Simple Summary
We use RNA-seq analysis to identify genes that may contribute to mutant p53-mediated prostate cancer initiation in a genetically engineered mouse model (B6.129S4-Trp53tm3.1Tyj/J). A total of 1378 differentially expressed genes, including wildtype p53 target genes (e.g. Cdkn1a, Bax, Bcl2, Kras, Mdm2), p53 gain-of-function-related genes (Mgmt, Id4), and prostate cancer-related genes (Cav-1, Raf1, Kras), were identified. Mice that were homozygous or heterozygous for the Trp53 R270H mutation developed grade one PIN lesions at 3 months and 5 months, respectively, whereas wildtype mice did not develop PIN. Immunohistochemical analysis revealed decreased levels of irradiation-mediated apoptosis in homozygous and heterozygous mice when compared to wildtype counterparts, and this aligned with observed differences in apoptosis-related gene expression.
Abstract
We previously demonstrated that the Trp53-R270H mutation can drive prostate cancer (CaP) initiation using the FVB.129S4 (Trp53tm3Tyj/wt); FVB.129S (Nkx3-1tm3(cre)Mmswt) genetically engineered mouse model (GEM). We now validate this finding in a different model (B6.129S4-Trp53tm3.1Tyj/J mice) and use RNA-sequencing (RNA-Seq) to identify genes which may contribute to Trp53 R270H-mediated prostate carcinogenesis. Wildtype (Trp53WT/WT), heterozygous (Trp53R270H/WT), and homozygous mice (Trp53R270H/R270H) were exposed to 5 Gy irradiation to activate and stabilize p53, and thereby enhance our ability to identify differences in transcriptional activity between the three groups of mice. Mouse prostates were harvested 6 h post-irradiation and processed for histological/immunohistochemistry (IHC) analysis or were snap-frozen for RNA extraction and transcriptome profiling. IHC analyses determined that presence of the Trp53-R270H mutation impacts apoptosis (lower caspase 3 activity) but not cell proliferation (Ki67). RNA-Seq analysis identified 1378 differentially expressed genes, including wildtype p53 target genes (E.g., Cdkn1a, Bax, Bcl2, Kras, Mdm2), p53 gain-of-function (GOF)-related genes (Mgmt, Id4), and CaP-related genes (Cav-1, Raf1, Kras). Further understanding the mechanisms which contribute to prostate carcinogenesis could allow for the development of improved preventive methods, diagnostics, and treatments for CaP.
1. Introduction
An estimated 1.5 million men are newly diagnosed with prostate cancer (CaP) each year (http://seer.cancer.gov/csr/ accessed on 13 December 2021). Understanding the mechanisms which contribute to CaP initiation is important because it can inform the development of chemoprevention strategies, which could reduce this number as well as support the development of improved diagnostic and prognostic biomarkers. CaP tumors are molecularly heterogeneous, indicating that CaP initiation can be caused by multiple different mechanisms [1,2]. In support of this, cell line and animal studies have identified several different genetic alterations which can independently play a causative role in CaP initiation. For example, loss of PTEN and amplification of Myc can both drive CaP initiation in mouse models [3]; these genetic alterations occur in up to 60 and 50% of CaP patients, respectively [4,5]. PTEN dysregulation promotes initiation by causing increased activity of the PI3K–Akt signaling pathway [3,6,7]. Amplification of Myc causes decreased Nkx3.1 expression and upregulation of PIM1 and EZH2, and thereby promotes increased cell plasticity [8]. Our group has previously demonstrated that the Trp53-R270H mutation can drive CaP initiation in FVB.129S4 (Trp53tm3Tyj/wt); FVB.129S (Nkx3-1tm3(cre)Mmswt) mice (Vinall et al., 2012). In this model, Trp53R270H/R270H mice developed prostatic intraepithelial neoplasia (PIN) as early as 6 weeks, and Trp53R270H/+ mice developed PIN as early as 3 months [9]. CaP was detected at 8 months in Trp53R270H/R270H mice. It is estimated that ~30% of CaP patients with early stage disease harbor Tp53 missense mutations, further supporting a role for p53 mutations in driving CaP initiation [10,11,12,13,14,15,16,17,18,19,20,21]. To our knowledge, the mechanisms by which p53 mutations drive CaP initiation remain completely unknown.
Many of the Tp53 missense mutations that are found in CaP patient tumors are gain-of-function (GOF) mutations, e.g., Tp53 R273H, G245S, R273C, and R248W (note that Trp53-R270H is the mouse equivalent of human Tp53-R273H). They are termed ‘GOF’ because the mutant proteins for which they encode can regulate the activity of genes that wildtype p53 does not usually regulate, e.g., BCL-XL and MDR1 [22]. These GOF mutations are similar to other p53 missense mutations, in as much as they still confer loss-of-function (LOF); they are also unable to regulate, or unable to fully regulate, the expression of wildtype p53 target genes, e.g., Cdkn1a, Bax, and Bbc3 [23]. This dual functionality that is displayed by Tp53 GOF mutants has led some researchers to suggest that Tp53 should be classified as both a proto-oncogene and tumor suppressor [24]. Multiple studies have investigated the role of p53 mutations in promoting CaP progression [23,25,26,27,28,29,30], and some of the underlying mechanisms by which Tp53 GOF mutations drive CaP progression have been elucidated. For example, our group has demonstrated that the Tp53 R273H mutation can drive castration resistant (CR) growth of LNCaP cells via increasing the transcription of H2 relaxin, a peptide hormone [23]. Increased expression of H2 relaxin causes increased activity of the Akt and NFkappaB signaling pathways and thereby promotes cell proliferation [29,30]. It is possible that some of these mechanisms could also contribute to Tp53 GOF mutant-driven CaP initiation.
The goal of the current study was to identify gene expression changes that may play a role in facilitating p53 GOF mutant-driven CaP initiation. A mouse model which harbors the Trp53 R270H mutation (B6.129S4-Trp53tm3.1Tyj/J) was used for these studies. Inclusion of mice that are heterozygous and homozygous for the Trp53 R270H mutation allowed for gene dosage effects to be determined and for parallels with early-stage human prostate carcinogenesis to be considered; the majority of CaP patients are heterozygous for Tp53 mutations and loss of heterozygosity (LOH) typically only occurs during late disease. The use of RNA-seq analysis provided an unbiased approach.
2. Materials and Methods
2.1. Mice
The Trp53 R270H mice (B6.129S4-Trp53tm3.1Tyj/J) used for this study were obtained from Jackson Labs (Bar Harbor, ME, USA). These mice were originally generated by the Tyler Jacks lab [31]. All animal procedures were reviewed and approved by the UC Davis Institutional Animal Care and Use Committee (IACUC, protocol approval # 18457).
2.2. Breeding and Genotyping
Breeding and genotyping was performed as previously described [9]. The following primers were used to distinguish between wildtype, heterozygous, and homozygous mice: F- 5′-agctagccaccatggcttgagtaa gtctgca-3′; R- 3′-cttggagacatagccacactg-3′ [32]. Amplification of the wildtype allele yielded a 290 bp PCR product, while amplification of the mutant p53 allele yielded a 330 bp PCR product.
2.3. Irradiation Studies
Mice that were ~3 months old were exposed to 5 Gy whole body irradiation (Shepard cesium source model 143-68 irradiator, San Fernando, CA, USA) to activate and stabilize p53, consequently increasing its expression. Mouse prostates were harvested 6 h post-irradiation and either processed for subsequent histological and/or immunohistochemistry (IHC) analysis or snap-frozen for subsequent RNA extraction and transcriptome profiling with RNA-Sequencing (RNA-Seq) analysis (please see below for details).
2.4. Histology and Immunohistochemistry
Standard histopathology and IHC was performed as described previously [33]. The presence and number of PIN lesions was assessed by the pathological analysis of hematoxylin and eosin (H&E)-stained sections. Primary antibodies used for IHC analyses were p53 (1:1000: Santa Cruz Biotech Inc., Santa Cruz, CA, USA), Ki67 Ab-4 (1:500; Neomarker, Fremont, CA, USA), and activated caspase 3 (1:1000; Promega, Madison, WI, USA). For the assessment of activated caspase 3 and Ki67 positivity, the total number of prostate epithelial cells versus the number of positive cells was assessed in 3 randomly selected fields of view (×20 magnification) and the percentage of positivity was calculated.
2.5. RNA Isolation and Sequencing Analysis
Samples were submitted to the UC Davis Comprehensive Cancer Center’s Genomics Shared Resource (GSR) for isolation of total cellular RNA and RNA-sequencing (RNA-Seq) analysis. Total cellular RNA was isolated from snap-frozen prostates using the TRIzol Reagent (Invitrogen, Waltham, MA, USA) followed by a clean-up with an RNeasy spin column (Qiagen, Hilden, Germany). Indexed, directional RNA-Seq libraries were prepared from 100 ng total RNA using the TruSeq Stranded mRNA (Illumina, San Diego, CA, USA) according to the manufacturer’s standard protocol. Two independent samples were run per group: wildtype mice (Trp53+/+), heterozygous mice (Trp53+/R270H), and homozygous mice (Trp53R270H/R270H). Subsequently, libraries were combined for multiplex sequencing on an Illumina HiSeq 4000 System (2 × 100 bp, paired-end; ≥30 × 106 reads per sample). A HISAT-Cufflinks workflow was utilized for spliced alignment of the sequence reads (FASTQ format) to the reference genome assembly (December 2011, GRCm38/mm10), transcript assembly, quantitation, and differential expression analysis [34,35]. Gene-level FPKM expression values were used for downstream statistical analyses. Principal component analysis (PCA), differential expression analyses, hierarchical clustering, and heatmap visualization were performed with the GeneSpring GX (Agilent Technologies, Inc., Santa Clara, CA, USA) and Partek Flow (Partek, Inc, Chesterfield, MO, USA) software packages.
3. Results
3.1. B6.129S4-Trp53tm3.1Tyj/J Mice Develop PIN as Early as 3 Months of Age
Prostatic intraepithelial neoplasia (PIN) lesions were observed in B6.129S4-Trp53tm3.1Tyj/J mice that were homozygous (Trp53R270H/R270H) or heterozygous (Trp53R270H/+) for the Trp53 R270H mutation (Figure 1A), indicating that the p53 R270H mutation can drive CaP initiation in this setting. At 3 months of age, areas of metaplasia and atypical hyperplasia were observed in heterozygous mice, and grade one PIN lesions were observed in homozygous mice. At 5 months of age, heterozygous mice had developed grade one PIN lesions and homozygous mice had developed grade three to four PIN lesions. No metaplasia, atypical hyperplasia, or PIN lesions were observed in wildtype mice at either the 3 or 5 month time points.
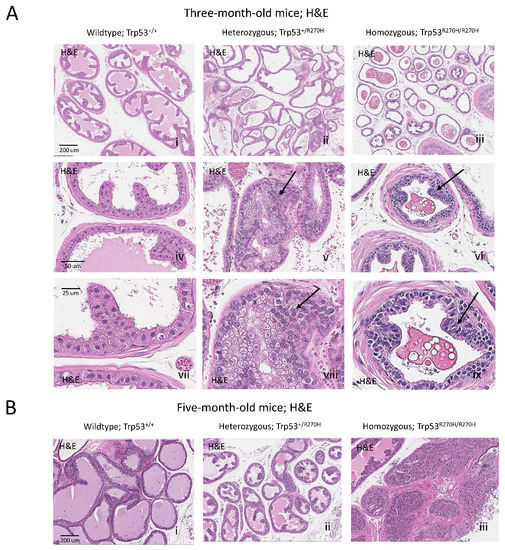
Figure 1.
The Trp53R270H mutant allele promotes the development of prostatic intraepithelial neoplasia (PIN). At ~3 months of age, areas of metaplasia and atypical hyperplasia (arrows) were observed in heterozygous mice (Trp53+/R270H; Aii,v,viii) and grade 1 PIN lesions were observed in homozygous mice (Trp53R270H/R270H; Aiii,vi,ix) but not wildtype mice (Trp53+/+; Ai,iv,vii). PIN lesions (arrows) were observed in older Trp53+/R270H and Trp53R270H/R270H; mice (~5 months of age, Bii,iii, respectively). At ~5 months of age, grade 1 and grade 3–4 PIN lesions were observed in heterozygous and homozygous mice, respectively (Bi,ii). PIN lesions were not observed in Trp53+/+ (wildtype) mice at either time point (Ai,Bi). The combined data further confirm that the Trp53R270H mutation can drive initiation of prostate carcinogenesis. Please note that a representative scale bar is included in the first image for each horizontal panel.
The ability of the p53 R270H mutation to drive CaP initiation in this setting, and the timing of PIN lesion incidence and PIN lesion grade aligns with what was observed by our group in a different Trp53 R270H GEM (FVB.129S4 (Trp53tm3Tyj); FVB.129S (Nkx3-1tm3(cre)Mms)). In our prior study, pre-PIN atypia and grade one PIN lesions were observed in homozygous mice as early as 5 weeks (note that this time point was not assessed in the current study) and grade three to four PIN lesions were observed at 6 months of age (compared to 5 months in the current model). In heterozygous mice, grade two PIN lesions were observed at 6 months of age (grade one PIN lesions were observed at 5 months in the current model).
Our finding that the Trp53 R270H mutation can drive CaP initiation in two independent GEMs, and the similarities in the timing of PIN incidence and PIN grade between the GEM strongly supports a causative role for this mutation in driving prostate carcinogenesis. Note that PIN grading was performed as described by Park et al. for both studies [33].
3.2. Exposure to 5 Gy Whole Body Irradiation Stabilizes p53 Expression in the Majority of Mouse Prostate Cells
We demonstrate that exposure to 5 Gy irradiation stabilized p53 in all three groups of mice (Trp53+/+ (wildtype), Trp53+/R270H (heterozygous), and Trp53R270H/R270H (homozygous for the Trp53 mutation)); IHC analysis showed that more than 90% of prostate cells were p53-positive 6 h post-irradiation (Supplementary Figure S1). This is important because increased stabilization of p53 equates to increased transcriptional activity of p53 [36,37,38], and so irradiating mice helped enhance our ability to identify the differential expression of p53 target genes between the three groups, i.e., enhanced our ability to determine the impact of the Trp53 R270H mutation on the expression levels of these target genes. Cellular levels of p53 are usually kept very low via MDM2-mediated ubiquitination and subsequent proteasomal degradation, and irradiation is one of many environmental triggers that causes the inhibition of p53 degradation, and thereby causes p53 stabilization and increased transcriptional activity [36,37,38]. It is noteworthy that the 5 Gy dosage and 6 h time point were chosen for this study based on data from Levine et al.; the Levine lab demonstrated that 5 Gy whole-body ionizing irradiation causes stabilization of p53 in mouse spleen, heart, lung, kidney, liver, and skin at the 6 h post-irradiation time point [39]. While they did not look at p53 levels in the prostate, the prostate is well known to be a radiosensitive tissue and so we expected to also see p53 stabilization in the prostate at this time point. Our IHC analysis confirmed this to the case.
3.3. The Trp53 R270H Mutant Is Able to Alter Gene Expression in Mouse Prostate Cells, and in Many Cases Gene Expression Levels Correlate with Trp53 R270H Gene Dosage
Three groups of mice (Trp53+/+ (wildtype), Trp53+/R270H (heterozygous), and Trp53R270H/R270H (homozygous for the Trp53 mutation)) were assessed as part of the RNA sequencing studies. Mice were exposed to 5 Gy irradiation and prostates were harvested for RNA extraction 6 h post-irradiation, i.e., at the time point that we had observed more than 90% IHC positivity for p53 for all three groups of mice (Supplementary Figure S1). Genes exhibiting differential expression (ANOVA, p < 0.05, +/−1.5-fold-change cut-off) across the three groups of mice were determined, and hierarchical clustering and heatmap visualization of the differentially expressed genes (DEGs) demonstrated that the mice within each group had very similar gene expression (Figure 2A,B). Assessment of the normalized expression values confirmed this (selected genes Table 1 and Table 2, entire dataset can be observed via this link; GEO accession GSE130440, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130440 accessed on 13 December 2021). The similarity of the RNA-Seq profiles within each group of mice indicate that our model is robust and gives confidence in the RNA-seq data produced. Statistically significant differences in gene expression between the three groups of mice were observed for 664 genes. Trp53 R270H gene dosage affects were observed for many genes, i.e., a further increase or decrease in gene expression relative to the wild type controls was observed in homozygous mice when compared to heterozygous mice (Table 1 and Table 2, Figure 3). This was not the case for all genes; in some instances, gene expression changed by a similar amount in both the Trp53 R270H heterozygous and homozygous mice relative to the wildtype mice, or only changed in the homozygous mice. Gene ontology analyses of these DEGs revealed a significant enrichment of genes belonging to the cell differentiation, apoptotic process, and cell proliferation categories (Figure 2C). Although the expression levels of DEGs were not always reflective of Trp53 genotype status, there was a strong concordance with regard to function, in that Trp53 R270H homozygous mice typically exhibited a higher number of DEGs in enriched GO categories when compared to that of heterozygous mice. For example, for the ‘apoptotic process’ category comparison of Trp53 R270H homozygous versus wildtype mice, we identified 34 differentially expressed genes, while the comparison of Trp53 R270H heterozygous versus wildtype mice identified only 22 differentially expressed genes. Additionally, in the ‘positive regulation of gene expression’ category, DEGs were only observed in Trp53 R270H homozygous versus wildtype mice (28 differentially expressed genes), not Trp53 R270H heterozygous versus wildtype mice.
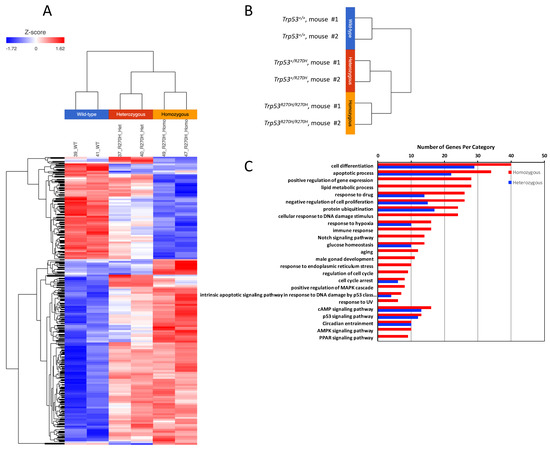
Figure 2.
RNA-seq analysis demonstrates that presence of the Trp53R270H mutant allele is able to alter the transcriptional response of prostate cells to 5 Gy radiation. A distinct difference in gene expression patterns was observed between wildtype mice (Trp53+/+) versus heterozygous mice (Trp53+/R270H) and between wildtype mice (Trp53+/+) versus homozygous mice (Trp53R270H/R270H); heatmap (A), hierarchical clustering (B), and gene ontology (C) analyses. Differences between heterozygous and homozygous mice were also observed.

Table 1.
Presence of the Trp53R270H mutant allele mediates alterations in the expression levels of validated transcriptional targets of wildtype p53. The expression of 16 genes that are known transcriptional targets of wildtype p53 was impacted by the presence of the Trp53R270H mutant allele. It is noteworthy that allele dosage effects were observed for some genes; Bax, Ccng1, Cdkn1a, Gdf15, Mdm2. Data shown are fragments per kilobase of exon model per million mapped reads (FPKM).
Table 2.
The Trp53R270 mutant allele mediates alterations in expression levels of validated transcriptional targets of the Trp53R270H allele as well as alterations in expression levels of genes associated with incidence of prostate cancer in patients. Two known transcriptional targets of Trp53R270H, Mgmt and Id4, demonstrated differential expression in wildtype (Trp53+/+) versus heterozygous (Trp53+/R270H) and/or or homozygous mice (Trp53R270H/R270H). A dose-dependent effect was observed for Mgmt. The expression of 4 genes which are associated with prostate cancer incidence and/or disease progression (Cdkn1a, Kras, Raf1, and Cav1) were impacted by the presence of the Trp53R270H allele. A gene dosage effect was observed for 1 of these, Cdkn1a. Data shown are fragments per kilobase of exon model per million mapped reads (FPKM).
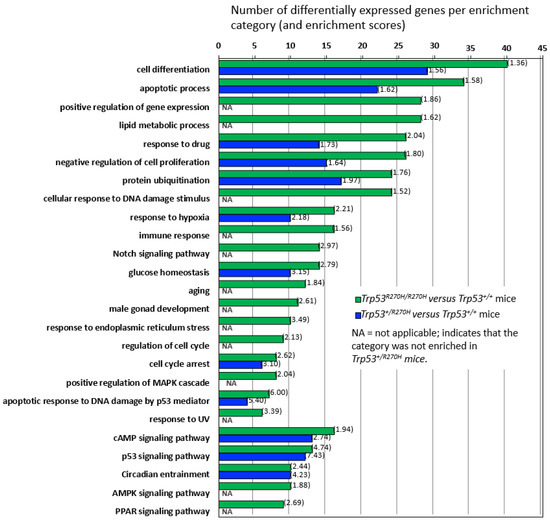
Figure 3.
Gene enrichment analysis and gene dosage effects. Comparison of differential gene expression in Trp53 R270H homozygous versus wildtype mice and in Trp53 R270H heterozygous versus wildtype mice by gene enrichment category demonstrates that gene dosage differences exist for many, but not all of the categories. Gene enrichment scores are shown in brackets.
3.4. The Trp53 R270H Mutant Impacts Expression Levels of Known LOF and GOF Genes, as Well as Known CaP-Related Genes
Comparison of our list of differentially expressed genes with a previously published list of validated potential transcriptional targets of wildtype p53 (122 genes) that included targets validated by three different types of assay, including ChIP assay [23]) determined that 16 of the 122 validated targets of wildtype p53 were differentially expressed in our model (13%); Acta1, Bax, Bbc3, Cav1, Ccng1, Cdkn1a, Ddit4, Gdf15, Hsp90ab1, Mdm2, Nos3, Ppm1j, Prkab1, Ptk2b, Tap1, and Tnfrsf10b (Table 1). Expression of nine of these genes (Bax, Bbc3, Ccng1, Cdkn1a, Ddit4, Gdf15, Mdm2, Tap1, Tnfrsf10b) was decreased in heterozygous mice and Trp53 R270H gene dosage affects were observed for 7 of the 16 differentially expressed genes; Bax, Bbc3, Ccng1, Cdkn1a, Ddit4, Gdf15, and Mdm2. The fact that some but not all genes showed dosage effects, and that the impact of the mutant on gene expression varied between genes, can be interpreted as meaning that the p53 R270H mutant, a mutation which is located in the DNA binding domain of p53, is able to bind with different affinities to different gene promoters and that the interaction can be dose dependent.
Comparison of our list of differentially expressed genes with a list of known Trp53 GOF genes (total of 28 genes, [22]) identified two Trp53 GOF genes that are differentially expressed in our model (2%); Mgmt and Id4 (Table 2). Gene dosage effects were observed for only one of these (Mgmt). Our list of differentially expressed genes was also compared with a list of genes that are differentially expressed in CaP patients and that are known to contribute to CaP progression [40,41]. This comparison identified only four genes; Cdkn1a, Kras, Raf1, and Cav1. Gene dosage effects were observed for one of these, Cdkn1a.
Heat map visualization of relative expression levels of genes listed in Table 1 and Table 2 (Figure 4) further confirms that gene dosage effects occur for many, but not all, of these genes, and that the relative impact of the p53 R270H mutant on gene expression varies widely between genes. This visualization also further confirmed that gene expression levels are very similar for each of the two mice in each group (Trp53+/+ (wildtype), Trp53+/R270H (heterozygous), and Trp53R270H/R270H (homozygous for the Trp53 mutation)).
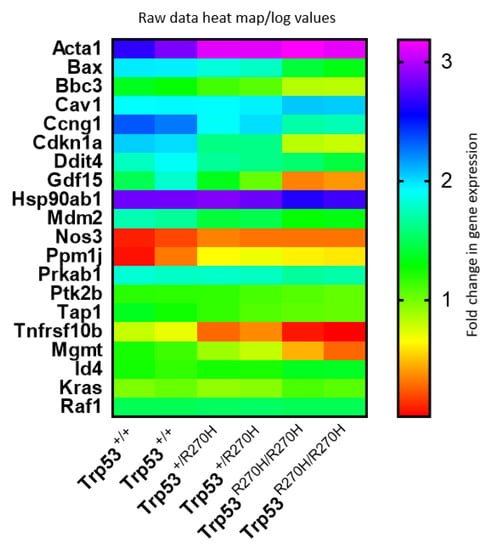
Figure 4.
Gene expression heat map. Comparison of gene expression log values between the three genotypes and between differentially genes reveals that gene dosage effects exist for some, but not all genes, and that the relative impact of the p53 R270H mutant on gene expression varies widely between genes. This figure was generated using log transformed raw data for differentially expressed genes.
3.5. The Trp53 R270H Mutation Causes Decreased Levels of Irradiation-Mediated Apoptosis but Does Not Appear to Impact Cell Proliferation
IHC analyses were conducted to determine the impact of the Trp53 R270H mutation on irradiation-mediated apoptosis and cell proliferation. Irradiation is well known to impact both of these processes via both p53-dependent and p53-independent processes [42]. Apoptosis was assessed via activated caspase IHC, and cell proliferation was assessed via Ki67 IHC. Approximately 5% of cells in prostates from wildtype mice (Trp53+/+) expressed active caspase 3, compared to only ~1% of cells in prostates from heterozygous mice (Trp53+/R270H) (Figure 5A). Less than 1% of cells in prostates from homozygous mice (Trp53R270H/R270H) expressed active caspase 3. No differences in the number of cells expressing Ki67 were observed; approximately 2–5% of cells were Ki67 positive in all three groups of mice (Figure 5B). These data indicate that the Trp53 R270H mutant impacts apoptosis related-pathways to a greater extent when compared to cell proliferation-related pathways. This finding aligns with the gene ontology analysis of our RNA-seq data, which show more apoptosis-related genes were differentially expressed in the homozygous and heterozygous mice when compared to cell proliferation-related genes (Figure 2B).
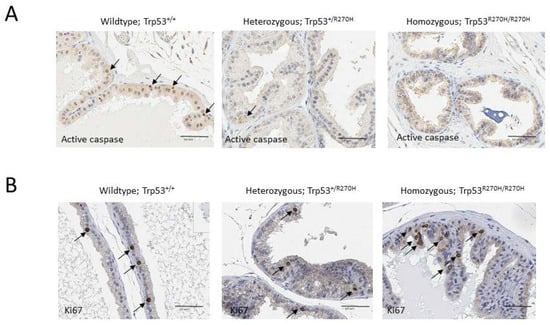
Figure 5.
Presence of the Trp53R270H mutant allele causes increased cell survival following irradiation but does not affect cell proliferation. All mice were exposed to 5 Gy whole body irradiation 6 h prior to time of necropsy. IHC analyses of revealed that ~5% of cells in prostates from wildtype mice (Trp53+/+) expressed active caspase 3 (arrows), compared to only ~1% of cells in prostates from heterozygous mice (Trp53+/R270H) (A). Less than 1% of cells in prostates from homozygous mice (Trp53R270H/R270H) expressed active caspase 3 (A). These data indicate that the Trp53R270H mutant allele promotes survival of prostate cells. In contrast, Ki67 IHC revealed similar levels of cell proliferation (~2–5%) in all three genotypes (B).
4. Discussion
We previously demonstrated that the Trp53 R270H mutation can drive CaP initiation using the FVB.129S4 (Trp53tm3Tyj); FVB.129S (Nkx3-1tm3(cre)Mms) GEM [9]. We now validated this finding in a different GEM: B6.129S4-Trp53tm3.1Tyj/J. In both models, heterozygous mice developed PIN as early as 3 months. The main goal of the current study was to identify genes that may play a role in this process. RNA sequencing analysis of homozygous, heterozygous, and wildtype mice prostate samples identified a total of 1378 differentially expressed genes, demonstrating that the Trp53 R270H mutation has a strong impact on gene expression. Sixteen of these differentially expressed genes are known transcriptional targets of wildtype p53, while two of them are known to be p53 GOF genes. To our knowledge, this is the first study to document the impact of mutant p53 on prostate cell gene expression in a GEM which develops PIN.
In addition to the GEM described in our previous and current study, there is only group who have generated a GEM to help determine the role of mutant p53 in driving CaP initiation. Elgavish et al. generated a GEM with prostate-specific expression of the Tp53 R273H mutant (a hotspot mutation in CaP patients) [43]. Tp53 R273H is the human equivalent of the mouse Trp53 R270H mutant. Their mutant mice developed grade three to four PINs at 52 weeks, and assessment of levels of apoptosis following castration revealed that apoptosis was reduced in mutant mice. These data align with ours; our mutant mice also developed PIN (albeit much earlier), and in our GEM the Trp53 R270H mutant caused decreased levels of apoptosis and decreased the expression of pro-apoptosis-related genes in response to irradiation. While other p53 GEMs exist, in these models, typically compound models, p53 expression has been silenced to mimic a loss of heterozygosity, a genetic alteration which is associated with late-stage CaP [44,45,46,47]. Generation and characterization of models which harbor p53 mutations, and placing focus on elucidating the role of p53 mutations in driving CaP incidence and progression is important because a significant number of CaP patients with early-stage disease harbor p53 mutations within their primary tumors, and p53 mutations have been found in human PIN lesions [48].
Two of the pro-apoptosis genes that we observed as being downregulated in our homozygous and heterozygous Trp53 R270H GEM are direct transcriptional targets of wildtype p53: Bax and Bbc3 (also known as Puma). Decreased Bax and/or Puma expression has been shown to be associated with increased pathological grade in CaP patients [49,50,51,52], and multiple CaP cell line and animal studies indicate that decreased expression of pro-apoptotic molecules can contribute to CaP progression and chemoresistance [53]. While a direct association between decreased Puma and/or Bax expression and CaP initiation has not been reported, it is well known that impaired cellular damage responses and dysregulation of apoptosis can contribute to the initiation of multiple cancer types, including CaP [54,55]. Several studies have demonstrated that environmental exposures, e.g., ingestion of environmental carcinogens or chronic inflammation, trigger apoptosis responses and are associated with a higher risk of CaP incidence [56,57]. While our mice were exposed to an extreme environmental trigger of apoptosis (5 Gy whole body irradiation), the combined data indicate it is possible that p53 mutant-mediated dysregulation of the apoptosis response to environmental carcinogens could potentially contribute to CaP initiation in some patients.
Dysregulation of the cell cycle is well known to contribute to cancer initiation [58]. In our study, the expression of cell proliferation-related genes was also impacted by presence of the Trp53 R270H mutant, but to a lesser extent when compared to apoptosis-related genes; the cell proliferation gene category ranked sixth in the gene ontology analyses while the apoptosis-related gene category ranked second. Alterations in proliferative index (Ki67) were not observed, further supporting a lesser role for the dysregulation of cell proliferation in mediating p53 mutant-driven CaP initiation. The most notable cell cycle molecule to be impacted by the Trp53 R270H mutant was Cdkn1a. Cdkn1a is a transcriptional target of wildtype p53 and its protein product, p21, mediates G1 cell cycle arrest, primarily through inhibition of Cdk molecules, e.g., Cdk1 and 2 proteins [59]. While a direct link between CaP initiation and decreased p21 has not been reported, loss of p21 is associated with cancer initiation in other cancer types, e.g., liver cancer [60]. Interestingly, p21 expression is frequently increased in CaP patients and is associated with progression and worse outcomes [61,62]. This paradoxical effect has been hypothesized to result from the p21-mediated inhibition of apoptosis, and studies indicate that p21 can act as a tumor suppressor or oncogene, depending on cell type and context [63]. Whether p21 is acting as a tumor suppressor or oncogene in our model remains to be determined.
Differential expression of two known GOF genes was also observed in our study; Mgmt and Id4. Mgmt (O6-methylguanine DNA methyltransferase) plays a key role in DNA repair; it removes DNA adducts that are added by alkylating agents, such as cisplatin and volatile N-nitroso compounds (found in high concentrations at rubber and metal factories), and thereby prevents the accumulation of mutations/genotoxicity [64,65]. In our GEM, the Trp53 R270H mutant causes the decreased expression of Mgmt, indicating that mutant mice will be more likely to accumulate DNA damage. It is possible that this mechanism contributes to mutant p53-initiated CaP. Expression levels of the other p53 GOF gene that our study identified, Id4, were elevated in mice that harbor the Trp53 R270H mutant. Id4 encodes for a DNA-binding protein inhibitor that can regulate cell proliferation, apoptosis, and differentiation. Interestingly, Id4 has been shown to act upstream of wildtype p53; it acetylates wildtype p53 and thereby enhances its transcriptional activity [66]. Furthermore, studies have shown that increasing Id4 blocks the tumorigenicity of CaP cells [67]. Based on this information, it would appear that increased expression of Id4 should therefore inhibit prostate carcinogenesis, i.e., the opposite of what we would expect in this setting.
The ability of the Trp53 R270H to drive breast and lung cancer initiation has been demonstrated in other GEM. Many consider the ability of the Trp53 R270H mutant to mediate cancer initiation is, in of itself, confirmation that this mutant can cause GOF, because p53-null mice do not develop breast or lung cancers [68,69,70]. In alignment with this, p53-null mice do not develop PIN or CaP in the absence of other genetic alterations [3]. Gene expression profiling studies have been performed in breast and lung cancer-specific p53-mutant GEMs and have also identified differential expression of apoptosis and cell cycle-related genes. Turrell et al. found that expression of Bbc3, Ccng1, and Cdkn1a were impacted by presence of the Trp53 R270H mutant in their lung cancer GEM [71], and Wijnhoven et al. showed that Bbc3 expression is impacted in their breast cancer GEM [70]. These data support our findings and indicate that further analysis of the role of Bbc3 and Cdkn1a in prostate carcinogenesis is warranted. To our knowledge, these breast and lung GEM studies did not identify dysregulation of p53 GOF genes.
It is noteworthy that the Trp53 R270H mutant mediated dose-dependent effects for some, but not all, wildtype p53 transcriptional target genes. For example, gene dosage effects were observed for Bax, Bbc3, and Cdkn1a; ~1.5-fold, ~1.6-fold, and ~2-fold decreases in these genes were observed in heterozygous mice, respectively, while ~3.6-fold, ~2.9-fold, and 14-fold decreases were observed in homozygous mice. The majority of CaP patients with early disease who harbor Tp53 mutations are heterozygous for the mutations and loss of heterozygosity (LOH) is typically only observed in advanced disease. Based on this and our gene expression profiling data, which show that apoptosis is dysregulated, it is likely that patients with LOH will have a worse response to cytotoxic chemotherapy. It is also noteworthy that only a small fraction of known transcriptional targets of wildtype p53 were significantly impacted by presence of the Trp53 R270H mutation; 16 out of 122 genes (13%). This has been shown to be the case in other studies and cancer types and it appears that the impact of mutant p53 is both cell type and context dependent.
A major limitation of this study is the lack of validation of differentially expressed genes. This will be an important next step and focus will be placed on validating the differential expression of the genes discussed above.
5. Conclusions
Our combined data confirm that the Trp53 R270H mutant can drive CaP initiation and indicate that the dysregulation of apoptosis plays a role in promoting mutant p53-mediated CaP initiation. Importantly, we identified several potential mediators of mutant p53-driven CaP, which thereby provide a starting point for future validation and mechanistic studies. Our molecular characterization of the Trp53 R270H GEM will help support its usage for diagnostic and prognostic biomarker development studies, as well as for pre-clinical studies of therapeutic agents.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology11020218/s1. Supplementary Figure S1: Immunohistochemical analysis confirms p53 stabilization in all 3 groups of mice following exposure to 5 Gy irradiation.
Author Contributions
Conceptualization, R.V. and A.B.; methodology, R.V., Q.C., G.T. and A.B.; software, R.V., R.R., A.D., C.G.T. and A.B.; formal analysis, R.V., R.R., A.D., C.G.T. and A.B.; resources, R.V., C.G.T. and A.B.; data curation, R.V., Q.C., A.D., C.G.T. and A.B.; writing—original draft preparation, R.V.; writing—review and editing, R.V., Q.C., G.T., R.R., A.D., C.G.T. and A.B.; supervision, R.V. and A.B.; project administration, R.V. and A.B.; funding acquisition, R.V. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this publication was supported by an internal grant to RV from California Northstate University College of Pharmacy. RNA sequencing studies were conducted by the Genomics Shared Resource (GSR) at the UC Davis Comprehensive Cancer Center. The GSR is supported by the UC Davis Comprehensive Cancer Center Support Grant (CCSG) awarded by the National Cancer Institute (NCI P30CA093373).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California Davis (protocol approval # 18457, approved on 8 January 2015).
Informed Consent Statement
Not applicable.
Data Availability Statement
RNA-seq data is available via the following link, GEO accession GSE130440, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130440, accessed on 13 December 2021).
Acknowledgments
The authors would like to thank Neil (Ed) Hubbard for his help with setting up the imaging studies for the IHC and H&E analysis.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Frank, S.; Nelson, P.; Vasioukhin, V. Recent advances in prostate cancer research: Large-scale genomic analyses reveal novel driver mutations and DNA repair defects. F1000Research 2018, 7, Rev-1173. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Bangma, C.H.; Bjartell, A.; Catto, J.W.; Culig, Z.; Gronberg, H.; Luo, J.; Visakorpi, T.; Rubin, M.A. The mutational landscape of prostate cancer. Eur. Urol. 2013, 64, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittmann, M.; Huang, J.; Radaelli, E.; Martin, P.; Signoretti, S.; Sullivan, R.; Simons, B.W.; Ward, J.M.; Robinson, B.D.; Chu, G.C.; et al. Animal models of human prostate cancer: The consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013, 73, 2718–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic rearrangements of PTEN in prostate Cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef] [Green Version]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Wang, S.; Garcia, A.J.; Wu, M.; Lawson, D.A.; Witte, O.N.; Wu, H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1480–1485. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and prostate cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Vinall, R.L.; Chen, J.Q.; Hubbard, N.E.; Sulaimon, S.S.; Shen, M.M.; Devere White, R.W.; Borowsky, A.D. Initiation of prostate cancer in mice by Tp53R270H: Evidence for an alternative molecular progression. Dis. Model. Mech. 2012, 5, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Downing, S.R.; Russell, P.J.; Jackson, P. Alterations of p53 are common in early stage prostate cancer. Can. J. Urol. 2003, 10, 1924–1933. [Google Scholar]
- Shi, X.B.; Di Mauro, S.M.; Highshaw, R.; Deitch, A.D.; Evans, C.P.; Gumerlock, P.H.; devere White, R.W. Application of a yeast assay to detect functional p53 mutations in archival prostate cancer tissue. Cancer Biother. Radiopharm. 2002, 17, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.H.; Cohen, M.B.; Robinson, R.A. p53 immunoreactivity in primary and metastatic prostatic adenocarcinoma. Mod. Pathol. 1995, 8, 462–466. [Google Scholar] [PubMed]
- Heidenberg, H.B.; Sesterhenn, I.A.; Gaddipati, J.P.; Weghorst, C.M.; Buzard, G.S.; Moul, J.W.; Srivastava, S. Alteration of the tumor suppressor gene p53 in a high fraction of hormone refractory prostate cancer. J. Urol. 1995, 154, 414–421. [Google Scholar] [CrossRef]
- Prendergast, N.J.; Atkins, M.R.; Schatte, E.C.; Paulson, D.F.; Walther, P.J. p53 immunohistochemical and genetic alterations are associated at high incidence with post-irradiated locally persistent prostate carcinoma. J. Urol. 1996, 155, 1685–1692. [Google Scholar] [CrossRef]
- Moul, J.W.; Bettencourt, M.C.; Sesterhenn, I.A.; Mostofi, F.K.; Mcleod, D.G.; Srivastava, S.; Bauer, J.J. Protein expression of p53, bcl-2, and KI-67 (MIB-1) as prognostic biomarkers in patients with surgically treated, clinically localized prostate cancer. Surgery 1996, 120, 159–166; discussion 166–157. [Google Scholar] [CrossRef]
- Bauer, J.J.; Sesterhenn, I.A.; Mostofi, F.K.; Mcleod, D.G.; Srivastava, S.; Moul, J.W. Elevated levels of apoptosis regulator proteins p53 and bcl-2 are independent prognostic biomarkers in surgically treated clinically localized prostate cancer. J. Urol. 1996, 156, 1511–1516. [Google Scholar] [CrossRef]
- Byrne, R.L.; Horne, C.H.; Robinson, M.C.; Autzen, P.; Apakama, I.; Bishop, R.I.; Neal, D.E.; Hamdy, F.C. The expression of waf-1, p53 and bcl-2 in prostatic adenocarcinoma. Br. J. Urol. 1997, 79, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Meyers, F.J.; Gumerlock, P.H.; Chi, S.G.; Borchers, H.; Deitch, A.D.; devere White, R.W. Very frequent p53 mutations in metastatic prostate carcinoma and in matched primary tumors. Cancer 1998, 83, 2534–2539. [Google Scholar] [CrossRef]
- Chi, S.G.; devere White, R.W.; Meyers, F.J.; Siders, D.B.; Lee, F.; Gumerlock, P.H. p53 in prostate cancer: Frequent expressed transition mutations. J. Natl. Cancer Inst. 1994, 86, 926–933. [Google Scholar] [CrossRef]
- Schlechte, H.; Lenk, S.V.; Loning, T.; Schnorr, D.; Rudolph, B.D.; Ditscherlein, G.; Loening, S.A. p53 tumour suppressor gene mutations in benign prostatic hyperplasia and prostate cancer. Eur. Urol. 1998, 34, 433–440. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschauer, L.; Muller, P.A. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem. Soc. Trans. 2016, 44, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2006, 25, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Bossi, G.; Marampon, F.; Maor-Aloni, R.; Zani, B.; Rotter, V.; Oren, M.; Strano, S.; Blandino, G.; Sacchi, A. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle 2008, 7, 1870–1879. [Google Scholar] [CrossRef] [Green Version]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower tgfbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef]
- Liu, S.; Vinall, R.L.; Tepper, C.; Shi, X.B.; Xue, L.R.; Ma, A.H.; Wang, L.Y.; Fitzgerald, L.D.; Wu, Z.; Gandour-Edwards, R.; et al. Inappropriate activation of androgen receptor by relaxin via beta-catenin pathway. Oncogene 2008, 27, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Vinall, R.L.; Mahaffey, C.M.; Davis, R.R.; Luo, Z.; Gandour-Edwards, R.; Ghosh, P.M.; Tepper, C.G.; de Vere White, R.W. Dual blockade of PKA and NF-kappab inhibits H2 relaxin-mediated castrate-resistant growth of prostate cancer sublines and induces apoptosis. Horm. Cancer 2011, 2, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, A.; Flores, E.R.; Miranda, B.; Hsieh, H.M.; van Oostrom, C.T.; Sage, J.; Jacks, T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA 2002, 99, 2948–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Walls, J.E.; Galvez, J.J.; Kim, M.; Abate-Shen, C.; Shen, M.M.; Cardiff, R.D. Prostatic intraepithelial neoplasia in genetically engineered mice. Am. J. Pathol. 2002, 161, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, stringtie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Mol. Cancer Res. 2008, 6, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [Green Version]
- Schoenborn, J.R.; Nelson, P.; Fang, M. Genomic profiling defines subtypes of prostate cancer with the potential for therapeutic stratification. Clin. Cancer Res. 2013, 19, 4058–4066. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.L.; Blum, J.M.; Kirsch, D.G. Role of p53 in regulating tissue response to radiation by mechanisms independent of apoptosis. Transl. Cancer Res. 2013, 2, 412–421. [Google Scholar] [PubMed]
- Elgavish, A.; Wood, P.A.; Pinkert, C.A.; Eltoum, I.E.; Cartee, T.; Wilbanks, J.; Mentor-Marcel, R.; Tian, L.; Scroggins, S.E. Transgenic mouse with human mutant p53 expression in the prostate epithelium. Prostate 2004, 61, 26–34. [Google Scholar] [CrossRef]
- Kim, J.; Roh, M.; Doubinskaia, I.; Algarroba, G.N.; Eltoum, I.E.A.; Abdulkadir, S.A. A mouse model of heterogeneous, c-MYC-initiated prostate cancer with loss of Pten and p53. Oncogene 2012, 31, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Wu, C.-J.; Jaskelioff, M.; Ivanova, E.; Kost-Alimova, M.; Protopopov, A.; Chu, G.C.; Wang, G.; Lu, X.; Labrot, E.S.; et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 2012, 148, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downing, S.R.; Jackson, P.; Russell, P.J. Mutations within the tumour suppressor gene p53 are not confined to a late event in prostate cancer progression. A review of the evidence. Urol. Oncol. 2001, 6, 103–110. [Google Scholar] [CrossRef]
- Torrealba, N.; Rodriguez-Berriguete, G.; Vera, R.; Fraile, B.; Olmedilla, G.; Martinez-Onsurbe, P.; Sanchez-Chapado, M.; Paniagua, R.; Royuela, M. Homeostasis: Apoptosis and cell cycle in normal and pathological prostate. Aging Male 2018, 23, 354–361. [Google Scholar] [CrossRef]
- Diallo, J.S.; Aldejmah, A.; Mouhim, A.F.; Peant, B.; Fahmy, M.A.; Koumakpayi, I.H.; Sircar, K.; Begin, L.R.; Mes-Masson, A.M.; Saad, F. NOXA and PUMA expression add to clinical markers in predicting biochemical recurrence of prostate cancer patients in a survival tree model. Clin. Cancer Res. 2007, 13, 7044–7052. [Google Scholar] [CrossRef] [Green Version]
- Mcdonnell, T.J.; Chari, N.S.; Cho-Vega, J.H.; Troncoso, P.; Wang, X.; Bueso-Ramos, C.E.; Coombes, K.; Brisbay, S.; Lopez, R.; Prendergast, G.; et al. Biomarker expression patterns that correlate with high grade features in treatment naive, organ-confined prostate cancer. BMC Med. Genom. 2008, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehinde, E.O.; Maghrebi, M.A.; Anim, J.T. The importance of determining the aggressiveness of prostate cancer using serum and tissue molecular markers. Can. J. Urol. 2008, 15, 3967–3974. [Google Scholar] [PubMed]
- Wang, G.; Reed, E.; Li, Q.Q. Apoptosis in prostate cancer: Progressive and therapeutic implications (Review). Int J. Mol. Med. 2004, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Kyprianou, N. Growth factor signalling in prostatic growth: Significance in tumour development and therapeutic targeting. Br. J. Pharmacol. 2006, 147 (Suppl 2), S144–S152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Dysregulation of apoptotic signaling in cancer: Molecular mechanisms and therapeutic opportunities. J. Cell Biochem. 2008, 104, 1124–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, P.L.; Martin, F.L. The initiation of breast and prostate cancer. Carcinogenesis 2002, 23, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Gronberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Willenbring, H.; Sharma, A.D.; Vogel, A.; Lee, A.Y.; Rothfuss, A.; Wang, Z.; Finegold, M.; Grompe, M. Loss of p21 permits carcinogenesis from chronically damaged liver and kidney epithelial cells despite unchecked apoptosis. Cancer Cell 2008, 14, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Baretton, G.B.; Klenk, U.; Diebold, J.; Schmeller, N.; Lohrs, U. Proliferation- and apoptosis-associated factors in advanced prostatic carcinomas before and after androgen deprivation therapy: Prognostic significance of p21/WAF1/CIP1 expression. Br. J. Cancer 1999, 80, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Sogawa, N.; Takiguchi, N.; Koda, K.; Oda, K.; Satomi, D.; Kato, K.; Ishikura, H.; Miyazaki, M. Value of expression of p21waf1/CIP1 as a prognostic factor in advanced middle and lower rectal cancer patients treated with preoperative radio-chemotherapy. Int. J. Oncol. 2002, 21, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002, 179, 1–14. [Google Scholar] [CrossRef]
- Fang, Q.; Kanugula, S.; Pegg, A.E. Function of domains of human O6-alkylguanine-DNA alkyltransferase. Biochemistry 2005, 44, 15396–15405. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P.M. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Morton, D.J.; Patel, D.; Joshi, J.; Hunt, A.; Knowell, A.E.; Chaudhary, J. ID4 regulates transcriptional activity of wild type and mutant p53 via K373 acetylation. Oncotarget 2017, 8, 2536–2549. [Google Scholar] [CrossRef] [Green Version]
- Komaragiri, S.K.; Bostanthirige, D.H.; Morton, D.J.; Patel, D.; Joshi, J.; Upadhyay, S.; Chaudhary, J. ID4 promotes AR expression and blocks tumorigenicity of PC3 prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 478, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Heinlein, C.; Krepulat, F.; Lohler, J.; Speidel, D.; Deppert, W.; Tolstonog, G.V. Mutant p53(R270H) gain of function phenotype in a mouse model for oncogene-induced mammary carcinogenesis. Int. J. Cancer 2008, 122, 1701–1709. [Google Scholar] [CrossRef]
- Wijnhoven, S.W.; Zwart, E.; Speksnijder, E.N.; Beems, R.B.; Olive, K.P.; Tuveson, D.A.; Jonkers, J.; Schaap, M.M.; van den Berg, J.; Jacks, T.; et al. Mice expressing a mammary gland-specific R270H mutation in the p53 tumor suppressor gene mimic human breast cancer development. Cancer Res. 2005, 65, 8166–8173. [Google Scholar] [CrossRef] [Green Version]
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev. 2017, 31, 1339–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).