
NF-κB Signaling and Inflammation—Drug Repurposing to Treat Inflammatory Disorders?



Simple Summary
Abstract
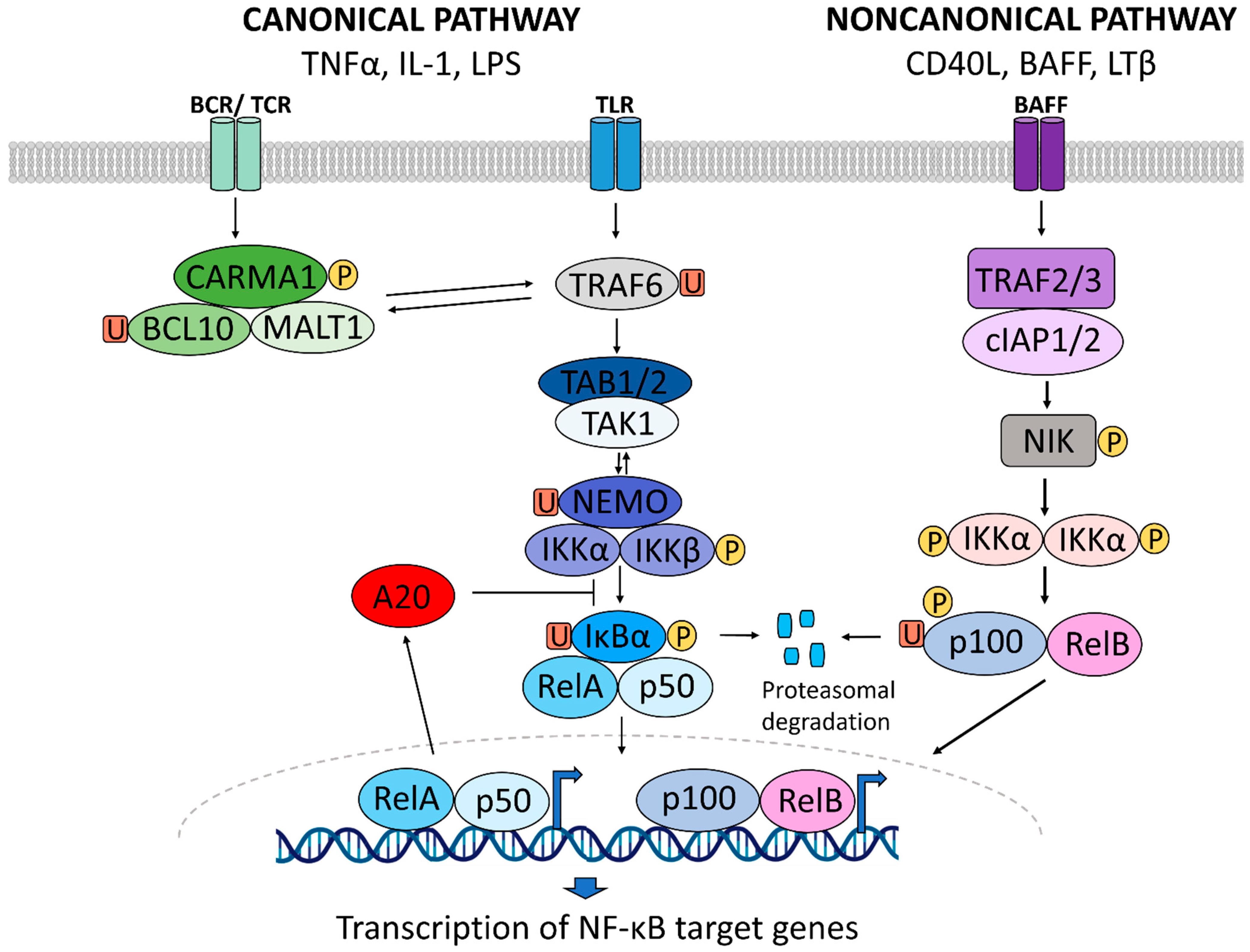
1. NF-κB Signaling in Inflammation
1.1. A Brief History of NF-κB Signaling in Inflammatory Diseases
1.2. NF-κB Signaling in Inflammatory Diseases
1.3. NF-κB Activity as a Druggable Target in Inflammatory Diseases
2. Drug Repurposing to Identify NF-κB Inhibitors
2.1. Why Do We Need New Anti-Inflammatory Drugs?
2.2. Approaches to Drug Repurposing
3. Computer-Based Drug Repurposing Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resource | Description | References |
|---|---|---|
| Databases and Libraries | ||
| Drug Repurposing Hub | Annotated library of FDA-approved drugs, drugs undergoing clinical trials, and preclinical tool compounds | [88] |
| DrugCentral | Online drug information resource containing 4714 drugs and 129,975 pharmaceuticals, providing up-to-date drug information, including a drug repositioning prioritization scheme for FDA-approved drugs | [89] |
| CheMBL | Database of bioactive molecules with druglike properties, containing chemical, bioactivity and genomic data to aid translation of genomic information into effective new drugs | [90,91] |
| ReDo_Trials Database | Database of active clincal trials investigating repurposed drugs for cancer therapy | [92] |
| RepoDB | Database of approved and failed drugs and their indications | [93] |
| ReFrame Database | Commercially available screening library of 12,000 molecules for use in high throughput cell-based repurposing assays | [94] |
| Zinc | Library of >700,000 small molecules for use in computational screening | [95] |
| COVID-19 Drug Repurposing Database (Excelra) | Commercially available database of approved drugs which can rapidly be entered into phase II or III trials against COVID-19 | [96] |
| DrugRepurposing Online (Nimedicus) | Commercially available database of 9040 candidate repurposing compounds annotated with indications and mechanisms | [97] |
| PROMISCUOUS | Publicly available database of 25,000 drugs annotated with drug-protein, protein-protein interactions, drug structural similarity and known side-effects | [98] |
| Methods | ||
| DeepDTNet | Deep learning system for identification of novel targets for drug repurposing in disease specific contexts | [99] |
| AOPDEF | Deep learning system identifying molecular targets among known drugs on two external validation sets | [100] |
| MBiRW | Computational method to identify novel indications for given drugs | [101] |
| KinderMiner | Text mining method to identify repurposing candidates | [102] |
| DrugQuest | Text mining method to identify simmilarities between DrugBank entries | [103] |
| Semantic Link Association Prediction (SLAP) | Statistical algorithm to predict novel drug-target pairs | [104] |
3.1. Pharmacophore Modeling-Based Drug Repurposing
3.2. Artificial Intelligence-Aided Drug Repurposing
4. Experimental Approaches to Drug Repurposing
4.1. Target-Based Approaches to Drug Repurposing
4.2. Phenotypic Screening Approaches to Drug Repurposing in Cell Lines and Model Organisms
5. NF-κB as a Potential Target for Drug Development in CNS Inflammation
5.1. NF-κB Activation in T Cells in MS/EAE
5.2. Role of NF-κB Activation in Macrophages and Microglia in MS
| Cell Type | Genotypic Alteration in NF-κB Signaling | Effect on Neuroinflammation | References |
|---|---|---|---|
| T cells | IKKβ deficient T cells | Resistance to EAE, impaired autoreactive T cell activation and expansion | [149] |
| p50 deficient | Attenuated EAE incidence and severity, impaired Th1 and Th2 differentiation | [150] | |
| c-Rel deficient | Resistance to EAE, defective Th1 and Th17 development | [148,151] | |
| MALT1 deficient | Protection from EAE, absence of demyelination, proinflammatory cytokines and immune cell infiltration into spinal cord. Effector function of autoreactive Th17 cells impaired | [165,166] | |
| CARMA1 deficient | Resistance to EAE, impaired Th17 differentiation | [167] | |
| IκBαΔN | Resistance to EAE, reduced Th17 differentiation | [167] | |
| NIK deficient | Protection from EAE due to DC function and independent from CD4+ T cell function | [168] | |
| NIK deficient | Resistance to EAE, impaired Th17 differentiation | [152] | |
| NIK deficient T cells | Attenuation of EAE, reduced generation of Th1 and Th17 cells, reduced immune cell infiltration | [153] | |
| Macrophages/ Microglia | IκBα deficient | Exacerbated EAE, increased immune cell infiltration and myeloid-derived proinflammatory cytokines | [160] |
| IKKβ deficient macrophages/ microglia | Attenuation of EAE, reduced immune cell infiltration, production of proinflammatory cytokines and permeability of the BBB. Increase in Tregs and decrease of Th1 and Th17 cells | [161,162] | |
| TAK1 deficient microglia | Reduced CNS inflammation and neurodegeneration, NF-κB inhibition | [163] | |
| TREM2 overexpressing myeloid precursor cells | Attenuation of EAE, reduced neurodegeneration, increase in anti-inflammatory cytokines and phagocytosis | [164] | |
| A20 deficient microglia | Aggravated EAE, Nrp3 inflammasome activation, increase in immune cell infiltration and proinflammatory cytokine production | [169] | |
| Astrocytes | IκBα overexpressing astrocytes | Attenuation of EAE, decreased immune cell infiltration and production of proinflammatory cytokines | [170,171,172] |
| IKKβ deficient astrocytes | Protection from myelin loss in cuprizone-induced inflammation model | [173] | |
| A20 deficient astrocytes | Aggravated EAE, increase in immune cell infiltration and proinflammatory cytokine production | [174,175] | |
| Oligodendrocytes | IκBαΔN in oligodendrocytes | Aggravated EAE, reduced remyelination and oligodendrocyte death in cuprizone-induced inflammation model | [176] |
| IKKβ deficient oligodendrocytes | No protection from demyelination in cuprizone-induced inflammation model | [173] | |
| Neurons | IKKβ deficient neurons | Aggravated EAE, increased Th1 infiltration and proinflammatory cytokine production. Reduced production of neuroprotective factors | [177] |
| IκBα overexpressing neurons | No effect on EAE progression or inflammation | [178] |
5.3. NF-κB Activation in CNS Cells in MS

5.4. Repurposing NF-κB Inhibitors to Treat CNS Inflammation
6. NF-κB as a Potential Target for Drug Development in Joint Inflammation
6.1. NF-κB Activation in Innate Immune Cells in RA

6.2. NF-κB Activation in T and B Cells in RA
6.3. Repurposing NF-κB Inhibitors to Treat Joint Inflammation
7. Drug Repurposing for Targeting Inflammation in COVID-19 Pneumonia
8. Problems with Progressing Repurposed Drugs to Clinical Applications
9. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Amiri, K.I.; Richmond, A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005, 24, 301–313. [Google Scholar] [CrossRef]
- Chen, F.; Demers, L.M.; Shi, X. Upstream signal transduction of NF-κB activation. Curr. Drug Targets-Inflamm. Allergy 2002, 1, 137–149. [Google Scholar] [CrossRef]
- Ben-Neriah, Y. Regulatory functions of ubiquitination in the immune system. Nat. Immunol. 2002, 3, 20–26. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Tchou-Wong, K.-M.; Rom, W.N. NF-kappaB in Lung Tumorigenesis. Cancers 2011, 3, 4258–4268. [Google Scholar] [CrossRef] [PubMed]
- Imbert, V.; Peyron, J.F. NF-κB in Hematological Malignancies. Biomedicines 2017, 5, 27. [Google Scholar] [CrossRef]
- Jiang, L.; Ren, L.; Zhang, X.; Chen, H.; Chen, X.; Lin, C.; Wang, L.; Hou, N.; Pan, J.; Zhou, Z.; et al. Overexpression of PIMREG promotes breast cancer aggressiveness via constitutive activation of NF-κB signaling. EBioMedicine 2019, 43, 188–200. [Google Scholar] [CrossRef]
- Jiang, L.; Wu, J.; Yang, Y.; Liu, L.; Song, L.; Li, J.; Li, M. Bmi-1 promotes the aggressiveness of glioma via activating the NF-kappaB/MMP-9 signaling pathway. BMC Cancer 2012, 12, 406. [Google Scholar] [CrossRef]
- Schön, M.; Wienrich, B.G.; Kneitz, S.; Sennefelder, H.; Amschler, K.; Vöhringer, V.; Weber, O.; Stiewe, T.; Ziegelbauer, K.; Schön, M.P. KINK-1, a novel small-molecule inhibitor of IKKbeta, and the susceptibility of melanoma cells to antitumoral treatment. J. Natl. Cancer Inst. 2008, 100, 862–875. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J.; Verma, I.M. Proposed NF-kappa B/I kappa B family nomenclature. Genes Dev. 1993, 7, 2063. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Gurunathan, S.; Winkles, J.A.; Ghosh, S.; Hayden, M.S. Regulation of fibroblast growth factor-inducible 14 (Fn14) expression levels via ligand-independent lysosomal degradation. J. Biol. Chem. 2014, 289, 12976–12988. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.C. NF-κB in inflammation and renal diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Schröfelbauer, B.; Polley, S.; Behar, M.; Ghosh, G.; Hoffmann, A. NEMO ensures signaling specificity of the pleiotropic IKKβ by directing its kinase activity toward IκBα. Mol. Cell 2012, 47, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Ley, S.C. New insights into NF-kappaB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef]
- Beinke, S.; Ley, S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004, 382 Pt 2, 393–409. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Sun, S.C.; Liu, Z.G. A special issue on NF-κB signaling and function. Cell Res. 2011, 21, 1–2. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.A.; Mitchell, J.P.; Cook, S.J. Inhibitory feedback control of NF-κB signalling in health and disease. Biochem. J. 2021, 478, 2619–2664. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef]
- Habib, A.A.; Chatterjee, S.; Park, S.-K.; Ratan, R.R.; Lefebvre, S.; Vartanian, T. The epidermal growth factor receptor engages receptor interacting protein and nuclear factor-κB (NF-κB)-inducing kinase to activate NF-κB: Identification of a novel receptor-tyrosine kinase signalosome. J. Biol. Chem. 2001, 276, 8865–8874. [Google Scholar] [CrossRef]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 is a C-terminal IκB kinase responsible for NF-κB activation during the UV response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65: Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Garbati, M.R. Inhibition of NF-κB signaling as a strategy in disease therapy. Curr. Top. Microbiol. Immunol. 2011, 349, 245–263. [Google Scholar] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Rainsford, K.D. Profile and mechanisms of gastrointestinal and other side effects of nonsteroidal anti-inflammatory drugs (NSAIDs). Am. J. Med. 1999, 107, 27S–35S; discussion 35S–36S. [Google Scholar] [CrossRef]
- Simon, L.S. Nonsteroidal anti-inflammatory drugs and their risk: A story still in development. Arthritis Res. Ther. 2013, 15 (Suppl. 3), S1. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Pavlaki, A.N.; Alexandra, M.A.M.; Chrousos, G.P. Glucocorticoid Therapy and Adrenal Suppression. In Feingold; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Oray, M.; Samra, K.A.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457. [CrossRef]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; de Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-Examining the Role of TNF in MS Pathogenesis and Therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-α Blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonté, C. Actions of the antihistaminergic clemastine on presymptomatic SOD1-G93A mice ameliorate ALS disease progression. J. Neuroinflamm. 2016, 13, 191. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Niu, J.; Hoi, K.K.; Zhao, C.; Caganap, S.D.; Henry, R.G.; Dao, D.Q.; Zollinger, D.R.; Mei, F.; Shen, Y.-A.A.; et al. Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Brain 2017, 141, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Leurs, R.; Church, M.K.; Taglialatela, M. H1-antihistamines: Inverse agonism, anti-inflammatory actions and cardiac effects. Clin. Exp. Allergy 2002, 32, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; He, Y.; Fan, S.; Sun, B. Clemastine rescues behavioral changes and enhances remyelination in the cuprizone mouse model of demyelination. Neurosci. Bull. 2015, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dupree, J.L.; Gacias, M.; Frawley, R.; Sikder, T.; Naik, P.; Casaccia, P. Clemastine Enhances Myelination in the Prefrontal Cortex and Rescues Behavioral Changes in Socially Isolated Mice. J. Neurosci. 2016, 36, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.-A.A.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.; Sagan, S.A.; Xiao, L.; Teuscher, C. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 2016, 5, e18246. [Google Scholar] [CrossRef]
- Su, W.J.; Zhang, T.; Jiang, C.L.; Wang, W. Clemastine Alleviates Depressive-Like Behavior Through Reversing the Imbalance of Microglia-Related Pro-inflammatory State in Mouse Hippocampus. Front. Cell. Neurosci. 2018, 12, 412. [Google Scholar] [CrossRef]
- Yuan, X.; Juan, Z.; Zhang, R.; Sun, X.; Yan, R.; Yue, F.; Huang, Y.; Yu, J.; Xia, X. Clemastine Fumarate Protects Against Myocardial Ischemia Reperfusion Injury by Activating the TLR4/PI3K/Akt Signaling Pathway. Front. Pharmacol. 2020, 11, 28. [Google Scholar] [CrossRef]
- Zada, D.; Tovin, A.; Lerer-Goldshtein, T.; Appelbaum, L. Pharmacological treatment and BBB-targeted genetic therapy for MCT8-dependent hypomyelination in zebrafish. Dis. Models Mech. 2016, 9, 1339–1348. [Google Scholar]
- Almatroodi, S.A.; Alrumaihi, F.; Alsahli, M.A.; Alhommrani, M.F.; Khan, A.; Rahmani, A.H. Curcumin, an Active Constituent of Turmeric Spice: Implication in the Prevention of Lung Injury Induced by Benzo(a) Pyrene (BaP) in Rats. Molecules 2020, 25, 724. [Google Scholar] [CrossRef]
- Chai, Y.-S.; Chen, Y.-Q.; Lin, S.-H.; Xie, K.; Wang, C.-J.; Yang, Y.-Z.; Xu, F. Curcumin regulates the differentiation of naïve CD4+T cells and activates IL-10 immune modulation against acute lung injury in mice. Biomed. Pharmacother. 2020, 125, 109946. [Google Scholar] [CrossRef] [PubMed]
- Soni, V.K.; Mehta, A.; Ratre, Y.K.; Tiwari, A.K.; Amit, A.; Singh, R.P.; Sonkar, S.C.; Chaturvedi, N.; Shukla, D.; Vishvakarma, N.K. Curcumin, a traditional spice component, can hold the promise against COVID-19? Eur. J. Pharmacol. 2020, 886, 173551. [Google Scholar] [CrossRef]
- Fu, Q.; Hue, J.; Li, S. Nonsteroidal Anti-Inflammatory Drugs Promote Axon Regeneration via RhoA Inhibition. J. Neurosci. 2007, 27, 4154–4164. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.A.; Liebscher, T.; Watzlawick, R.; Martus, P.; Laufer, S.; Blex, C.; Schindler, R.; Jungehulsing, G.J.; Knüppel, S.; Kreutzträger, M.; et al. SCISSOR-Spinal Cord Injury Study on Small molecule-derived Rho inhibition: A clinical study protocol. BMJ Open 2016, 6, e010651. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, M.J.; Cook, J.L. Nonsteroidal Anti-Inflammatory Drugs and Their Neuroprotective Role After an Acute Spinal Cord Injury: A Systematic Review of Animal Models. Glob. Spine J. 2021, 11, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.F.; Claudino, R.F.; Dutra, R.C.; Marcon, R.; Calixto, J.B. Omega-3 fatty acid-derived mediators 17 (R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J. Immunol. 2011, 187, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-W.; Lee, S.-M. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861 Pt A, 1025–1035. [Google Scholar] [CrossRef]
- Ren, Y.-Z.; Zhang, B.-Z.; Zhao, X.-J.; Zhang, Z.-Y. Resolvin D1 ameliorates cognitive impairment following traumatic brain injury via protecting astrocytic mitochondria. J. Neurochem. 2020, 154, 530–546. [Google Scholar] [CrossRef]
- Wang, B.; Gong, X.; Wan, J.-y.; Zhang, L.; Zhang, Z.; Li, H.-z.; Min, S. Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm. Pharmacol. Ther. 2011, 24, 434–441. [Google Scholar] [CrossRef]
- Wei, C.; Guo, S.; Liu, W.; Jin, F.; Wei, B.; Fan, H.; Su, H.; Liu, J.; Zhang, N.; Fang, D.; et al. Resolvin D1 ameliorates Inflammation-Mediated Blood-Brain Barrier Disruption after Subarachnoid Hemorrhage in rats by Modulating A20 and NLRP3 Inflammasome. Front. Pharmacol. 2021, 11, 10734. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.; Lee, S.; Lee, J.; Kim, K.; Lee, D. Inferring new drug indications using the complementarity between clinical disease signatures and drug effects. J. Biomed. Inform. 2016, 59, 248–257. [Google Scholar] [PubMed]
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Low, Z.Y.; Farouk, I.A.; Lal, S.K. Drug Repositioning: New Approaches and Future Prospects for Life-Debilitating Diseases and the COVID-19 Pandemic Outbreak. Viruses 2020, 12, 1058. [Google Scholar]
- Prakash, A.V.; Park, J.W.; Seong, J.-W.; Kang, T.J. Repositioned Drugs for Inflammatory Diseases such as Sepsis, Asthma, and Atopic Dermatitis. Biomol. Ther. 2020, 28, 222–229. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug-disease relationships for computational drug repositioning. Brief. Bioinform. 2011, 12, 303–311. [Google Scholar]
- Sam, E.; Athri, P. Web-based drug repurposing tools: A survey. Brief. Bioinform. 2017, 20, 299–316. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef]
- Baig, M.S.; Roy, A.; Saqib, U.; Rajpoot, S.; Srivastava, M.; Naim, A.; Liu, D.; Saluja, R.; Faisal, S.M.; Pan, Q.; et al. Repurposing Thioridazine (TDZ) as an anti-inflammatory agent. Sci. Rep. 2018, 8, 12471. [Google Scholar] [CrossRef]
- Feinberg, S.M.; Saadabadi, A.F.K. Thioridazine. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459140/ (accessed on 2 February 2022).
- Anthony, N.G.; Baiget, J.; Berretta, G.; Boyd, M.; Breen, D.; Edwards, J.; Gamble, C.; Gray, A.I.; Harvey, A.L.; Hatziieremia, S.; et al. Inhibitory Kappa B Kinase α (IKKα) Inhibitors That Recapitulate Their Selectivity in Cells against Isoform-Related Biomarkers. J. Med. Chem. 2017, 60, 7043–7066. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-molecule inhibitors of IκB kinase (IKK) and IKK-related kinases. Pharm. Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear Factor κB (NF-κB)–Mediated Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Bologa, C.G.; Holmes, J.; Bocci, G.; Wilson, T.B.; Nguyen, D.-T.; Curpan, R.; Halip, L.; Bora, A.; Yang, J.J.; et al. DrugCentral 2021 supports drug discovery and repositioning. Nucleic Acids Res. 2020, 49, D1160–D1169. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, L.; Giambartolomei, C.; Pereira, A.C.; Gaulton, A.; Posner, D.C.; Swanson, S.A.; Ho, Y.-L.; Iyengar, S.K.; Kosik, N.M.; Vujkovic, M.; et al. Actionable druggable genome-wide Mendelian randomization identifies repurposing opportunities for COVID-19. Nat. Med. 2021, 27, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; de Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Pantziarka, P.; Vandeborne, L.; Bouche, G. A Database of Drug Repurposing Clinical Trials in Oncology. Front. Pharmacol. 2021, 12, 790952. [Google Scholar] [CrossRef]
- Brown, A.S.; Patel, C.J. A standard database for drug repositioning. Sci. Data 2017, 4, 170029. [Google Scholar] [CrossRef]
- Janes, J.; Young, M.E.; Chen, E.; Rogers, N.H.; Burgstaller-Muehlbacher, S.; Hughes, L.D.; Love, M.S.; Hull, M.V.; Kuhen, K.L.; Woods, A.K.; et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2018, 115, 10750–10755. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Drug Repurposing Database. 2020. Available online: https://www.excelra.com/covid-19-drug-repurposing-database/ (accessed on 12 February 2022).
- Drug Repurposing Online. Available online: https://drugrepurposing.info/ (accessed on 12 February 2022).
- von Eichborn, J.; Murgueitio, M.S.; Dunkel, M.; Koerner, S.; Bourne, P.E.; Preissner, R. PROMISCUOUS: A database for network-based drug-repositioning. Nucleic Acids Res. 2010, 39 (Suppl. 1), D1060–D1066. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Hou, Y.; Zhang, P.; Li, L.; Li, J.; Huang, L.F.; Lewis, S.J.; Nussinov, R.; Cheng, F. Network-based prediction of drug–target interactions using an arbitrary-order proximity embedded deep forest. Bioinformatics 2020, 36, 2805–2812. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef]
- Kuusisto, F.; Steill, J.; Kuang, Z.; Thomson, J.; Page, D.; Stewart, R. A Simple Text Mining Approach for Ranking Pairwise Associations in Biomedical Applications. AMIA Jt. Summits Transl. Sci. Proc. 2017, 2017, 166–174. [Google Scholar]
- Papanikolaou, N.; Pavlopoulos, G.A.; Theodosiou, T.; Vizirianakis, I.S.; Iliopoulos, I. DrugQuest-a text mining workflow for drug association discovery. BMC Bioinform. 2016, 17 (Suppl. 5), 182. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ding, Y.; Wild, D.J. Assessing drug target association using semantic linked data. PLoS Comput. Biol. 2012, 8, e1002574. [Google Scholar] [CrossRef]
- Luu, T. Pharmacophore-Guided Virtual Screening for Drug Repurposing. 2020. Available online: https://blogs.3ds.com/biovia/pharmacophore-guided-virtual-screening-for-drug-repurposing/ (accessed on 2 February 2022).
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.M.H.; Abdel-Maksoud, M.S.; Hassan, R.M.; Mersal, K.I.; Ammar, U.M.; Se-In, C.; He-Soo, H.; Kim, H.K.; Lee, A.; Lee, K.T.; et al. Design, synthesis and anti-inflammatory activity of imidazol-5-yl pyridine derivatives as p38α/MAPK14 inhibitor. Bioorg. Med. Chem. 2021, 31, 115969. [Google Scholar] [CrossRef] [PubMed]
- Kanan, T.; Kanan, D.; Erol, I.; Yazdi, S.; Stein, M.; Durdagi, S. Targeting the NF-κB/IκBα complex via fragment-based E-Pharmacophore virtual screening and binary QSAR models. J. Mol. Graph. Model. 2019, 86, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Balaramnavar, V.M.; Ahmad, K.; Saeed, M.; Ahmad, I.; Kamal, M.; Jawed, T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. [Google Scholar] [CrossRef]
- Rampogu, S.; Lee, K.W. Pharmacophore Modelling-Based Drug Repurposing Approaches for SARS-CoV-2 Therapeutics. Front. Chem. 2021, 9, 636362. [Google Scholar] [CrossRef]
- Sundaresan, L.; Giri, S.; Singh, H.; Chatterjee, S. Repurposing of thalidomide and its derivatives for the treatment of SARS-CoV-2 infections: Hints on molecular action. Br. J. Clin. Pharmacol. 2021, 87, 3835–3850. [Google Scholar] [CrossRef]
- Rajan, S.; Satish, S.; Shankar, K.; Pandeti, S.; Varshney, S.; Srivastava, A.; Kumar, D.; Gupta, A.; Gupta, S.; Choudhary, R.; et al. Aegeline inspired synthesis of novel β3-AR agonist improves insulin sensitivity in vitro and in vivo models of insulin resistance. Metabolism 2018, 85, 1–13. [Google Scholar] [CrossRef]
- Chen, C.; Qi, F.; Shi, K.; Li, Y.; Li, J.; Chen, Y.; Pan, J.; Zhou, T.; Lin, X.; Zhang, J.; et al. Thalidomide combined with low-dose short-term glucocorticoid in the treatment of critical Coronavirus Disease 2019. Clin. Transl. Med. 2020, 10, e35. [Google Scholar] [CrossRef]
- Li, Y.; Shi, K.; Qi, F.; Yu, Z.; Chen, C.; Pan, J.; Wu, G.; Chen, Y.; Li, J.; Chen, Y.; et al. Thalidomide combined with short-term low-dose glucocorticoid therapy for the treatment of severe COVID-19: A case-series study. Int. J. Infect. Dis. 2021, 103, 507–513. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology—Patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Ozanne, J.; Prescott, A.R.; Clark, K. The clinically approved drugs dasatinib and bosutinib induce anti-inflammatory macrophages by inhibiting the salt-inducible kinases. Biochem. J. 2015, 465, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, A.; Padey, B.; Terrier, O.; Rosa-Calatrava, M. Drug Repurposing Approaches for the Treatment of Influenza Viral Infection: Reviving Old Drugs to Fight against a Long-Lived Enemy. Front. Immunol. 2019, 10, 531. [Google Scholar] [CrossRef] [PubMed]
- D’Aura Swanson, C.; Paniagua, R.T.; Lindstrom, T.M.; Robinson, W.H. Tyrosine kinases as targets for the treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 317–324. [Google Scholar] [CrossRef]
- Keating, G.M. Dasatinib: A review in chronic myeloid leukaemia and Ph+ acute lymphoblastic leukaemia. Drugs 2017, 77, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Meeker, N.D.; Trede, N.S. Immunology and zebrafish: Spawning new models of human disease. Dev. Comp. Immunol. 2008, 32, 745–757. [Google Scholar] [CrossRef]
- Traver, D.; Herbomel, P.; Patton, E.; Murphey, R.D.; Yoder, J.; Litman, G.; Catic, A.; Amemiya, C.; Zon, L.; Trede, N. The zebrafish as a model organism to study development of the immune system. Adv. Immunol. 2003, 81, 253–330. [Google Scholar]
- Guo, K.; Bu, X.; Yang, C.; Cao, X.; Bian, H.; Zhu, Q.; Zhu, J.; Zhang, D. Treatment Effects of the Second-Generation Tyrosine Kinase Inhibitor Dasatinib on Autoimmune Arthritis. Front. Immunol. 2019, 9, 3133. [Google Scholar] [CrossRef]
- Hekim, C.; Ilander, M.; Yan, J.; Michaud, E.; Smykla, R.; Vähä-Koskela, M.; Savola, P.; Tähtinen, S.; Saikko, L.; Hemminki, A. Dasatinib changes immune cell profiles concomitant with reduced tumor growth in several murine solid tumor models. Cancer Immunol. Res. 2017, 5, 157–169. [Google Scholar] [CrossRef]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef]
- Korashy, H.M.; Rahman, A.M.; Kassem, M.G. Dasatinib. Profiles Drug Subst. Excip. Relat. Methodol. 2014, 39, 205–237. [Google Scholar]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Nierode, G.; Kwon, P.S.; Dordick, J.S.; Kwon, S.J. Cell-Based Assay Design for High-Content Screening of Drug Candidates. J. Microbiol. Biotechnol. 2016, 26, 213–225. [Google Scholar] [CrossRef]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef]
- Xie, Y.; Meijer, A.H.; Schaaf, M.J.M. Modeling Inflammation in Zebrafish for the Development of Anti-inflammatory Drugs. Front. Cell Dev. Biol. 2020, 8, 620984. [Google Scholar] [CrossRef]
- Hall, C.J.; Wicker, S.M.; Chien, A.-T.; Tromp, A.; Lawrence, L.M.; Sun, X.; Krissansen, G.W.; Crosier, K.E.; Crosier, P.S. Repositioning drugs for inflammatory disease–fishing for new anti-inflammatory agents. Dis. Models Mech. 2014, 7, 1069–1081. [Google Scholar] [CrossRef]
- Herbomel, P.; Thisse, B.; Thisse, C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development 1999, 126, 3735–3745. [Google Scholar] [CrossRef]
- Hermann, A.C.; Millard, P.J.; Blake, S.L.; Kim, C.H. Development of a respiratory burst assay using zebrafish kidneys and embryos. J. Immunol. Methods 2004, 292, 119–129. [Google Scholar] [CrossRef]
- Lam, S.H.; Chua, H.L.; Gong, Z.; Lam, T.J.; Sin, Y.M. Development and maturation of the immune system in zebrafish, Danio rerio: A gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 2004, 28, 9–28. [Google Scholar] [CrossRef]
- Page, D.M.; Wittamer, V.; Bertrand, J.Y.; Lewis, K.L.; Pratt, D.N.; Delgado, N.; Schale, S.E.; McGue, C.; Jacobsen, B.H.; Doty, A.; et al. An evolutionarily conserved program of B-cell development and activation in zebrafish. Blood 2013, 122, e1–e11. [Google Scholar] [CrossRef]
- White, D.T.; Saxena, M.T.; Mumm, J.S. Let’s get small (and smaller): Combining zebrafish and nanomedicine to advance neuroregenerative therapeutics. Adv. Drug Deliv. Rev. 2019, 148, 344–359. [Google Scholar] [CrossRef]
- Rihel, J.; Prober, D.A.; Arvanites, A.; Lam, K.; Zimmerman, S.; Jang, S.; Haggarty, S.J.; Kokel, D.; Rubin, L.L.; Peterson, R.T.; et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 2010, 327, 348–351. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet 2013, 45, 1353–1360. [Google Scholar]
- Hussman, J.P.; Beecham, A.H.; Schmidt, M.; Martin, E.R.; McCauley, J.L.; Vance, J.M.; Haines, J.L.; Pericak-Vance, M.A. GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. 2016, 17, 305–312. [Google Scholar] [CrossRef]
- Miterski, B.; Böhringer, S.; Klein, W.; Sindern, E.; Haupts, M.; Schimrigk, S.; Epplen, J.T. Inhibitors in the NFκB cascade comprise prime candidate genes predisposing to multiple sclerosis, especially in selected combinations. Genes Immun. 2002, 3, 211–219. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-κB: Roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef]
- Ruan, Q.; Chen, Y.H. Nuclear factor-κB in immunity and inflammation: The Treg and Th17 connection. Adv. Exp. Med. Biol. 2012, 946, 207–221. [Google Scholar]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef]
- Nomura, T.; Abe, Y.; Kamada, H.; Shibata, H.; Kayamuro, H.; Inoue, M.; Kawara, T.; Arita, S.; Furuya, T.; Yamashita, T.; et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J. Control Release 2011, 149, 8–14. [Google Scholar] [CrossRef]
- Petrasch, S. Follicular dendritic cells in malignant lymphomas. Curr. Top. Microbiol. Immunol. 1995, 201, 189–203. [Google Scholar]
- van Doorn, R.; Pinheiro, M.A.L.; Kooij, G.; Lakeman, K.; Hof, B.v.; van der Pol, S.M.; Geerts, D.; van Horssen, J.; van der Valk, P.; van der Kam, E.; et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J. Neuroinflamm. 2012, 9, 133. [Google Scholar] [CrossRef]
- Chen, G.; Hardy, K.; Pagler, E.; Ma, L.; Lee, S.; Gerondakis, S.; Daley, S.; Shannon, M.F. The NF-κB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 4483–4491. [Google Scholar] [CrossRef]
- Greve, B.; Weissert, R.; Hamdi, N.; Bettelli, E.; Sobel, R.A.; Coyle, A.; Kuchroo, V.K.; Rajewsky, K.; Schmidt-Supprian, M. IκB Kinase 2/β Deficiency Controls Expansion of Autoreactive T Cells and Suppresses Experimental Autoimmune Encephalomyelitis. J. Immunol. 2007, 179, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, B.; Samoilova, E.B.; Liu, T.-S.T.; Rostami, A.; Chen, Y. Experimental Autoimmune Encephalomyelitis in NF-κB- Deficient Mice: Roles of NF-κB in the Activation and Differentiation of Autoreactive T Cells. J. Immunol. 1999, 163, 2937–2943. [Google Scholar]
- Hilliard, B.A.; Mason, N.; Xu, L.; Sun, J.; Lamhamedi-Cherradi, S.E.; Liou, H.C.; Hunter, C.; Chen, Y.H. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J. Clin. Investig. 2002, 110, 843–850. [Google Scholar] [CrossRef]
- Jin, W.; Zhou, X.F.; Yu, J.; Cheng, X.; Sun, S.C. Regulation of Th17 cell differentiation and EAE induction by MAP3K NIK. Blood 2009, 113, 6603–6610. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Zhou, X.; Xie, X.; Chen, X.; Jie, Z.; Zou, Q.; Hu, H.; Zhu, L.; Cheng, X.; et al. Cell intrinsic role of NF-κB-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci. Rep. 2016, 6, 22115. [Google Scholar] [CrossRef]
- Yue, Y.; Stone, S.; Lin, W. Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen. Res. 2018, 13, 1507–1515. [Google Scholar]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Hayes, C.E.; Spanier, J.A. Chapter 10-Multiple Sclerosis in Women: Vitamin D and Estrogen Synergy for Autoimmune T-Cell Regulation and Demyelinating Disease Prevention. In Nutrition and Lifestyle in Neurological Autoimmune Diseases; Watson, R.R., Killgore, W.D.S., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 81–107. [Google Scholar]
- Phelan, J. Generating EAE Mouse Models of Multiple Sclerosis. 2016. Available online: https://www.taconic.com/taconic-insights/neuroscience/eae-mouse-models-of-multiple-sclerosis.html (accessed on 2 February 2022).
- Blank, T.; Prinz, M. NF-κB signaling regulates myelination in the CNS. Front. Mol. Neurosci. 2014, 7, 47. [Google Scholar] [CrossRef]
- Leibowitz, S.M.; Yan, J. NF-κB Pathways in the Pathogenesis of Multiple Sclerosis and the Therapeutic Implications. Front. Mol. Neurosci. 2016, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Thöne, J.; Lee, D.-H.; Rupec, R.A.; Gold, R.; Linker, R.A. Constitutive activity of NF-kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J. Neuroinflamm. 2012, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Decker, Y.; Schnöder, L.; Schottek, A.; Li, D.; Menger, M.D.; Fassbender, K.; Liu, Y. Deficiency of IκB Kinase β in Myeloid Cells Reduces Severity of Experimental Autoimmune Encephalomyelitis. Am. J. Pathol. 2016, 186, 1245–1257. [Google Scholar] [CrossRef]
- Lee, M.J.; Bing, S.J.; Choi, J.; Jang, M.; Lee, G.; Lee, H.; Chang, B.S.; Jee, Y.; Lee, S.J.; Cho, I.-H. IKKβ-mediated inflammatory myeloid cell activation exacerbates experimental autoimmune encephalomyelitis by potentiating Th1/Th17 cell activation and compromising blood brain barrier. Mol. Neurodegener. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Müller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef]
- Takahashi, K.; Prinz, M.; Stagi, M.; Chechneva, O.; Neumann, H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 2007, 4, e124. [Google Scholar] [CrossRef]
- Brüstle, A.; Brenner, D.; Knobbe, C.B.; Lang, P.A.; Virtanen, C.; Hershenfield, B.M.; Reardon, C.; Lacher, S.M.; Ruland, J.; Ohashi, P.S.; et al. The NF-κB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J. Clin. Investig. 2012, 122, 4698–4709. [Google Scholar] [CrossRef]
- Mc Guire, C.; Wieghofer, P.; Elton, L.; Muylaert, D.; Prinz, M.; Beyaert, R.; van Loo, G. Paracaspase MALT1 deficiency protects mice from autoimmune-mediated demyelination. J. Immunol. 2013, 190, 2896–2903. [Google Scholar] [CrossRef]
- Molinero, L.L.; Cubre, A.; Mora-Solano, C.; Wang, Y.; Alegre, M.L. T cell receptor/CARMA1/NF-κB signaling controls T-helper (Th) 17 differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 18529–18534. [Google Scholar] [CrossRef]
- Hofmann, J.; Mair, F.; Greter, M.; Schmidt-Supprian, M.; Becher, B. NIK signaling in dendritic cells but not in T cells is required for the development of effector T cells and cell-mediated immune responses. J. Exp. Med. 2011, 208, 1917–1929. [Google Scholar] [CrossRef]
- Voet, S.; Guire, C.M.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Walle, L.V.; Jordao, M.J.C.; et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef]
- Brambilla, R.; Dvoriantchikova, G.; Barakat, D.; Ivanov, D.; Bethea, J.R.; Shestopalov, V.I. Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J. Neuroinflamm. 2012, 9, 213. [Google Scholar] [CrossRef]
- Brambilla, R.; Morton, P.D.; Ashbaugh, J.J.; Karmally, S.; Lambertsen, K.L.; Bethea, J.R. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia 2014, 62, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic Inhibition of Astroglial NF-κB Improves Functional Outcome in Experimental Autoimmune Encephalomyelitis by Suppressing Chronic Central Nervous System Inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar] [CrossRef]
- Raasch, J.; Zeller, N.; van Loo, G.; Merkler, D.; Mildner, A.; Erny, D.; Knobeloch, K.P.; Bethea, J.R.; Waisman, A.; Knust, M.; et al. IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain 2011, 134 Pt 4, 1184–1198. [Google Scholar] [CrossRef]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef]
- Wang, X.; Deckert, M.; Xuan, N.T.; Nishanth, G.; Just, S.; Waisman, A.; Naumann, M.; Schlüter, D. Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-κB-and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol. 2013, 126, 711–724. [Google Scholar] [CrossRef]
- Stone, S.; Jamison, S.; Yue, Y.; Durose, W.; Schmidt-Ullrich, R.; Lin, W. NF-κB Activation Protects Oligodendrocytes against Inflammation. J. Neurosci. 2017, 37, 9332–9344. [Google Scholar] [CrossRef]
- Emmanouil, M.; Taoufik, E.; Tseveleki, V.; Vamvakas, S.-S.; Tselios, T.; Karin, M.; Lassmann, H.; Probert, L. Neuronal I kappa B kinase beta protects mice from autoimmune encephalomyelitis by mediating neuroprotective and immunosuppressive effects in the central nervous system. J. Immunol. 2009, 183, 7877–7889. [Google Scholar] [CrossRef]
- Lee, D.H.; Kubera, K.; Rosenthal, B.; Kaltschmidt, B.; Kaltschmidt, C.; Gold, R.; Linker, R.A. Neuronal NF-κB ablation does not influence neuro-axonal degeneration in experimental autoimmune demyelination. J. Neuroimmunol. 2012, 246, 38–42. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; de Lorenzi, R.; Schmidt, H.; Huth, M.; Mildner, A.; Schmidt-Supprian, M.; Lassmann, H.; Prinz, M.R.; Pasparakis, M. Inhibition of transcription factor NF-κB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat. Immunol. 2006, 7, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, B.; Stegagno, C.; Cannella, B.; Rizzuto, N.; Moretto, G.; Raine, C.S. Activation of NF-κB and c-jun transcription factors in multiple sclerosis lesions: Implications for oligodendrocyte pathology. Am. J. Pathol. 1999, 155, 1433–1438. [Google Scholar] [CrossRef]
- Gupta, A.S.; Biswas, D.D.; Brown, L.S.N.; Mockenhaupt, K.; Marone, M.; Hoskins, A.; Siebenlist, U.; Kordula, T. A detrimental role of RelB in mature oligodendrocytes during experimental acute encephalomyelitis. J. Neuroinflamm. 2019, 16, 161. [Google Scholar] [CrossRef]
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-κB) in multiple sclerosis pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef]
- Bärnthaler, T.; Jandl, K.; Sill, H.; Uhl, B.; Schreiber, Y.; Grill, M.; Thomas, D.; Schicho, R.; Marsche, G.; Frank, S.; et al. Imatinib stimulates prostaglandin E(2) and attenuates cytokine release via EP4 receptor activation. J. Allergy Clin. Immunol. 2019, 143, 794–797.e10. [Google Scholar] [CrossRef]
- Ciarcia, R.; Vitiello, M.T.; Galdiero, M.; Pacilio, C.; Iovane, V.; d’Angelo, D.; Pagnini, D.; Caparrotti, G.; Conti, D.; Tomei, V.; et al. Imatinib treatment inhibit IL-6, IL-8, NF-KB and AP-1 production and modulate intracellular calcium in CML patients. J. Cell Physiol. 2012, 227, 2798–2803. [Google Scholar] [CrossRef]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Ludwiczek, S.; Enrich, B.; Gastl, G.; Weiss, G.; Tilg, H. The kinase inhibitor imatinib mesylate inhibits TNF-α production in vitro and prevents TNF-dependent acute hepatic inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 13622–13627. [Google Scholar] [CrossRef]
- Feng, J.; Misu, T.; Fujihara, K.; Sakoda, S.; Nakatsuji, Y.; Fukaura, H.; Kikuchi, S.; Tashiro, K.; Suzumura, A.; Ishii, N.; et al. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Mult. Scler. J. 2004, 10, 494–498. [Google Scholar] [CrossRef]
- Ghazi, A.; Abood, S.H.; Alaqouli, H.; Hadi, N.; Janabi, S.A.M.A.M. Ibudilast and octreotide can ameliorate acute pancreatitis via downregulation of the inflammatory cytokines and Nuclear Factor- Kappa B expression. Ann. Trop. Med. Public Health 2019, 22, 1–7. [Google Scholar] [CrossRef]
- Kiebala, M.; Maggirwar, S.B. Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells. PLoS ONE 2011, 6, e18633. [Google Scholar] [CrossRef] [PubMed]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Lin, J.-X.; Ihara, M.; Ohtani, R.; Shibata, M. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Res. 2003, 992, 53–59. [Google Scholar] [CrossRef]
- Chang, B.Y.; Huang, M.M.; Francesco, M.; Chen, J.; Sokolove, J.; Magadala, P.; Robinson, W.H.; Buggy, J.J. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 2011, 13, R115. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, C.E.; Purvis, G.S.D.; Collotta, D.; Chiazza, F.; Wissuwa, B.; al Zoubi, S.; Stiehler, L.; Martin, L.; Coldewey, S.M.; Collino, M.; et al. Bruton’s Tyrosine Kinase Inhibition Attenuates the Cardiac Dysfunction Caused by Cecal Ligation and Puncture in Mice. Front. Immunol. 2019, 10, 2129. [Google Scholar] [CrossRef]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Collino, M.; Aranda-Tavio, H.; Chiazza, F.; O’Riordan, C.E.; Zeboudj, L.; Mohammad, S.; Collotta, D.; Verta, R.; Guisot, N.E.S.; et al. Inhibition of Bruton’s TK regulates macrophage NF-κB and NLRP3 inflammasome activation in metabolic inflammation. Br. J. Pharmacol. 2020, 177, 4416–4432. [Google Scholar] [CrossRef]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef]
- Fissolo, N.; Kraus, M.; Reich, M.; Ayturan, M.; Overkleeft, H.; Driessen, C.; Weissert, R. Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur. J. Immunol. 2008, 38, 2401–2411. [Google Scholar] [CrossRef]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-κB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef]
- Lassoued, S.; Moyano, C.; Beldjerd, M.; Pauly, P.; Lassoued, D.; Billey, T. Bortezomib improved the joint manifestations of rheumatoid arthritis in three patients. Jt. Bone Spine 2019, 86, 381–382. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Chen, M.; Kuang, L. Bortezomib followed by autologous stem cell transplantation in a patient with rheumatoid arthritis: A case report and review of the literature. Medicine 2016, 95, e5760. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Gonzalez, E.; Visekruna, A.; Kühl, A.A.; Loddenkemper, C.; Mollenkopf, H.; Kaufmann, S.H.; Steinhoff, U.; Joeris, T. Targeting the proteasome: Partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut 2010, 59, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, J.H.; Park, Y.B.; Lee, S.K. Bortezomib attenuates murine collagen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, S.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther. 2015, 17, 17. [Google Scholar] [CrossRef]
- Cooper, M.J.; Cox, N.J.; Zimmerman, E.I.; Dewar, B.J.; Duncan, J.S.; Whittle, M.C.; Nguyen, T.A.; Jones, L.S.; Roy, S.G.; Smalley, D.M.; et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE 2013, 8, e66755. [Google Scholar] [CrossRef]
- Fei, F.; Yu, Y.; Schmitt, A.; Chen, J.; Chen, B.; Rojewski, M.; Ringhoffer, M.; von Harsdorf, S.; Greiner, J.; Goetz, M.; et al. Tyrosine Kinase Inhibitors Dasatinib, Nilotinib and Imatinib Have an Impact on Both CD8+ T Lymphocytes and CD4+CD25+FoxP3+ Regulatory T Cells by Downregulation of the NF-κB Pathway. Blood 2007, 110, 2368. [Google Scholar] [CrossRef]
- Fei, F.; Yu, Y.; Schmitt, A.; Rojewski, M.T.; Chen, B.; Greiner, J.; Götz, M.; Bunjes, D.; Schmitt, M. Effects of nilotinib on regulatory T cells: The dose matters. Mol. Cancer 2010, 9, 22. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2020, 384, 693–704. [Google Scholar]
- Abani, O.; Abbas, A.; Abbas, F.; Abbas, M.; Abbasi, S.; Abbass, H.; Abbott, A.; Abdallah, N.; Abdelaziz, A.; Abdelfattah, M.; et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-κB activity through induction of IκB synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Kandasamy, M. NF-κB signalling as a pharmacological target in COVID-19: Potential roles for IKKβ inhibitors. Naunyn Schmiedebergs. Arch. Pharmacol. 2021, 394, 561–567. [Google Scholar] [CrossRef]
- Cavalli, G.; de Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Din, C.T.; Boffini, N. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Dimopoulos, G.; de Mast, Q.; Markou, N.; Theodorakopoulou, M.; Komnos, A.; Mouktaroudi, M.; Netea, M.G.; Spyridopoulos, T.; Verheggen, R.J.; Hoogerwerf, J. Favorable anakinra responses in severe COVID-19 patients with secondary hemophagocytic lymphohistiocytosis. Cell Host Microbe 2020, 28, 117–123.e1. [Google Scholar] [CrossRef] [PubMed]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.-M.; Bézie, Y.; Laplanche, S. Anakinra for severe forms of COVID-19: A cohort study. Lancet Rheumatol. 2020, 2, e393–e400. [Google Scholar] [CrossRef]
- Navarro-Millán, I.; Sattui, S.E.; Lakhanpal, A.; Zisa, D.; Siegel, C.H.; Crow, M.K. Use of anakinra to prevent mechanical ventilation in severe COVID-19: A case series. Arthritis Rheumatol. 2020, 72, 1990–1997. [Google Scholar] [CrossRef]
- Pontali, E.; Volpi, S.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Tricerri, F.; Angelelli, A.; Caorsi, R.; Feasi, M.; Calautti, F. Safety and efficacy of early high-dose IV anakinra in severe COVID-19 lung disease. J. Allergy Clin. Immunol. 2020, 146, 213–215. [Google Scholar] [CrossRef]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of the macrophage activation syndrome: Re-analysis of a prior Phase III trial. Crit. Care Med. 2016, 44, 275. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002, 4 (Suppl. 3), S265–S272. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H.; Asanuma, M.; Ogawa, N.; Nishibayashi, S.; Inoue, H. High DNA-binding activity of transcription factor NF-kappa B in synovial membranes of patients with rheumatoid arthritis. Biochem. Mol. Biol. Int. 1995, 37, 827–832. [Google Scholar]
- Gilston, V.; Jones, H.W.; Soo, C.C.; Coumbe, A.; Blades, S.; Kaltschmidt, C.; Baeuerle, P.A.; Morris, C.J.; Blake, D.R.; Winyard, P.G. NF-kappa B activation in human knee-joint synovial tissue during the early stage of joint inflammation. Biochem. Soc. Trans. 1997, 25, 518s. [Google Scholar] [CrossRef] [PubMed]
- Marok, R.; Winyard, P.; Coumbe, A.; Kus, M.; Gaffney, K.; Blades, S.; Mapp, P.; Morris, C.; Blake, D.; Kaltschmidt, C. Activation of the transcription factor nuclear factor-κB in human inflamed synovial tissue. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1996, 39, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Handel, M.L.; McMorrow, L.B.; Gravallese, E.M. Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum. 1995, 38, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Miagkov, A.V.; Kovalenko, D.V.; Brown, C.E.; Didsbury, J.R.; Cogswell, J.P.; Stimpson, S.A.; Baldwin, A.S.; Makarov, S.S. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc. Natl. Acad. Sci. USA 1998, 95, 13859–13864. [Google Scholar] [CrossRef]
- Palombella, V.J.; Conner, E.M.; Fuseler, J.W.; Destree, A.; Davis, J.M.; Laroux, F.S.; Wolf, R.E.; Huang, J.; Brand, S.; Elliott, P.J.; et al. Role of the proteasome and NF-kappaB in streptococcal cell wall-induced polyarthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 15671–15676. [Google Scholar] [CrossRef]
- Tak, P.P.; Gerlag, D.M.; Aupperle, K.R.; van de Geest, D.A.; Overbeek, M.; Bennett, B.L.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001, 44, 1897–1907. [Google Scholar] [CrossRef]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef]
- Davignon, J.L.; Hayder, M.; Baron, M.; Boyer, J.F.; Constantin, A.; Apparailly, F.; Poupot, R.; Cantagrel, A. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology 2013, 52, 590–598. [Google Scholar] [CrossRef]
- Baum, R.; Gravallese, E.M. Bone as a Target Organ in Rheumatic Disease: Impact on Osteoclasts and Osteoblasts. Clin. Rev. Allergy Immunol. 2016, 51, 1–15. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Simmonds, R.E.; Foxwell, B.M. Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatology 2008, 47, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.G.; Gullick, N.J.; Kelly, S.; Pitzalis, C.; Lord, G.M.; Kirkham, B.W.; Taams, L.S. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc. Natl. Acad. Sci. USA 2009, 106, 6232–6237. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, B.; Burkhardt, H.; Dübel, S.; Voll, R.E. Cell-Type Targeted NF-kappaB Inhibition for the Treatment of Inflammatory Diseases. Cells 2020, 9, 1627. [Google Scholar] [CrossRef] [PubMed]
- Tateiwa, D.; Yoshikawa, H.; Kaito, T. Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review. Cells 2019, 8, 818. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.-Y.; Tee, S.Z.-Y.; Wong, M.M.-T.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Dong, C. TH17 cells in development: An updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 2008, 8, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef]
- Teng, M.W.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef]
- Thompson, N.; Isenberg, D.A.; Jury, E.C.; Ciurtin, C. Exploring BAFF: Its expression, receptors and contribution to the immunopathogenesis of Sjögren’s syndrome. Rheumatology 2016, 55, 1548–1555. [Google Scholar] [CrossRef]
- Wei, F.; Chang, Y.; Wei, W. The role of BAFF in the progression of rheumatoid arthritis. Cytokine 2015, 76, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.J.; Yoon, B.Y.; Jhun, J.Y.; Oh, H.J.; Min, S.W.; Cho, M.L.; Park, S.H.; Kim, H.Y.; Min, J.K. Regulation of B cell activating factor (BAFF) receptor expression by NF-ΚB signaling in rheumatoid arthritis B cells. Exp. Mol. Med. 2011, 43, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Nejatbakhsh Samimi, L.; Farhadi, E.; Tahmasebi, M.N.; Jamshidi, A.; Vaziri, A.S.; Mahmoudi, M. NF-κB signaling in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Autoimmun. Highlights 2020, 11, 11. [Google Scholar] [CrossRef]
- Tas, S.W.; de Jong, E.C.; Hajji, N.; May, M.J.; Ghosh, S.; Vervoordeldonk, M.J.; Tak, P.P. Selective inhibition of NF-κB in dendritic cells by the NEMO-binding domain peptide blocks maturation and prevents T cell proliferation and polarization. Eur. J. Immunol. 2005, 35, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Vomero, M.; Barbati, C.; Colasanti, T.; Perricone, C.; Novelli, L.; Ceccarelli, F.; Spinelli, F.R.; di Franco, M.; Conti, F.; Valesini, G. Autophagy and rheumatoid arthritis: Current knowledges and future perspectives. Front. Immunol. 2018, 9, 1577. [Google Scholar] [CrossRef]
- Tas, S.W.; Vervoordeldonk, M.J.; Hajji, N.; Schuitemaker, J.H.; van der Sluijs, K.F.; May, M.J.; Ghosh, S.; Kapsenberg, M.L.; Tak, P.P.; de Jong, E.C. Noncanonical NF-κB signaling in dendritic cells is required for indoleamine 2, 3-dioxygenase (IDO) induction and immune regulation. Blood J. Am. Soc. Hematol. 2007, 110, 1540–1549. [Google Scholar] [CrossRef]
- Dhar, A.; Chawla, M.; Chattopadhyay, S.; Oswal, N.; Umar, D.; Gupta, S.; Bal, V.; Rath, S.; George, A.; Arimbasseri, G.A.; et al. Role of NF-kappaB2-p100 in regulatory T cell homeostasis and activation. Sci. Rep. 2019, 9, 13867. [Google Scholar] [CrossRef]
- Murray, S.E. Cell-intrinsic role for NF-kappa B-inducing kinase in peripheral maintenance but not thymic development of Foxp3+ regulatory T cells in mice. PLoS ONE 2013, 8, e76216. [Google Scholar] [CrossRef]
- Gardam, S.; Sierro, F.; Basten, A.; Mackay, F.; Brink, R. TRAF2 and TRAF3 Signal Adapters Act Cooperatively to Control the Maturation and Survival Signals Delivered to B Cells by the BAFF Receptor. Immunity 2008, 28, 391–401. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Bittner, Z.; Liu, X.; Dang, T.-M.; Radsak, M.P.; Brunner, C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front. Immunol. 2017, 8, 1454. [Google Scholar] [CrossRef]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Sochorová, K.R.; Horváth, R.; Rozková, D.; Litzman, J.I.; Bartunková, J.I.; Sedivá, A.; Spísek, R. Impaired Toll-like receptor 8–mediated IL-6 and TNF-α production in antigen-presenting cells from patients with X-linked agammaglobulinemia. Blood 2006, 109, 2553–2556. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mukherjee, S.; Raje, N.; Schoonmaker, J.A.; Liu, J.C.; Hideshima, T.; Wein, M.N.; Jones, D.C.; Vallet, S.; Bouxsein, M.L.; Pozzi, S. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J. Clin. Investig. 2008, 118, 491–504. [Google Scholar] [CrossRef]
- Garrett, I.; Chen, D.; Gutierrez, G.; Zhao, M.; Escobedo, A.; Rossini, G.; Harris, S.; Gallwitz, W.; Kim, K.; Hu, S. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J. Clin. Investig. 2003, 111, 1771–1782. [Google Scholar] [CrossRef]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood J. Am. Soc. Hematol. 2007, 110, 334–338. [Google Scholar] [CrossRef]
- Zangari, M.; Esseltine, D.; Lee, C.-K.; Barlogie, B.; Elice, F.; Burns, M.J.; Kang, S.-H.; Yaccoby, S.; Najarian, K.; Richardson, P.; et al. Response to bortezomib is associated to osteoblastic activation in patients with multiple myeloma. Br. J. Haematol. 2005, 131, 71–73. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Gao, Y.M.; Xu, G.; Wang, B.; Liu, B.C. Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J. Intern. Med. 2020, 289, 147–161. [Google Scholar] [CrossRef]
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The Role and Therapeutic Potential of NF-kappa-B Pathway in Severe COVID-19 Patients. Inflammopharmacology 2020, 29, 91–100. [Google Scholar] [CrossRef]
- Liao, Q.J.; Ye, L.B.; Timani, K.A.; Zeng, Y.C.; She, Y.L.; Ye, L.; Wu, Z.H. Activation of NF-kappaB by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim. Biophys. Sin. 2005, 37, 607–612. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Cai, S.; Zhuang, Z.; Zhao, Z.; Jin, S.; Xie, W.; Zhou, L.; Zhang, L.; Zhao, J.; et al. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-κB hyper-activation and inflammation. Signal Transduct. Target. Ther. 2021, 6, 167. [Google Scholar] [CrossRef]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus. Res. 2009, 142, 19–27. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Singh, A.K.; Majumdar, S.; Singh, R.; Misra, A. Role of corticosteroid in the management of COVID-19: A systemic review and a Clinician’s perspective. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 971–978. [Google Scholar] [CrossRef]
- Cavalli, G.; Farina, N.; Campochiaro, C.; de Luca, G.; Della-Torre, E.; Tomelleri, A.; Dagna, L. Repurposing of Biologic and Targeted Synthetic Anti-Rheumatic Drugs in COVID-19 and Hyper-Inflammation: A Comprehensive Review of Available and Emerging Evidence at the Peak of the Pandemic. Front. Pharmacol. 2020, 11, 2111. [Google Scholar] [CrossRef]
- Weisberg, E.; Parent, A.; Yang, P.L.; Sattler, M.; Liu, Q.; Liu, Q.; Wang, J.; Meng, C.; Buhrlage, S.J.; Gray, N.; et al. Repurposing of Kinase Inhibitors for Treatment of COVID-19. Pharm. Res. 2020, 37, 167. [Google Scholar] [CrossRef]
- Bahadoram, M.; Keikhaei, B.; Saeedi-Boroujeni, A.; Mahmoudian-Sani, M.-R. Chloroquine/hydroxychloroquine: An inflammasome inhibitor in severe COVID-19? Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Li, J.; Li, H.; Song, J.; Zhou, Y.; Lu, R.; Liu, B.; Pang, Y.; Zhang, P.; Chen, J.; et al. Renoprotective effects of artemisinin and hydroxychloroquine combination therapy on IgA nephropathy via suppressing NF-κB signaling and NLRP3 inflammasome activation by exosomes in rats. Biochem. Pharmacol. 2019, 169, 113619. [Google Scholar] [CrossRef]
- Tang, T.-T.; Lv, L.-L.; Pan, M.-M.; Wen, Y.; Wang, B.; Li, Z.-L.; Wu, M.; Wang, F.-M.; Crowley, S.D.; Liu, B.-C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef]
- Lenzer, J. COVID-19: US gives emergency approval to hydroxychloroquine despite lack of evidence. BMJ 2020, 369, m1335. [Google Scholar] [CrossRef]
- Mahase, E. Hydroxychloroquine for Covid-19: The end of the line? BMJ 2020, 369, m2378. [Google Scholar] [CrossRef]
- Cavalcanti, A.B.; Zampieri, F.G.; Rosa, R.G.; Azevedo, L.C.P.; Veiga, V.C.; Avezum, A.; Damiani, L.P.; Marcadenti, A.; Kawano-Dourado, L.; Lisboa, T.; et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate COVID-19. N. Engl. J. Med. 2020, 383, 2041–2052. [Google Scholar] [CrossRef]
- Consortium, W.S.T. Repurposed antiviral drugs for COVID-19—Interim WHO SOLIDARITY trial results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Group, R.C. Effect of hydroxychloroquine in hospitalized patients with COVID-19. N. Engl. J. Med. 2020, 383, 2030–2040. [Google Scholar]
- Jorge, A. Hydroxychloroquine in the prevention of COVID-19 mortality. Lancet Rheumatol. 2021, 3, e2–e3. [Google Scholar] [CrossRef]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.H.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Outcomes of Hydroxychloroquine Usage in United States Veterans Hospitalized with COVID-19. Med 2020, 1, 114–127.e3. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Tran, V.-T.; Roumier, M.; Chabrol, A.; Paule, R.; Guillaud, C.; Fois, E.; Lepeule, R.; Szwebel, T.-A.; Lescure, F.-X. Clinical efficacy of hydroxychloroquine in patients with COVID-19 pneumonia who require oxygen: Observational comparative study using routine care data. BMJ 2020, 369, m1844. [Google Scholar] [CrossRef]
- Mitjà, O.; Corbacho-Monné, M.; Ubals, M.; Tebé, C.; Peñafiel, J.; Tobias, A.; Ballana, E.; Alemany, A.; Riera-Martí, N.; Pérez, C.A.; et al. Hydroxychloroquine for Early Treatment of Adults With Mild Coronavirus Disease 2019: A Randomized, Controlled Trial. Clin. Infect. Dis. 2020, 73, e4073–e4081. [Google Scholar] [CrossRef]
- Mullard, A. RECOVERY 1 year on: A rare success in the COVID-19 clinical trial landscape. Nat. Rev. Drug Discov. 2021, 20, 336–337. [Google Scholar] [CrossRef]
- Pathak, D.S.K.; Salunke, D.A.A.; Thivari, D.P.; Pandey, A.; Nandy, D.K.; Harish, V.K.R.D.; Pandey, D.S.; Chawla, D.J.; Mujawar, D.J.; Dhanwate, D.A.; et al. No benefit of hydroxychloroquine in COVID-19: Results of Systematic Review and Meta-Analysis of Randomized Controlled Trials. Diabetes Metab. Syndr. 2020, 14, 1673–1680. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Dufort, E.M.; Udo, T.; Wilberschied, L.A.; Kumar, J.; Tesoriero, J.; Weinberg, P.; Kirkwood, J.; Muse, A.; DeHovitz, J. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York State. JAMA 2020, 323, 2493–2502. [Google Scholar] [CrossRef]
- Skipper, C.P.; Pastick, K.A.; Engen, N.W.; Bangdiwala, A.S.; Abassi, M.; Lofgren, S.M.; Williams, D.A.; Okafor, E.C.; Pullen, M.F.; Nicol, M.R.; et al. Hydroxychloroquine in Nonhospitalized Adults With Early COVID-19: A Randomized Trial. Ann. Intern. Med. 2020, 173, 623–631. [Google Scholar] [CrossRef]
- Chatre, C.; Roubille, F.; Vernhet, H.; Jorgensen, C.; Pers, Y.M. Cardiac Complications Attributed to Chloroquine and Hydroxychloroquine: A Systematic Review of the Literature. Drug Saf. 2018, 41, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Tleyjeh, I.M.; Kashour, Z.; AlDosary, O.; Riaz, M.; Tlayjeh, H.; Garbati, M.A.; Tleyjeh, R.; Al-Mallah, M.H.; Sohail, M.R.; Gerberi, D.; et al. Cardiac Toxicity of Chloroquine or Hydroxychloroquine in Patients With COVID-19: A Systematic Review and Meta-regression Analysis. Mayo Clin. Proc. Innov. Qual. Outcomes 2021, 5, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.V.; Makunts, T.; Moumedjian, T.; Issa, M.A.; Abagyan, R. Cardiac adverse events associated with chloroquine and hydroxychloroquine exposure in 20 years of drug safety surveillance reports. Sci. Rep. 2020, 10, 19199. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, A.; Kirresh, A.; Conway, S.; White, L.; Ahmad, M.; Little, C. Hydroxychloroquine use in COVID-19: Is the risk of cardiovascular toxicity justified? Open Heart 2020, 7, e001362. [Google Scholar] [CrossRef] [PubMed]
- FDA. Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Chloroquine and Hydroxychloroquine. 15 June 2020. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and (accessed on 4 February 2021).
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-κB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef]
- Bennett, J.; Capece, D.; Begalli, F.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. NF-κB in the crosshairs: Rethinking an old riddle. Int. J. Biochem. Cell Biol. 2018, 95, 108–112. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef]
- Hsu, L.-C.; Enzler, T.; Seita, J.; Timmer, A.M.; Lee, C.-Y.; Lai, T.-Y.; Yu, G.-Y.; Lai, L.-C.; Temkin, V.; Sinzig, U. IL-1β-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKβ. Nat. Immunol. 2011, 12, 144–150. [Google Scholar] [CrossRef]
- Sehnert, B.; Burkhardt, H.; Wessels, J.T.; Schröder, A.; May, M.J.; Vestweber, D.; Zwerina, J.; Warnatz, K.; Nimmerjahn, F.; Schett, G.; et al. NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 16556–16561. [Google Scholar] [CrossRef] [PubMed]




| Stimulus | Receptor | NF-κB Pathway |
|---|---|---|
| LPS | TLR4 | Canonical |
| TNF-α | TNFR1 | Canonical |
| IL-1 | IL-1R | Canonical |
| BAFF | BAFFR | Noncanonical |
| CD40L | CD40 | Noncanonical |
| RANKL | RANK | Noncanonical |
| LTβ | LTβR | Noncanonical |
| TNF | TNFR2 | Canonical/Noncanonical |
| TWEAK | Fn14 | Canonical/Noncanonical |
| EGF | EGFR | Atypical |
| UV | CK2 | Atypical |
| Compound | Original Indication | New Indication | Comments | References |
|---|---|---|---|---|
| Clemastine fumarate | Allergic reactions | MS | Promotes oligodendrocyte precursor cell differentiation and therefore myelin repair, reduces neuroinflammation in ALS model (inhibits NF-κB) | [51,52,53,54,55,56,57,58,59,60,61] |
| Curcumin | Dietary supplement | COVID-19 | Protects and promotes repair of alveolar ATII cells in inflammatory lung injury model, increases Tregs, IL-10 and M2 macrophages in acute lung injury model, protects from cardiovascular injuries, reduces inflammation by inhibiting NF-κB | [62,63,64] |
| Ibuprofen | Pain relief | SCI | Promotes axon growth and motor function improvement in spinal cord injury models by RhoA inhibition | [65,66,67] |
| Indomethacin | Pain relief | SCI | Promotes axon growth in spinal cord lesion model by RhoA inhibition | [65,67] |
| Resolvin D1 | - | Liver injury | Protects from IR-induced sterile liver inflammation by promoting M2 phenotype and efferocytosis in Kupffer cells, protects astrocytes and ameliorates cognitive impairment after TBI, inhibits inflammation and NF-κB signaling | [68,69,70,71,72] |
| New Indication | Drug | Original Implication | Effect on NF-κB Signaling | Effect On Inflammation | References |
|---|---|---|---|---|---|
| MS | Imatinib mesylate | Cancer (CML, ALL, GIST, HES, CEL) | Inhibits IκB phosphorylation and DNA binding of NF-κB | Attenuates inflammation and enhances BBB integrity in EAE, Phase II clinical trial for MS | [183,184,185] |
| Clemastine | Relief of allergy symptoms | Decreases NF-κB activity and TLR4 expression | Promotes oligodendrocyte differentiation and remyelination in EAE/MS, inhibits inflammation and microglial M1-like activation | [52,53,55,56,57,58,61] | |
| Ibudilast | Asthma, stroke | Inhibits NF-κB activity (possibly by preventing nuclear translocation) | Reduces inflammation in rats with chronic cerebral reperfusion and MS patients, Phase II clinical trial for MS | [186,187,188,189] | |
| Topotecan | Cancer (ovarian cancer, lung cancer, SCLC) | Inhibits IKKβ and thus IκBα degradation | Attenuates inflammation in EAE | [99] | |
| RA | Ibrutinib | Cancer (MCL, CLL, WM) | Inhibits NF-κB nuclear translocation | Anti-inflammatory effects in models of RA, sepsis and diabetes | [190,191,192,193] |
| Bortezomib | Cancer (MM, MCL) | Proteasome inhibitor, prevents degradation of IκBα | Anti-inflammatory effects in models of MS, RA, lupus erythematosus and colitis, promotes osteoblast activation and RA pathogenesis, Phase II clinical trial for RA | [194,195,196,197,198,199] [200,201] | |
| TDZ | Schizophrenia, psychosis | Inhibits IKKβ phosphorylation and IκBα degradation | Attenuates inflammation in endotoxemia model | [83] | |
| Dasatinib | Cancer (CML, ALL) | Inhibits phosphorylation of IKKα, p65/p100/p105 and c-Rel | Inhibits inflammation and bone erosion in CIA and human FLS, increases IL-10 in CIA | [122,202,203,204] | |
| COVID-19 | Dexamethasone | Inflammatory conditions (RA, asthma, allergies etc.) | Induces the expression of IκBα | Reduces mortality in later stage COVID-19 patients | [205,206,207,208] |
| Anakinra | Relief of RA symptoms | Prevents activation of IL-1R | Reduces hyperinflammation and mortality and improves clinical signs of COVID-19 | [209,210,211,212,213,214] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberti, A.; Chaffey, L.E.; Greaves, D.R. NF-κB Signaling and Inflammation—Drug Repurposing to Treat Inflammatory Disorders? Biology 2022, 11, 372. https://doi.org/10.3390/biology11030372
Roberti A, Chaffey LE, Greaves DR. NF-κB Signaling and Inflammation—Drug Repurposing to Treat Inflammatory Disorders? Biology. 2022; 11(3):372. https://doi.org/10.3390/biology11030372
Chicago/Turabian StyleRoberti, Annabell, Laura Elizabeth Chaffey, and David R. Greaves. 2022. "NF-κB Signaling and Inflammation—Drug Repurposing to Treat Inflammatory Disorders?" Biology 11, no. 3: 372. https://doi.org/10.3390/biology11030372
APA StyleRoberti, A., Chaffey, L. E., & Greaves, D. R. (2022). NF-κB Signaling and Inflammation—Drug Repurposing to Treat Inflammatory Disorders? Biology, 11(3), 372. https://doi.org/10.3390/biology11030372