
S-15176 Difumarate Salt Can Impair Mitochondrial Function through Inhibition of the Respiratory Complex III and Permeabilization of the Inner Mitochondrial Membrane

 ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Rat Thymocytes and Measurement of Mitochondrial Membrane Potential
2.3. Isolation of Mitochondria from Rat Liver
2.4. Measurements of Mitochondrial Bioenergetics
2.5. Assessment of H2O2 Production by Liver Mitochondria
2.6. Quantification of Mitochondrial Calcium Retention Capacity Index
2.7. Mitochondrial Swelling Assay
2.8. Statistical Analysis
3. Results
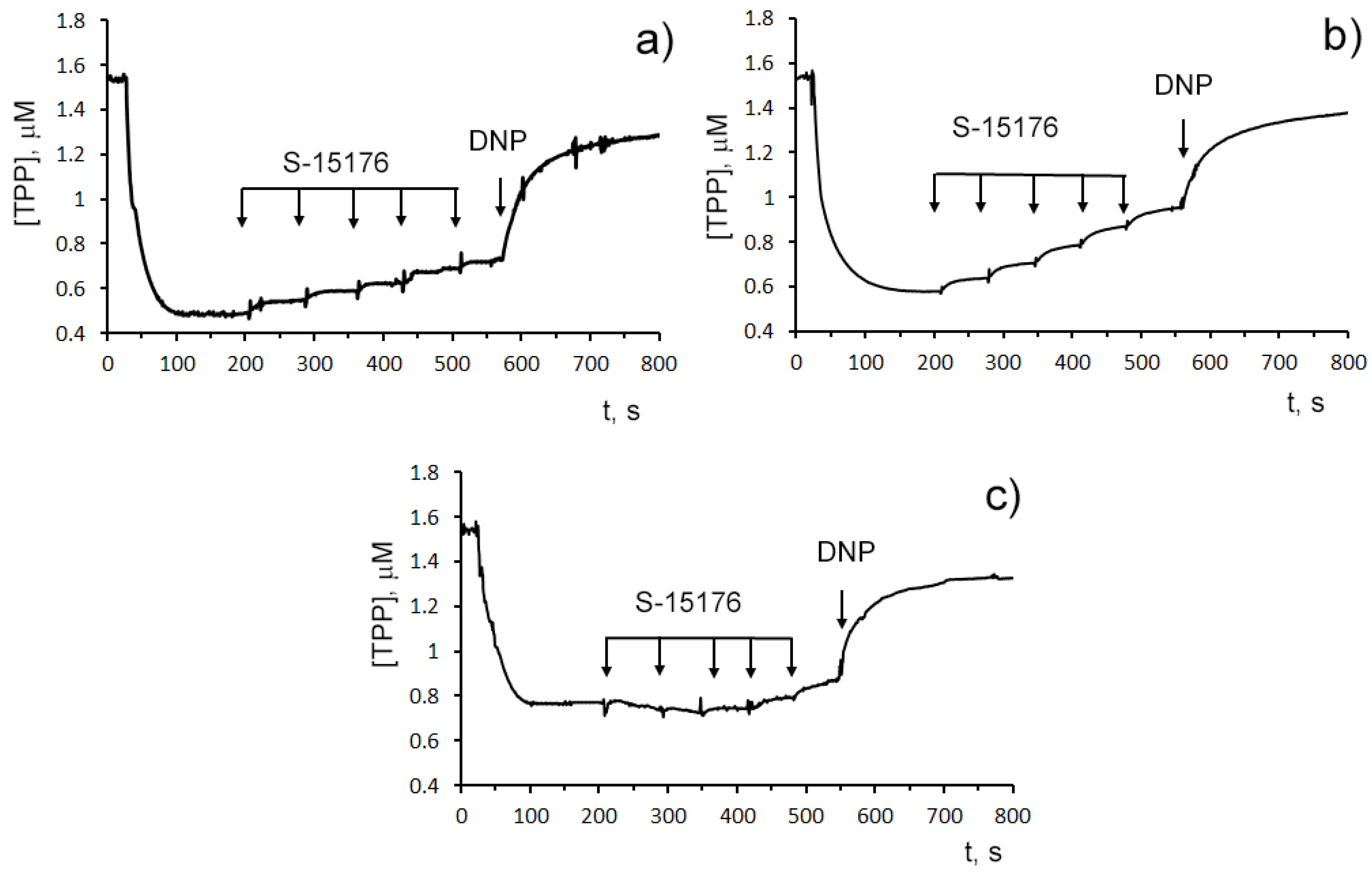
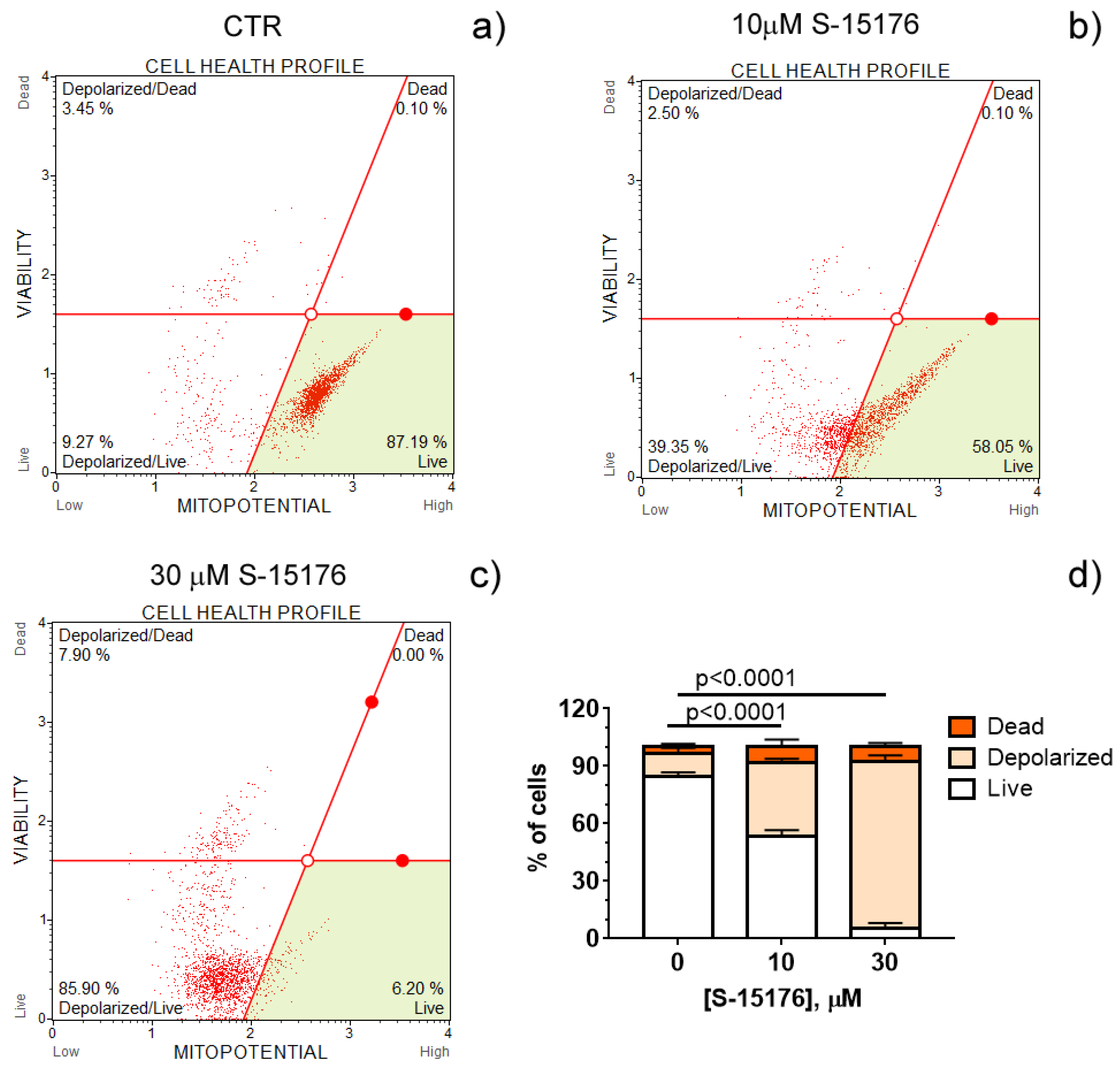
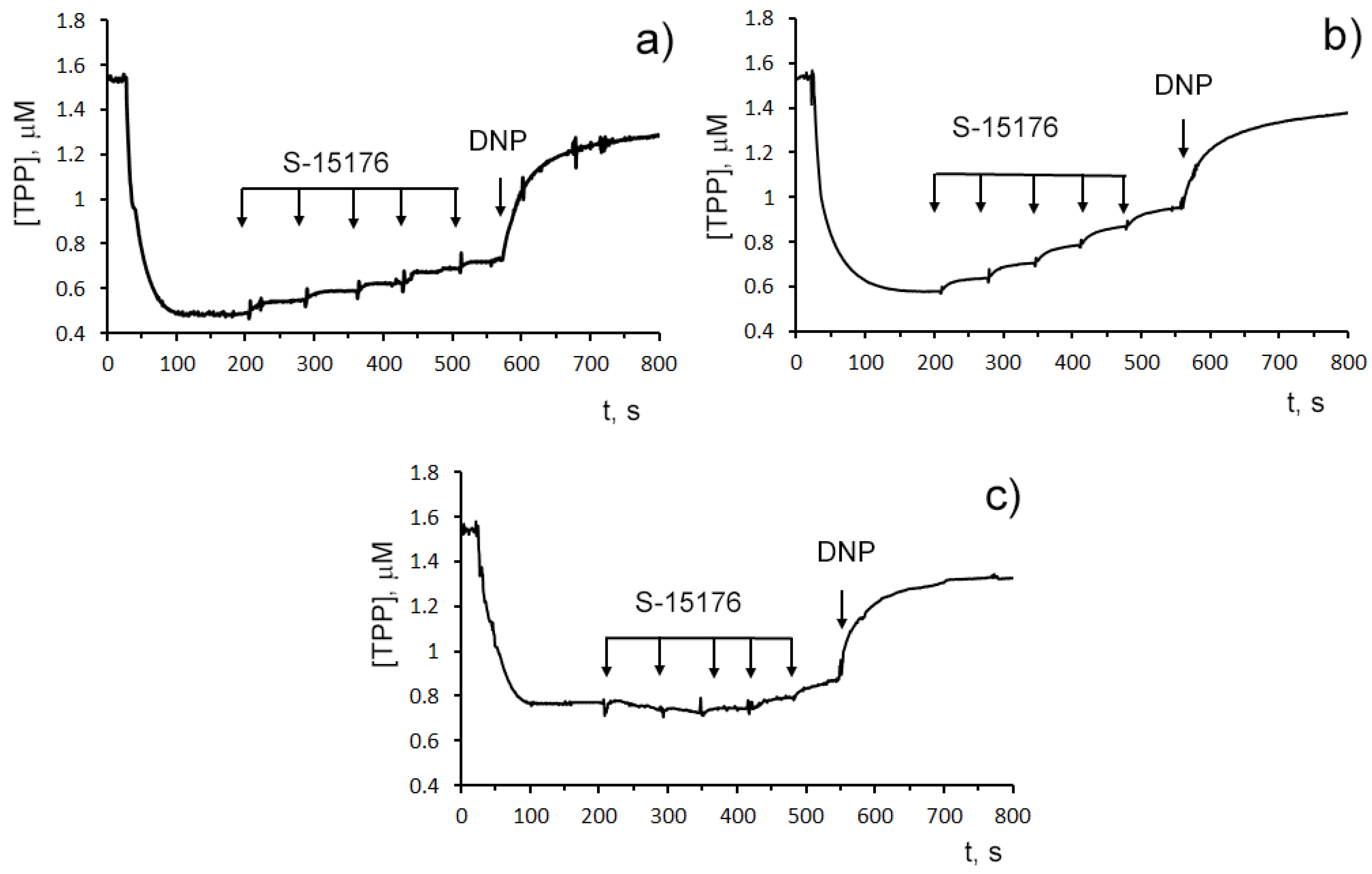
3.1. S-15176 Difumarate Salt Promotes Mitochondrial Depolarization in Rat Thymocytes
3.2. S-15176 Inhibits ADP- and 2,4-Dinitrophenol- Stimulated Mitochondrial Respiration Due to Suppression of the Enzymatic Activity of the Respiratory Complex III
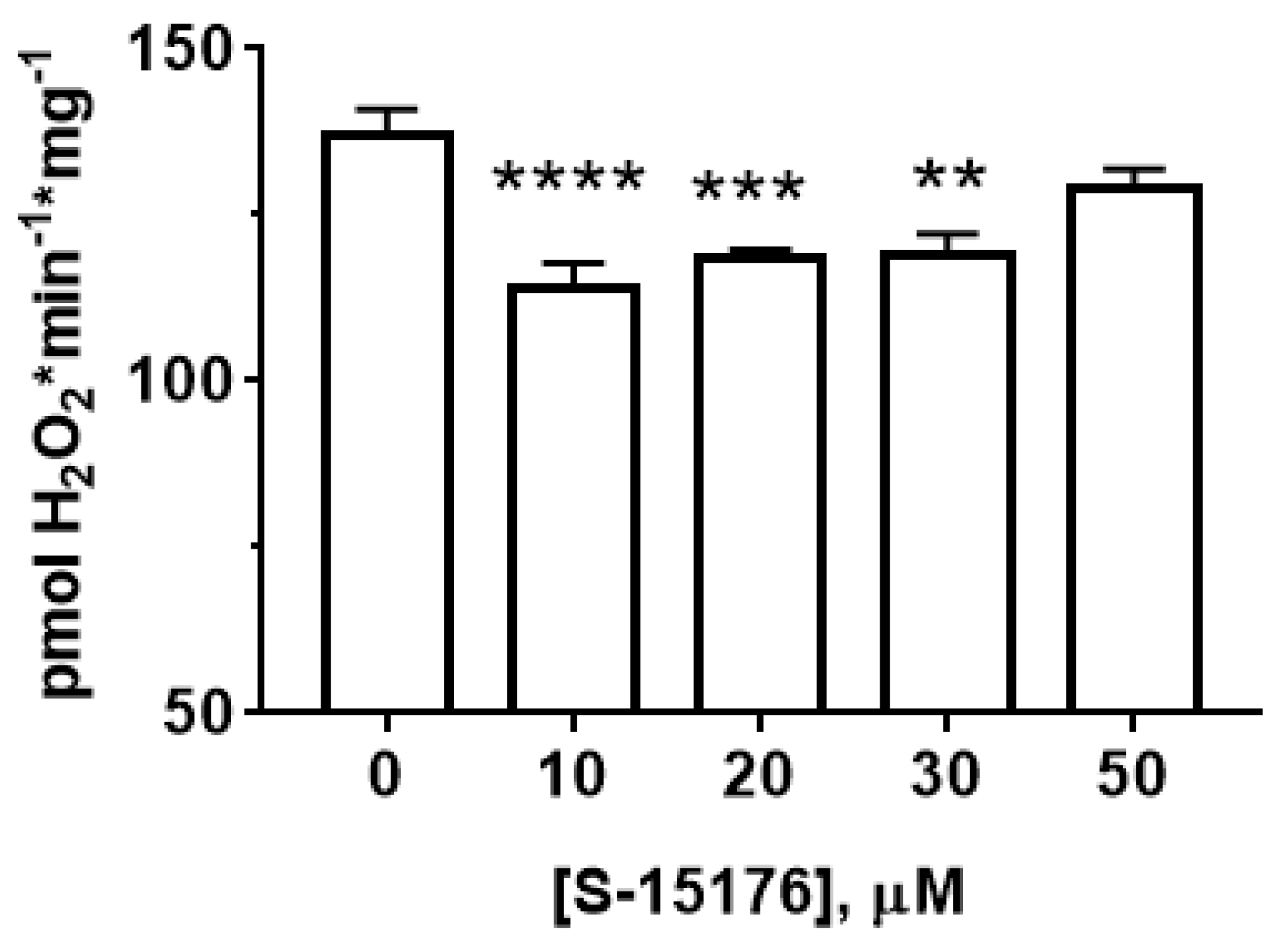
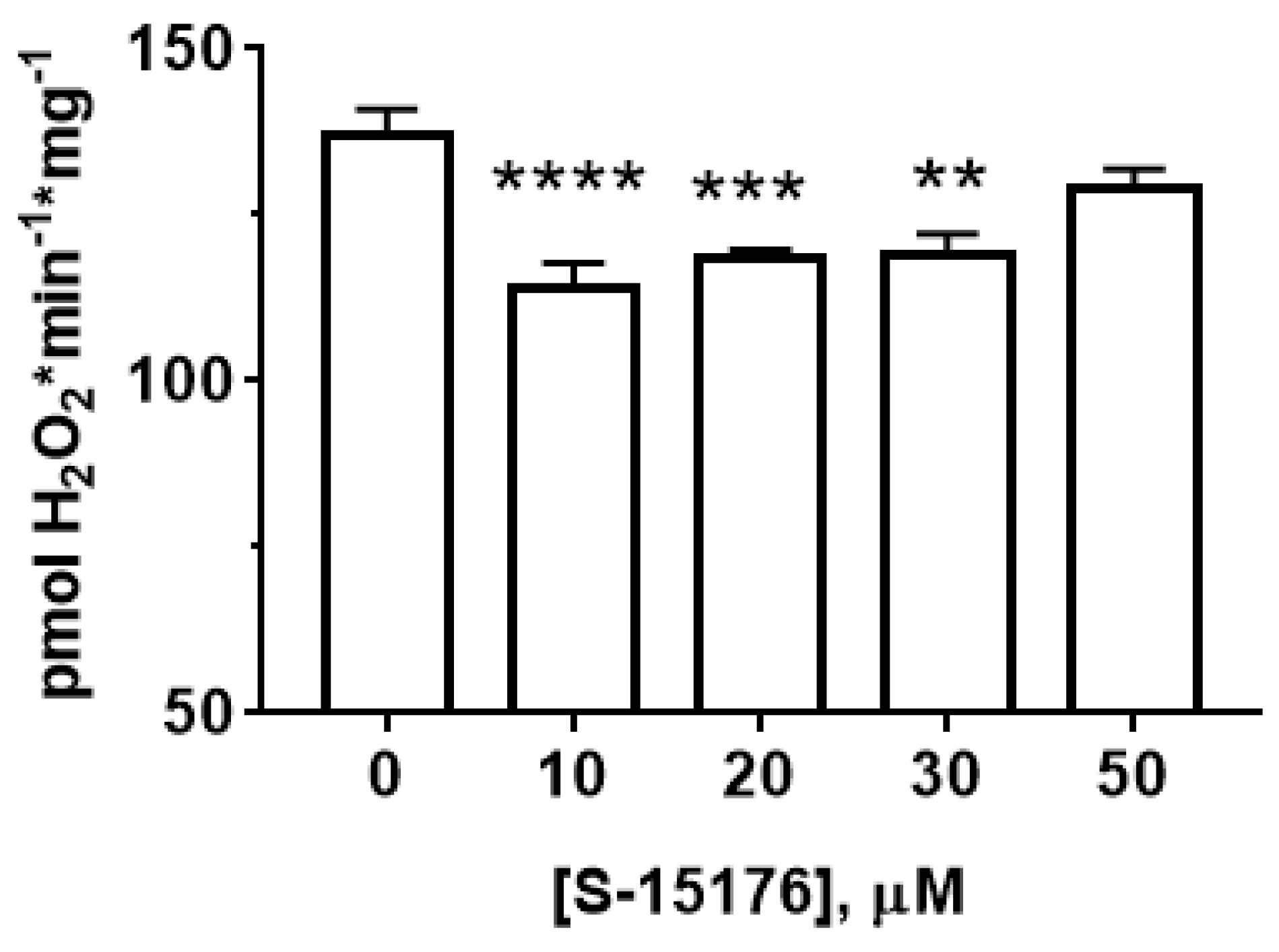
3.3. S-15176 Inhibits H2O2 Production by Rat Liver Mitochondria
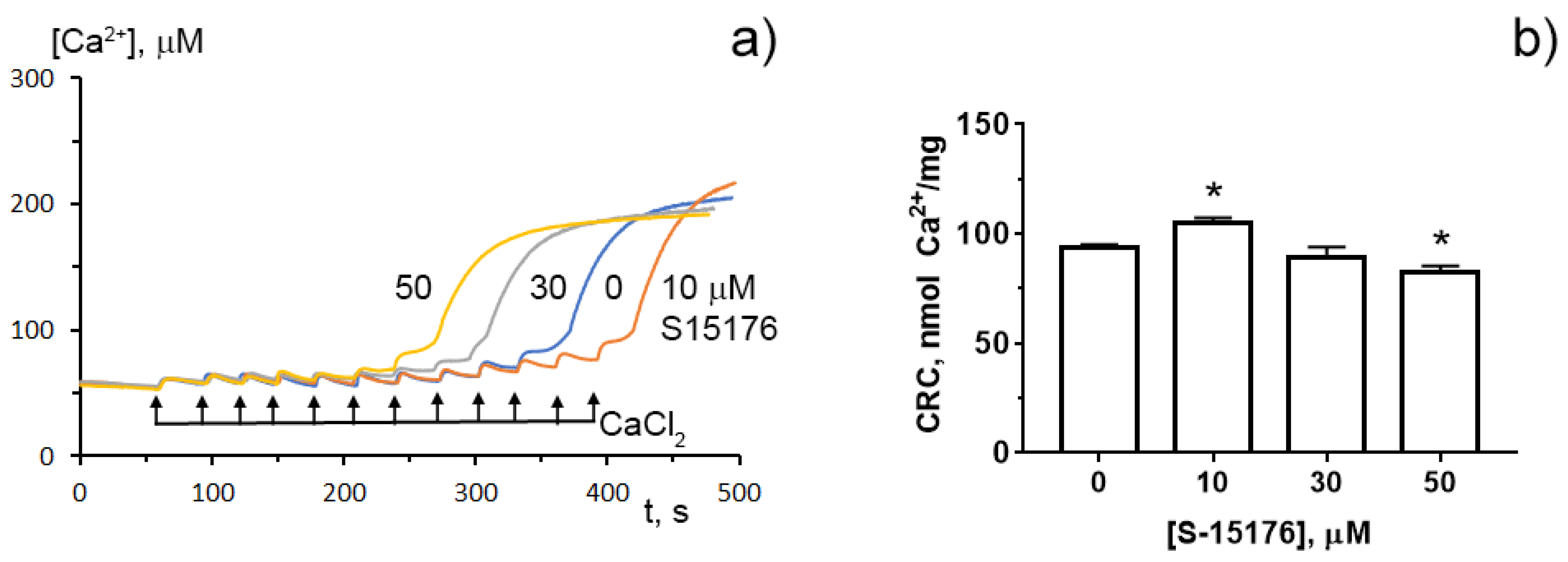
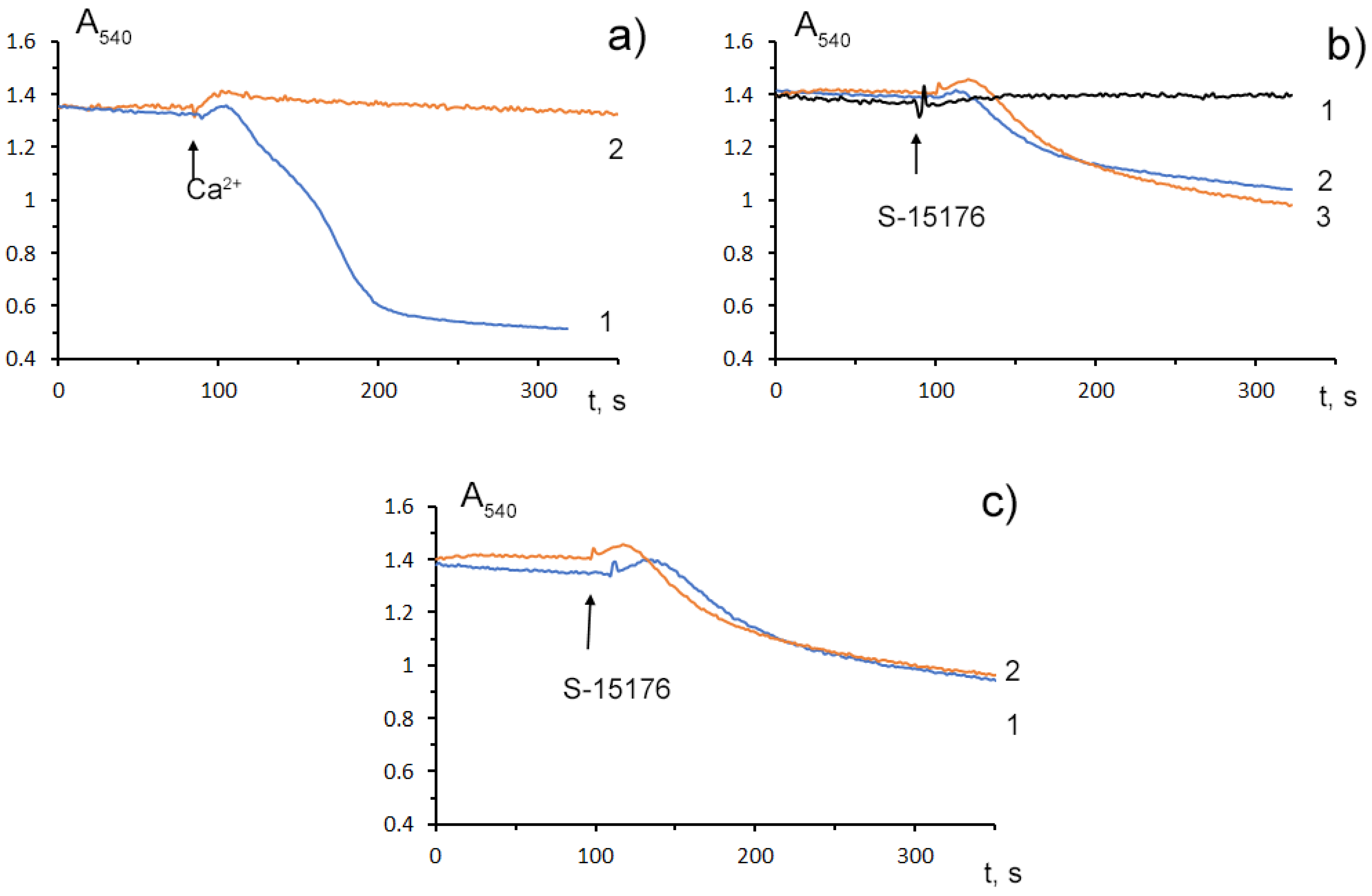
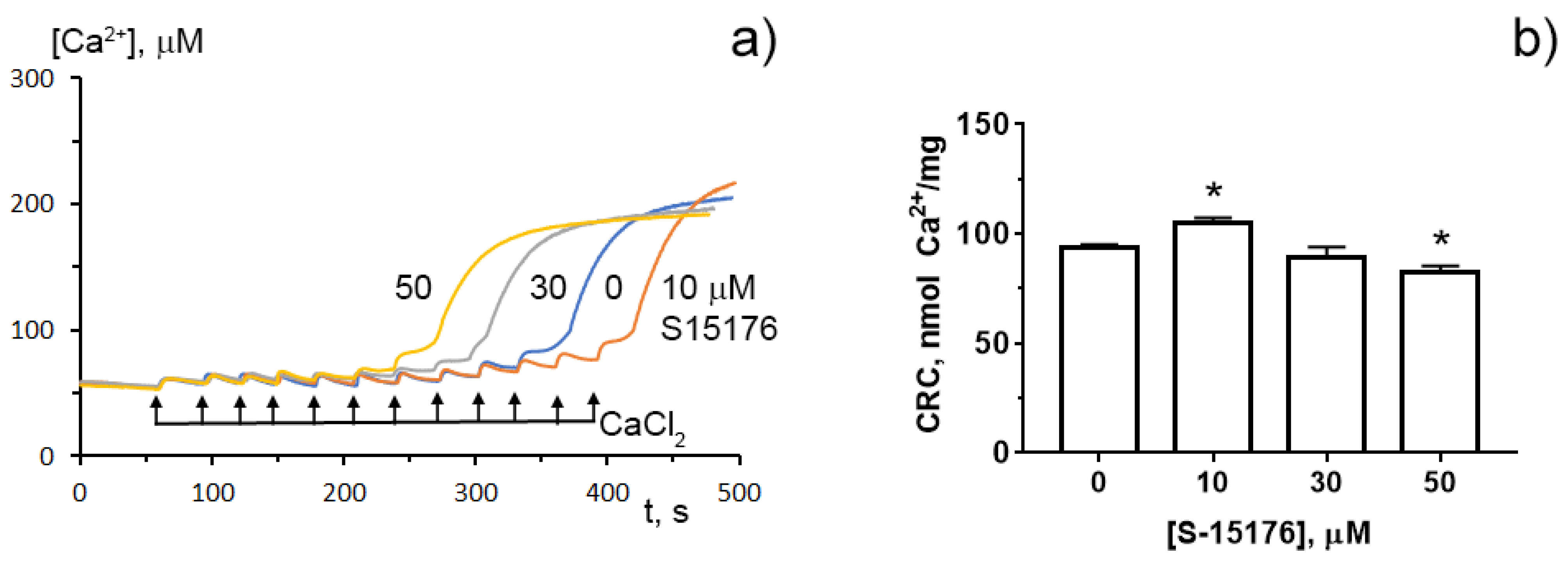
3.4. S-15176 at Low Concentrations Suppress the Opening of the Ca2+-Dependent MPT Pore and, at High Concentrations, the Agent Itself Contributes to the Permeabilization of the Mitochondrial Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, C.; Jin, C.; He, X.; Xiang, M. The efficacy of trimetazidine in non-ischemic heart failure patients: A meta-analysis of randomized controlled trials. Rev. Cardiovasc. Med. 2021, 22, 1451–1459. [Google Scholar] [CrossRef]
- Kallistratos, M.S.; Poulimenos, L.E.; Giannitsi, S.; Tsinivizov, P.; Manolis, A.J. Trimetazidine in the Prevention of Tissue Ischemic Conditions. Angiology 2019, 70, 291–298. [Google Scholar] [CrossRef]
- Marzilli, M.; Vinereanu, D.; Lopaschuk, G.; Chen, Y.; Dalal, J.J.; Danchin, N.; Etriby, E.; Ferrari, R.; Gowdak, L.H.; Lopatin, Y.; et al. Trimetazidine in cardiovascular medicine. Int. J. Cardiol. 2019, 293, 39–44. [Google Scholar] [CrossRef]
- Gupta, K.; Pandey, S.; Bagang, N.; Mehra, K.; Singh, G. Trimetazidine an emerging paradigm in renal therapeutics: Preclinical and clinical insights. Eur. J. Pharmacol. 2021, 913, 174624. [Google Scholar] [CrossRef]
- Abu-Amara, M.; Gurusamy, K.; Hori, S.; Glantzounis, G.; Fuller, B.; Davidson, B.R. Systematic review of randomized controlled trials of pharmacological interventions to reduce ischaemia-reperfusion injury in elective liver resection with vascular occlusion. HPB 2010, 12, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Guarini, G.; Huqi, A.; Morrone, D.; Capozza, P.F.G.; Marzilli, M. Trimetazidine and Other Metabolic Modifiers. Eur. Cardiol. 2018, 13, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Mahajan, A.U. Current Clinical Evidence of Trimetazidine in the Management of Heart Disease in Patients with Diabetes. J. Assoc. Physicians India 2020, 68, 46–50. [Google Scholar]
- Rosano, G.M.; Vitale, C.; Volterrani, M.; Fini, M. Metabolic therapy for the diabetic patients with ischaemic heart disease. Coron. Artery Dis. 2005, 98, S17–S21. [Google Scholar] [CrossRef]
- Danchin, N.; Marzilli, M.; Parkhomenko, A.; Ribeiro, J.P. Efficacy comparison of trimetazidine with therapeutic alternatives in stable angina pectoris: A network meta-analysis. Cardiology 2011, 120, 59–72. [Google Scholar] [CrossRef]
- Fragasso, G.; Rosano, G.; Baek, S.H.; Sisakian, H.; Di Napoli, P.; Alberti, L.; Calori, G.; Kang, S.M.; Sahakyan, L.; Sanosyan, A.; et al. Effect of partial fatty acid oxidation inhibition with trimetazidine on mortality and morbidity in heart failure: Results from an international multicentre retrospective cohort study. Int. J. Cardiol. 2013, 163, 320–325. [Google Scholar] [CrossRef]
- Vitale, C.; Spoletini, I.; Malorni, W.; Perrone-Filardi, P.; Volterrani, M.; Rosano, G.M. Efficacy of trimetazidine on functional capacity in symptomatic patients with stable exertional angina--the VASCO-angina study. Int. J. Cardiol. 2013, 168, 1078–1081. [Google Scholar] [CrossRef]
- Kantor, P.F.; Lucien, A.; Kozak, R.; Lopaschuk, G.D. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 2000, 86, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Dedkova, E.N.; Seidlmayer, L.K.; Blatter, L.A. Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J. Mol. Cell Cardiol. 2013, 59, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Argaud, L.; Gomez, L.; Gateau-Roesch, O.; Couture-Lepetit, E.; Loufouat, J.; Robert, D.; Ovize, M. Trimetazidine inhibits mitochondrial permeability transition pore opening and prevents lethal ischemia-reperfusion injury. J. Mol. Cell Cardiol. 2005, 39, 893–899. [Google Scholar] [CrossRef]
- Morota, S.; Månsson, R.; Hansson, M.J.; Kasuya, K.; Shimazu, M.; Hasegawa, E.; Yanagi, S.; Omi, A.; Uchino, H.; Elmér, E. Evaluation of putative inhibitors of mitochondrial permeability transition for brain disorders—Specificity vs. toxicity. Exp Neurol. 2009, 218, 353–362. [Google Scholar] [CrossRef]
- Hamdan, M.; Urien, S.; Le Louet, H.; Tillement, J.P.; Morin, D. Inhibition of mitochondrial carnitine palmitoyltransferase-1 by a trimetazidine derivative, S-15176. Pharmacol. Res. 2001, 44, 99–104. [Google Scholar] [CrossRef]
- Elimadi, A.; Jullien, V.; Tillement, J.P.; Morin, D.S. S-15176 inhibits mitochondrial permeability transition via a mechanism independent of its antioxidant properties. Eur. J. Pharmacol. 2003, 468, 93–101. [Google Scholar] [CrossRef]
- Kawashima, S.; Yamamoto, T.; Horiuchi, Y.; Fujiwara, K.; Gouda, S.; Yoshimura, Y.; Yamamoto, A.; Inotani, Y.; Yamashita, K.; Kitamura, S.; et al. S-15176 and its methylated derivative suppress the CsA-insensitive mitochondrial permeability transition and subsequent cytochrome c release induced by silver ion, and show weak protonophoric activity. Mol. Cell Biochem. 2011, 358, 45–51. [Google Scholar] [CrossRef]
- Erdoğan, H.; Tunçdemir, M.; Kelten, B.; Akdemir, O.; Karaoğlan, A.; Taşdemiroğlu, E. The Effects of Difumarate Salt S-15176 after Spinal Cord Injury in Rats. J. Korean Neurosurg Soc. 2015, 57, 445–454. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef]
- Elimadi, A.; Sapena, R.; Settaf, A.; Le Louet, H.; Tillement, J.; Morin, D. Attenuation of liver normothermic ischemia-reperfusion injury by preservation of mitochondrial functions with S-15176, a potent trimetazidine derivative. Biochem. Pharmacol. 2001, 62, 509–516. [Google Scholar] [CrossRef]
- Settaf, A.; Zahidy, M.; Elimadi, A.; Sapena, R.; Alsamad, I.A.; Tillement, J.; Morin, D. S-15176 reduces the hepatic injury in rats subjected to experimental ischemia and reperfusion. Eur. J. Pharmacol. 2000, 406, 281–292. [Google Scholar] [CrossRef]
- Rupp, H.; Zarain-Herzberg, A.; Maisch, B. The use of partial fatty acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz 2002, 27, 621–636. [Google Scholar] [CrossRef]
- Morin, D.; Zini, R.; Berdeaux, A.; Tillement, J.P. Effect of the mitochondrial transition pore inhibitor, S-15176, on rat liver mitochondria: ATP synthase modulation and mitochondrial uncoupling induction. Biochem. Pharmacol. 2006, 72, 911–918. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Penkov, N.V.; Nedopekina, D.A.; Sharapov, V.A.; Khoroshavina, E.I.; Davletshin, E.V.; Belosludtseva, N.V.; Spivak, A.Y.; et al. Mitochondria-targeted prooxidant effects of betulinic acid conjugated with delocalized lipophilic cation F16. Free Radic. Biol. Med. 2021, 168, 55–69. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Agafonov, A.V.; Pavlik, L.L.; Dubinin, M.V. Effect of bedaquiline on the functions of rat liver mitochondria. Biochim. Biophys. Acta Biomembr. 2019, 1861, 288–297. [Google Scholar] [CrossRef]
- Pollard, A.K.; Craig, E.L.; Chakrabarti, L. Mitochondrial complex I activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS ONE 2016, 11, e0157405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Catalán, M.; Olmedo, I.; Faúndez, J.; Jara, J.A. Medicinal chemistry targeting mitochondria: From new vehicles and pharmacophore groups to old drugs with mitochondrial activity. Int. J. Mol. Sci. 2020, 21, 8684. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Aspects Med. 2010, 31, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J. Drug-induced mitochondrial toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef]
- Schuurman, H.-J.; Loveren, H.V.; Rozing, J.; Vos, J.G. Chemicals trophic for the thymus: Risk for immunodeficiency and autoimmunity. Int. J. Immunopharm. 1992, 14, 369–375. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Mironova, G.D.; Pavlov, E.V. Mitochondrial cyclosporine A-independent palmitate/Ca2+-induced permeability transition pore (PA-mPT Pore) and its role in mitochondrial function and protection against calcium overload and glutamate toxicity. Cells 2021, 10, 125. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S-15176, μM | V Respiration, nmol O2 × min−1 × mg−1 Protein | ||||
|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | RCR | |
| Glutamate + malate | |||||
| 0 | 3.1 ± 0.2 | 21.5 ± 1.1 | 3.5 ± 0.1 | 21.8 ± 1.0 | 5.9 ± 0.1 |
| 10 | 4.0 ± 0.3 * | 18.1 ± 0.8 * | 3.9 ± 0.2 | 17.9 ± 0.9 * | 4.7 ± 0.1 *** |
| 30 | 4.5 ± 0.3 ** | 16.0 ± 0.2 ** | 5.9 ± 0.1 ** | 15.9 ± 0.2 ** | 2.7 ± 0.1 *** |
| Succinate | |||||
| 0 | 6.0 ± 0.3 | 32.8 ± 0.6 | 6.9 ± 0.2 | 47.2 ± 1.2 | 4.8 ± 0.1 |
| 10 | 7.7 ± 0.2 ** | 30.7 ± 0.6 * | 8.4 ± 0.3 ** | 44.1 ± 0.8 | 3.7 ± 0.2 ** |
| 30 | 8.9 ± 0.2 ** | 29.3 ± 0.9 * | 10.8 ± 0.3 ** | 40.9 ± 1.4 * | 2.7 ± 0.1 *** |
| Ascorbate + TMPD | |||||
| 0 | 25.4 ± 0.6 | 34.9 ± 1.0 | 23.7 ± 0.4 | 38.9 ± 1.3 | 1.5 ± 0.1 |
| 10 | 27.1 ± 0.8 | 35.3 ± 0.8 | 25.0 ± 0.5 | 37.7 ± 0.9 | 1.4 ± 0.1 |
| 30 | 29.0 ± 0.5 ** | 35.7 ± 0.5 | 26.7 ± 0.3 * | 36.4 ± 1.1 | 1.3 ± 0.1 * |
| Values in % of Activity Compared with the Control (100%) | |||
|---|---|---|---|
| CI | CII | CIII | CIV |
| 102.7 ± 1.9 | 99.4 ± 3.2 | 63.3 ± 1.4 * | 105.1 ± 3.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtseva, N.V.; Starinets, V.S.; Semenova, A.A.; Igoshkina, A.D.; Dubinin, M.V.; Belosludtsev, K.N. S-15176 Difumarate Salt Can Impair Mitochondrial Function through Inhibition of the Respiratory Complex III and Permeabilization of the Inner Mitochondrial Membrane. Biology 2022, 11, 380. https://doi.org/10.3390/biology11030380
Belosludtseva NV, Starinets VS, Semenova AA, Igoshkina AD, Dubinin MV, Belosludtsev KN. S-15176 Difumarate Salt Can Impair Mitochondrial Function through Inhibition of the Respiratory Complex III and Permeabilization of the Inner Mitochondrial Membrane. Biology. 2022; 11(3):380. https://doi.org/10.3390/biology11030380
Chicago/Turabian StyleBelosludtseva, Natalia V., Vlada S. Starinets, Alena A. Semenova, Anastasia D. Igoshkina, Mikhail V. Dubinin, and Konstantin N. Belosludtsev. 2022. "S-15176 Difumarate Salt Can Impair Mitochondrial Function through Inhibition of the Respiratory Complex III and Permeabilization of the Inner Mitochondrial Membrane" Biology 11, no. 3: 380. https://doi.org/10.3390/biology11030380
APA StyleBelosludtseva, N. V., Starinets, V. S., Semenova, A. A., Igoshkina, A. D., Dubinin, M. V., & Belosludtsev, K. N. (2022). S-15176 Difumarate Salt Can Impair Mitochondrial Function through Inhibition of the Respiratory Complex III and Permeabilization of the Inner Mitochondrial Membrane. Biology, 11(3), 380. https://doi.org/10.3390/biology11030380