerosis. Clin. Chim. Acta; Int. J. Clin. Chem. 2021, 517, 31–40. [Google Scholar] [CrossRef]
- Kola, B.; Boscaro, M.; Rutter, G.A.; Grossman, A.B.; Korbonits, M. Expanding role of AMPK in endocrinology. Trends Endocrinol. Metab. TEM 2006, 17, 205–215. [Google Scholar] [CrossRef]
- Konishi, M.; Haraguchi, G.; Ohigashi, H.; Ishihara, T.; Saito, K.; Nakano, Y.; Isobe, M. Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc. Res. 2011, 89, 309–319. [Google Scholar] [CrossRef]
- Hada, Y.; Yamauchi, T.; Waki, H.; Tsuchida, A.; Hara, K.; Yago, H.; Miyazaki, O.; Ebinuma, H.; Kadowaki, T. Selective purification and characterization of adiponectin multimer species from human plasma. Biochem. Biophys. Res. Commun. 2007, 356, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Liu, M.; Liu, X.; Dong, L.Q.; Glickman, R.D.; Slaga, T.J.; Zhou, Z.; Liu, F. Up-regulation of adiponectin by resveratrol: The essential roles of the Akt/FOXO1 and AMP-activated protein kinase signaling pathways and DsbA-L. J. Biol. Chem. 2011, 286, 60–66. [Google Scholar] [CrossRef]
- Li, Y.; Wang, P.; Zhuang, Y.; Lin, H.; Liu, L.; Meng, Q.; Cui, T.; Liu, J.; Li, Z. Activation of AMPK by berberine promotes adiponectin multimerization in 3T3-L1 adipocytes. FEBS Lett. 2011, 585, 1735–1740. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Viollet, B.; Andreelli, F.; Frosig, C.; Birk, J.B.; Schjerling, P.; Vaulont, S.; Richter, E.A.; Wojtaszewski, J.F. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 2004, 279, 1070–1079. [Google Scholar] [CrossRef]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.; Li, C.; Wang, J.; Wu, A.; Zhang, T.; Ma, Z.; Chai, L.; Zhang, D.; Zhao, Y.; Nie, B.; et al. Yiqi Huoxue preserves heart function by upregulating the Sigma-1 receptor in rats with myocardial infarction. Exp. Ther. Med. 2021, 22, 1308. [Google Scholar] [CrossRef] [PubMed]
- Hingst, J.R.; Kjobsted, R.; Birk, J.B.; Jorgensen, N.O.; Larsen, M.R.; Kido, K.; Larsen, J.K.; Kjeldsen, S.A.S.; Fentz, J.; Frosig, C.; et al. Inducible deletion of skeletal muscle AMPKalpha reveals that AMPK is required for nucleotide balance but dispensable for muscle glucose uptake and fat oxidation during exercise. Mol. Metab. 2020, 40, 101028. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.; Ren, J.M.; Cadman, K.S.; Moore, I.K.; Perret, P.; Pypaert, M.; Young, L.H.; Semenkovich, C.F.; Shulman, G.I. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1340–E1346. [Google Scholar] [CrossRef]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, H.; Liu, P.; Wang, H.; Liu, J.; Gao, C.; Liu, Y.; Lian, K.; Yang, L.; Sun, L.; et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1alpha signaling contributing to increased vulnerability in diabetic heart. Basic Res. Cardiol. 2013, 108, 329. [Google Scholar] [CrossRef]
- Oka, S.I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1alpha Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef]
- Hu, X.; Xu, X.; Lu, Z.; Zhang, P.; Fassett, J.; Zhang, Y.; Xin, Y.; Hall, J.L.; Viollet, B.; Bache, R.J.; et al. AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension 2011, 58, 696–703. [Google Scholar] [CrossRef]
- Bhat, S.; Chin, A.; Shirakabe, A.; Ikeda, Y.; Ikeda, S.; Zhai, P.; Hsu, C.P.; Sayed, D.; Abdellatif, M.; Byun, J.; et al. Recruitment of RNA Polymerase II to Metabolic Gene Promoters Is Inhibited in the Failing Heart Possibly Through PGC-1alpha (Peroxisome Proliferator-Activated Receptor-gamma Coactivator-1alpha) Dysregulation. Circ. Heart Fail. 2019, 12, e005529. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Li, L.; Siegler, D.; Siegler, B.H.; Knapp, F.; Hanna, J.; Aslam, M.; Kracht, M.; Schulz, R.; Rohrbach, S. CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response. Cells 2020, 9, 1229. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef] [PubMed]
- Guevara, R.; Valle, A.; Gianotti, M.; Roca, P.; Oliver, J. Gender-dependent differences in serum profiles of insulin and leptin in caloric restricted rats. Horm. Metab. Res. 2008, 40, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Honjoh, S.; Ihara, A.; Kajiwara, Y.; Yamamoto, T.; Nishida, E. The Sexual Dimorphism of Dietary Restriction Responsiveness in Caenorhabditis elegans. Cell Rep. 2017, 21, 3646–3652. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Oliver, J.; Roca, P.; Garcia-Palmer, F.J. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Trouwborst, I.; Goossens, G.H.; Astrup, A.; Saris, W.H.M.; Blaak, E.E. Sexual Dimorphism in Body Weight Loss, Improvements in Cardiometabolic Risk Factors and Maintenance of Beneficial Effects 6 Months after a Low-Calorie Diet: Results from the Randomized Controlled DiOGenes Trial. Nutrients 2021, 13, 1588. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.R.; Patel, H.N.; Mehta, L.S.; Sanghani, R.M.; Lundberg, G.P.; Lewis, S.J.; Mendelson, M.A.; Wood, M.J.; Volgman, A.S.; Mieres, J.H. Sex Differences in Ischemic Heart Disease: Advances, Obstacles, and Next Steps. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e004437. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Ferdinandy, P.; Botker, H.E.; Brundel, B.; Collins, P.; Davidson, S.M.; den Ruijter, H.M.; Engel, F.B.; Gerdts, E.; Girao, H.; et al. Improving translational research in sex-specific effects of comorbidities and risk factors in ischaemic heart disease and cardioprotection: Position paper and recommendations of the ESC Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2021, 117, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Meana, M.; Boengler, K.; Garcia-Dorado, D.; Hausenloy, D.J.; Kaambre, T.; Kararigas, G.; Perrino, C.; Schulz, R.; Ytrehus, K. Ageing, sex, and cardioprotection. Br. J. Pharmacol. 2020, 177, 5270–5286. [Google Scholar] [CrossRef] [PubMed]
- Palee, S.; Minta, W.; Mantor, D.; Sutham, W.; Jaiwongkam, T.; Kerdphoo, S.; Pratchayasakul, W.; Chattipakorn, S.C.; Chattipakorn, N. Combination of exercise and calorie restriction exerts greater efficacy on cardioprotection than monotherapy in obese-insulin resistant rats through the improvement of cardiac calcium regulation. Metab. Clin. Exp. 2019, 94, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.P.; Albert, S.G.; Reeds, D.N.; Kress, K.S.; McDaniel, J.L.; Klein, S.; Villareal, D.T. Effects of matched weight loss from calorie restriction, exercise, or both on cardiovascular disease risk factors: A randomized intervention trial. Am. J. Clin. Nutr. 2016, 104, 576–586. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Accession Number | Sense | Antisense |
|---|---|---|---|
| BNP rat | NM_031545 | CTCAAAGGACCAAGGCCC TAC | CTGCCCAAAGCAGCTTGAAC |
| BNP mouse | NM_008726 | CTGAAGGTGCTGTCCCAGAT | CCTTGGTCCTTCAAGAGCTG |
| ANP mouse | NM_008725 | ATGGGCTCCTTCTCCATC | GTGTTGGACACCGCACTGTA |
| ANP rat | NM_012612.2 | GAGCGAGCAGACCGATGAA | GATCTATCGGAGGGGTCCCA |
| beta-MHC mouse | NM_080728.2 | GAGAAGATGTGCCGGACCTT | GGACAGCTCCCCATTCTCTG |
| beta-MHC rat | NM_017240.2 | TGCTCTACAATCTCAAGGAGAGGT | AGGCGTTGTCAGAGATGGAGA |
| mt. DNA rat | KM577634.1 | AGTGAAGGGGCGGATCATA | GAGGTCACCCCAACCGAAAT |
| beta-globin rat | NM_001113223.1 | GCCTGTGGGGAAAGGTGAATG | CTTCACCTGGGGGTTACCCAT |
| HPRT rat | NM_012583.2 | ACCAGTCAACGGGGGACATA | ATTTTGGGGCTGTACTGCTTGA |
| HPRT mouse | NM_013556.2 | GATCAGTCAACGGGGGACAT | AGAGGTCCTTTTCACCAGCAA |
| GAPDH rat | NM_017008.4 | CACCATCTTCCAGGAGCGAG | GAAGGGGCGGAGATGATGAC |
| GAPDH mouse | BC023196.2 | CATCACCATCTTCCAGGAGCG | CGTTTGGCTCCACCCTTCAA |
| 18S rRNA | NR_046237 | TGGAGCGATTTGTCTGGTTA | ACGCCACTTGTCCCTCTAAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niemann, B.; Pan, R.; Issa, H.; Simm, A.; Schulz, R.; Rohrbach, S. AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium. Biology 2022, 11, 448. https://doi.org/10.3390/biology11030448
Niemann B, Pan R, Issa H, Simm A, Schulz R, Rohrbach S. AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium. Biology. 2022; 11(3):448. https://doi.org/10.3390/biology11030448
Chicago/Turabian StyleNiemann, Bernd, Ruping Pan, Hassan Issa, Andreas Simm, Rainer Schulz, and Susanne Rohrbach. 2022. "AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium" Biology 11, no. 3: 448. https://doi.org/10.3390/biology11030448
APA StyleNiemann, B., Pan, R., Issa, H., Simm, A., Schulz, R., & Rohrbach, S. (2022). AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium. Biology, 11(3), 448. https://doi.org/10.3390/biology11030448