
A Unified Model of Age-Related Cardiovascular Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Current Approaches to Cardiovascular Disease Intervention
2. Future Approaches to Cardiovascular Disease Intervention
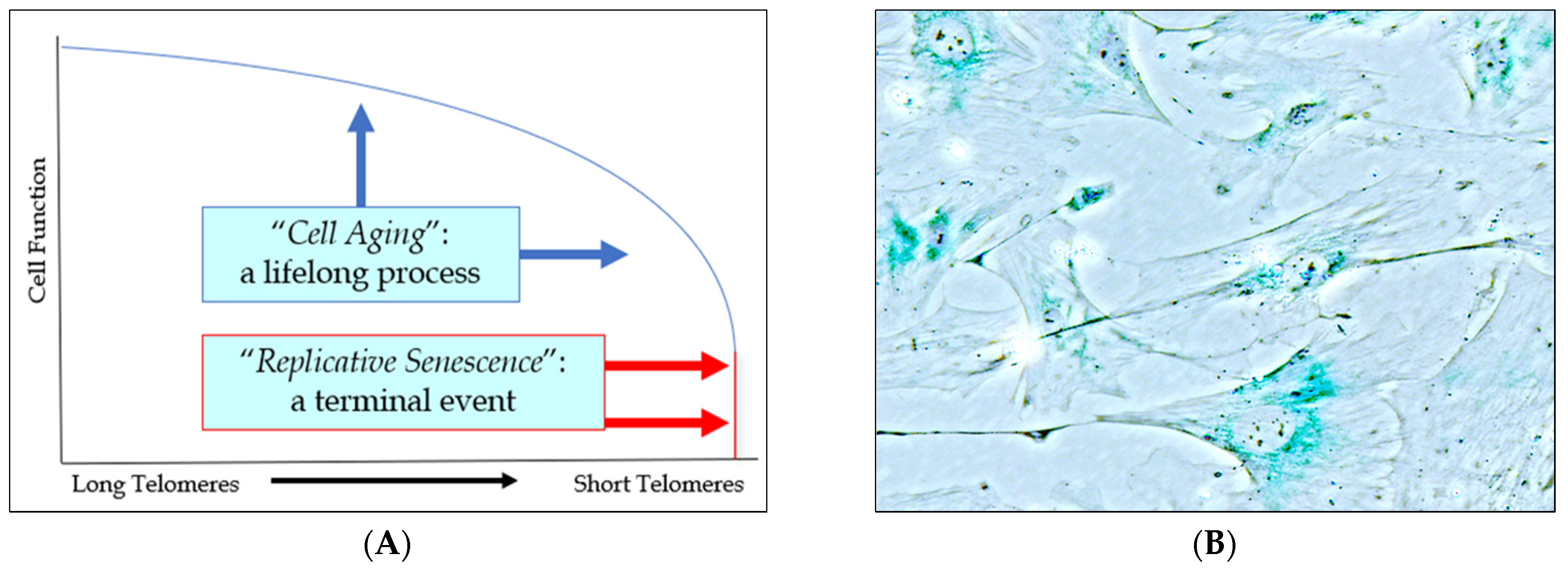
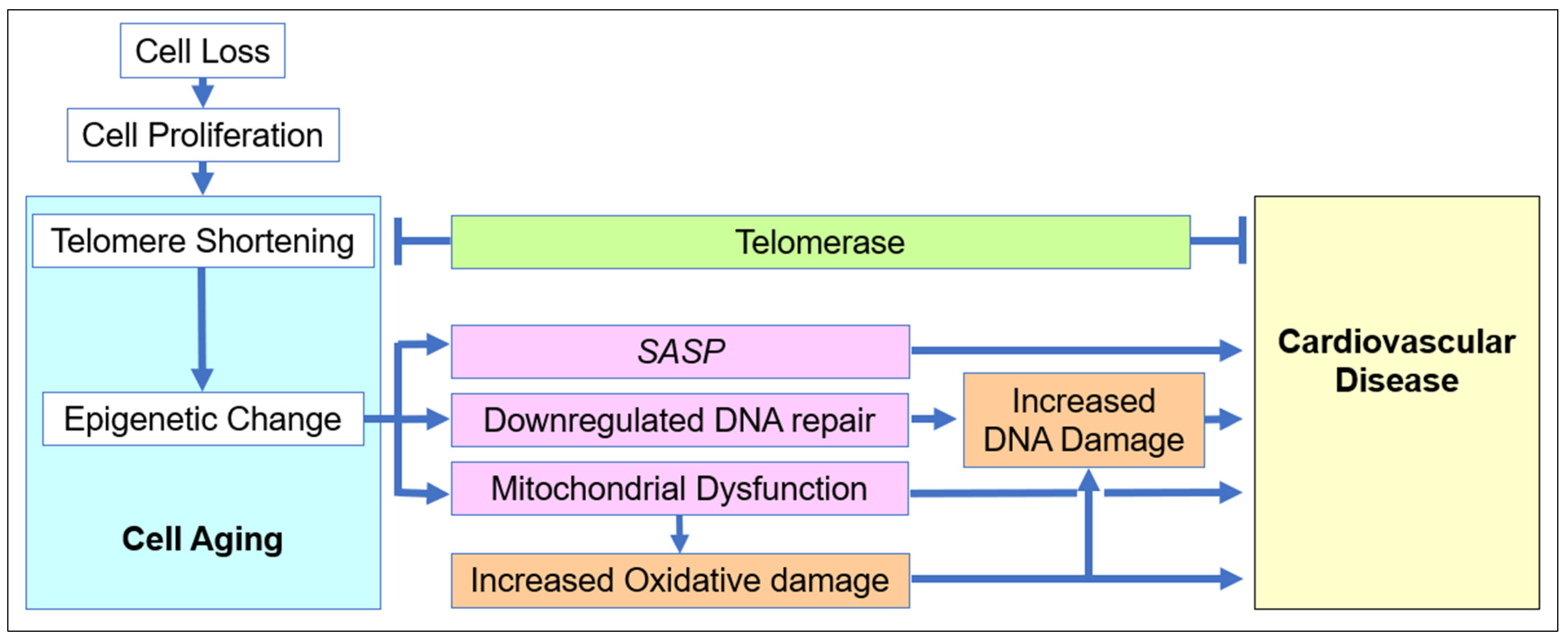
2.1. The Role of Cell Aging in Age-Related Cardiovascular Disease
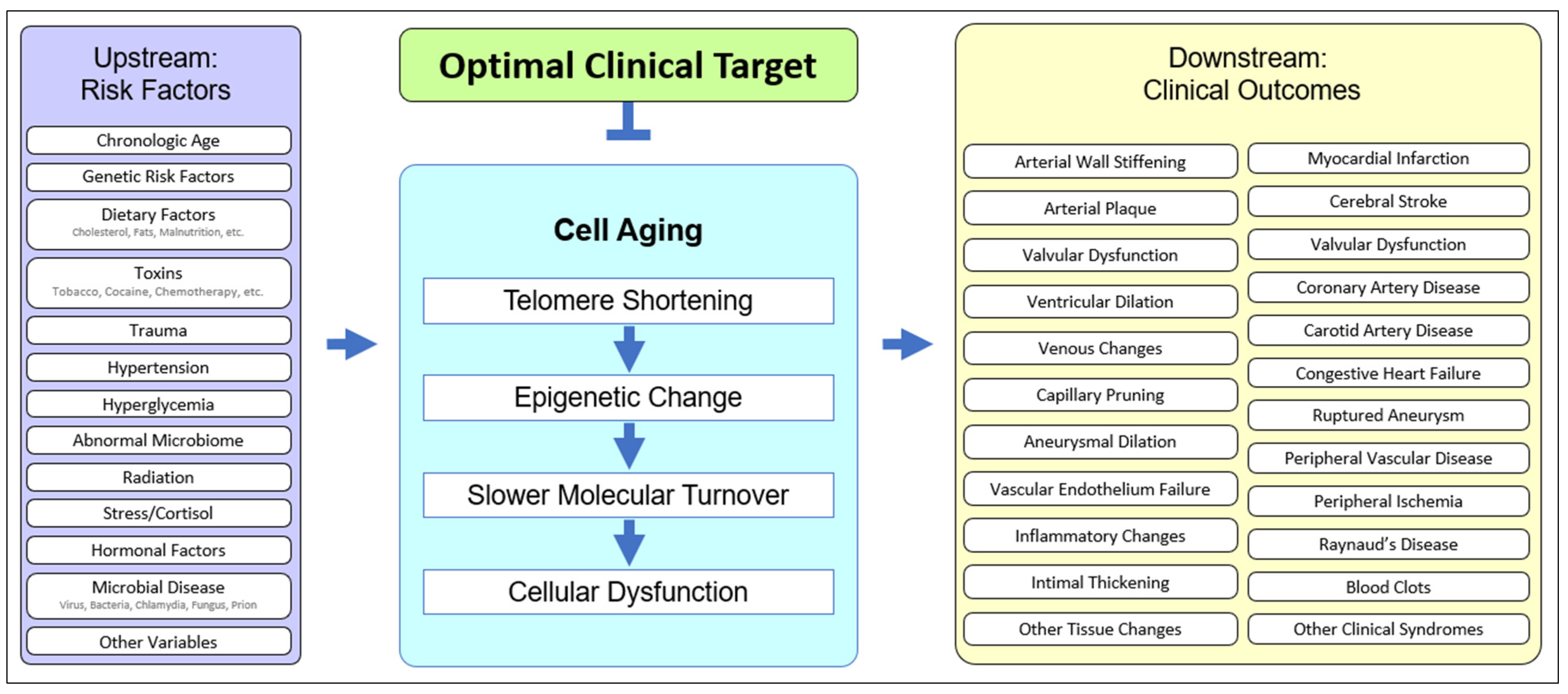
2.2. Upstream Risk Factors in Cardiovascular Disease
2.3. Downstream Outcomes in Cardiovascular Disease
2.4. Heterogeneity of Clinical Presentations in Cardiovascular Disease
3. Perspective on Optimal Clinical Intervention
3.1. Component Interventions in Cardiovascular Disease
3.2. Systems Interventions in Cardiovascular Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises. Part I: Aging Arteries: A “Set Up” for Vascular Disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises. Part II: The Aging Heart in Health: Links to Heart Disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.cardiosmart.org/news/2019/12/latest-statistics-say-nearly-half-of-americans-have-some-form-of-heart-disease (accessed on 15 October 2022).
- Yazdanyar, A.; Newman, A.B. The burden of cardiovascular disease in the elderly: Morbidity, mortality, and costs. Clin. Geriatr. Med. 2009, 25, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Landau, S.M.; Harrison, T.M. A Link Between Cardiovascular Risk Management and Alzheimer Disease Is Still Elusive. JAMA Neurol. 2021, 78, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.championadvocates.org/en/champion-advocates-programme/the-costs-of-cvd (accessed on 15 October 2022).
- Available online: https://www.who.int/health-topics/cardiovascular-diseases (accessed on 15 October 2022).
- Available online: https://healthmetrics.heart.org/wp-content/uploads/2017/10/Cardiovascular-Disease-A-Costly-Burden.pdf (accessed on 15 October 2022).
- Available online: https://report.nih.gov/categorical_spending.aspx (accessed on 15 October 2022).
- Ford, E.S.; Capewell, S. Proportion of the decline in cardiovascular mortality disease due to prevention versus treatment: Public health versus clinical care. Annu. Rev. Public Health 2011, 32, 5–22. [Google Scholar] [CrossRef]
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable risk factors, cardiovascular disease and mortality in 155, 722 individuals from 21 high-, middle-, and low-income countries. Lancet 2020, 395, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.heart.org/-/media/Files/About-Us/Policy-Research/Fact-Sheets/Public-Health-Advocacy-and-Research/Investing-in-Our-Hearts--NIH-Fact-Sheet.pdf (accessed on 28 November 2022).
- Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6642725/ (accessed on 15 October 2022).
- Available online: https://www.hopkinsmedicine.org/gim/research/content/cvd.html (accessed on 1 May 2022).
- Lloyd-Jones, D.M.; Hong, Y.; Labarthe, D.; Mozaffarian, D.; Appel, L.J.; Van Horn, L.; Greenlund, K.; Daniels, S.; Nichol, G.; Tomaselli, G.F.; et al. Defining and Setting National Goals for Cardiovascular Health Promotion and Disease Reduction. Circulation 2010, 121, 586–613. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M. Lifestyle Strategies for Risk Factor Reduction, Prevention, and Treatment of Cardiovascular Disease. Am. J. Lifestyle Med. 2018, 13, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, N.; Knight, J.; Mizdrak, A.; Blakely, T.; Wilson, N. Preventive Pharmacotherapy for Cardiovascular Disease: A Modelling Study Considering Health Gain, Costs, and Cost-Effectiveness when Stratifying by Absolute Risk. Sci. Rep. 2019, 9, 19562. [Google Scholar] [CrossRef] [PubMed]
- Davidson, L.J.; Davidson, C.J. Transcatheter Treatment of Valvular Heart Disease: A Review. JAMA 2021, 325, 2480–2494. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.health.harvard.edu/blog/mitraclip-valve-repair-device-offers-new-treatment-option-for-some-with-severe-mitral-regurgitation-2019042416495 (accessed on 15 October 2022).
- Cesaro, A.; Schiavo, A.; Moscarella, E.; Coletta, S.; Conte, M.; Gragnano, F.; Fimiani, F.; Monda, E.; Caiazza, M.; Limongelli, G.; et al. Lipoprotein(a): A genetic marker for cardiovascular disease and target for emerging therapies. J. Cardiovasc. Med. 2021, 3, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Decoding genetic risks of heart disease. EbioMedicine 2019, 40, 1–2. [CrossRef] [PubMed]
- Jia, X.; Baig, M.M.; Mirza, F.; GholamHosseini, H. A Cox-Based Risk Prediction Model for Early Detection of Cardiovascular Disease: Identification of Key Risk Factors for the Development of a 10-Year CVD Risk Prediction. Adv. Prev. Med. 2019, 2019, 8392348. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Krishnamurthi, R.V.; Parmar, P.; Norrving, B.; Mensah, G.A.; Bennett, D.A.; Barker-Collo, S.; Moran, A.E.; Sacco, R.L.; Truelsen, T.; et al. Update on the Global Burden of Ischemic and Hemorrhagic Stroke in 1990–2013, The GBD 2013 Study. Neuroepidemiology 2015, 45, 161–176. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Hu, F.B.; Manson, J.E.; Rimm, E.B.; Willett, W.C. Primary prevention of coronary heart disease in women through diet and lifestyle. N. Engl. J. Med. 2000, 343, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kurth, T.; Moore, S.C.; Gaziano, J.M.; Kase, C.S.; Stampfer, M.J.; Berger, K.; Buring, J.E. Healthy lifestyle and the risk of stroke in women. Arch. Intern. Med. 2006, 166, 1403–1409. [Google Scholar] [CrossRef]
- Sidney, S.; Quesenberry CPJr Jaffe, M.G.; Sorel, M.; Nguyen-Huynh, M.N.; Kushi, L.H.; Go, A.S.; Rana, J.S. Recent Trends in Cardiovascular Mortality in the United States and Public Health Goals. JAMA Cardiol. 2016, 1, 594–599. [Google Scholar] [CrossRef]
- Wald, N.J.; Simmonds, M.; Morris, J.K. Screening for Future Cardiovascular Disease Using Age Alone Compared with Multiple Risk Factors and Age. PLoS ONE 2011, 6, e18742. [Google Scholar] [CrossRef] [PubMed]
- Murabito, J.M.; Pencina, M.J.; Nam, B.H.; D’Agostino, R.B.; Wang, T.J.; Lloyd-Jones, D.; Wilson, P.W.F.; O’Donnell, C.J. Sibling Cardiovascular Disease as a Risk Factor for Cardiovascular Disease in Middle-aged Adults. JAMA 2005, 294, 3117–3123. [Google Scholar] [CrossRef] [PubMed]
- Valerio, L.; Peters, R.J.; Zwinderman, A.H.; Pinto-Sietsma, S.J. Association of Family History With Cardiovascular Disease in Hypertensive Individuals in a Multiethnic Population. J. Am. Heart Assoc. 2016, 5, e004260. [Google Scholar] [CrossRef] [PubMed]
- Savji, N.; Rockman, C.B.; Skolnick, A.H.; Guo, Y.; Adelman, M.A.; Riles, T.; Berger, J.S. Association between advanced age and vascular disease in different arterial territories: A population database of over 3.6 million subjects. J. Am. Coll. Cardiol. 2013, 61, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Whisnant, J.P.; Sicks, J.D.; O’Fallon, W.M.; Wiebers, D.O. Stroke incidence, prevalence, and survival: Secular trends in Rochester, Minnesota, through 1989. Stroke 1996, 27, 373–380. [Google Scholar]
- Wolf, P.A.; D’Agostino, R.B.; O’Neal, M.A.; Sytkowski, P.; Kase, C.S.; Belanger, A.J.; Kannel, W.B. Secular trends in stroke incidence and mortality. The Framingham Study. Stroke 1992, 23, 1551–1555. [Google Scholar] [CrossRef]
- Boniewska-Bernacka, E.; Pańczyszyn, A.; Klinger, M. Telomeres and telomerase in risk assessment of cardiovascular diseases. Exp. Cell Res. 2020, 397, 112361. [Google Scholar] [CrossRef]
- Li, A.; Koch, Z.; Ideker, T. Epigenetic aging: Biological age prediction and informing a mechanistic theory of aging. J. Internal Med. 2022, 292, 733–744. [Google Scholar] [CrossRef]
- Nie, C.; Li, Y.; Li, R.; Yan, Y.; Zhang, D.; Li, T.; Li, Z.; Sun, Y.; Zhen, H.; Ding, J.; et al. Distinct biological ages of organs and systems identified from a multi-omics study. Cell Rep. 2022, 38, 110459. [Google Scholar] [CrossRef]
- MacNamara, J.; Eapen, D.J.; Quyyumi, A.; Sperling, L. Novel biomarkers for cardiovascular risk assessment: Current status and future directions. Future Med. 2015, 11, 597–613. [Google Scholar] [CrossRef]
- Soler-Botija, C.; Carolina Gálvez-Montón, C.; Bayés-Genís, A. Epigenetic Biomarkers in Cardiovascular Diseases. Front. Genet. 2019, 10, 950. [Google Scholar] [CrossRef]
- Laurent Metzinger, L.; de Franciscis, S.; Serra, R. The Management of Cardiovascular Risk through Epigenetic Biomarkers. BioMed Res. Int. 2017, 2017, 9158572. [Google Scholar] [CrossRef]
- JorgeDíaz-Garzón, J.; Fernández–Calle, P.; Minchinela, J.; Aarsand, A.K.; Bartlett, W.A.; Aslan, B.; Boned, B.; Braga, F.; Carobene, A.; Coskun, A.; et al. Biological variation data for lipid cardiovascular risk assessment biomarkers. A systematic review applying the biological variation data critical appraisal checklist (BIVAC). Clin. Chim. Acta 2019, 495, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Gilstrap, L.G.; Wang, T.J. Biomarkers and Cardiovascular Risk Assessment for Primary Prevention: An Update. Clin. Chem. 2012, 58, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.H.; Cameron-Smith, D.; Wessner, B.; Franzke, B. Biomarkers of Aging: From Function to Molecular Biology. Nutrients 2016, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Yetim, E.; Topcuoglu, M.A.; Kutlay, N.Y.; Tukun, A.; KaOguz, K.K.; Arsava, E.M. The association between telomere length and ischemic stroke risk and phenotype. Sci. Rep. 2021, 11, 10967. [Google Scholar] [CrossRef]
- Piplani, S.; Prabhu, M.; Alemao, N.N.; Akash, C.; Ram, P.; Ambar, S.; Kumbar, V.; Chugh, Y.; Raychauduri, S.P.; Chugh, S.K. Conventional Risk Factors, Telomere Length, and Ischemic Heart disease: Insights into the Mediation Analysis. Genome Integr. 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Capewell, S.; Beaglehole, R.; Seddon, M.; McMurray, J. Explanation for the decline in coronary heart disease mortality rates in Auckland, New Zealand, between 1982 and 1993. Circulation 2000, 102, 1511–1516. [Google Scholar] [CrossRef]
- Ahmad, Y.; Lansky, A.J.; Velazquez, E.J. Current Landscape and Future Directions of Coronary Revascularization in Patients with Heart Failure. JAMA Cardiol. 2022, 7, 577–578. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives postmitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Childs, B.G.; Li, H.; van Deursen, J.M. Senescent cells: A therapeutic target for cardiovascular disease. J. Clin. Investig. 2018, 128, 1217–1228. [Google Scholar] [CrossRef]
- Fossel, M. A Unified Model of Dementias and Age-Related Neurodegeneration. Alzheimer’s Dement. 2020, 16, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Vasileiou, P.V.S.; Papaspyropoulos, A.; Hazapis, O.; Petty, R.; Demaria, M.; Gorgoulis, V.G. Cellular senescence and cardiovascular diseases: Moving to the “heart” of the problem. Physiol. Rev. 2022, 103, 609–647. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Fossel, M. Telomerase and the aging cell: Implications for human health. JAMA 1998, 279, 1732–1735. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.; Fossel, M. Telomeres, cancer, and aging: Altering the human lifespan. JAMA 1997, 278, 1345–1348. [Google Scholar] [CrossRef]
- Hayflick, L. The greatest risk factor for the leading cause of death is ignored. Biogerontology 2020, 22, 133–141. [Google Scholar] [CrossRef]
- Fossel, M. Cells, Aging, and Human Disease; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Chan, M.; Yuan, H.; Soifer, I.; Maile, T.M.; Wang, R.Y.; Ireland, A.; O’Brien, J.J.; Goudeau, J.; Chan, L.J.G.; Vijay, T.; et al. Novel insights from a multiomics dissection of the Hayflick limit. eLife 2022, 11, e70283. [Google Scholar] [CrossRef]
- Ogrodnik, M. Cellular aging beyond cellular senescence: Markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell 2021, 20, e13338. [Google Scholar] [CrossRef]
- Dong, X.; Sun, S.; Zhang, L.; Kim, S.; Tu, Z.; Montagna, C.; Maslov, A.Y.; Suh, Y.; Wang, T.; Campisi, J.; et al. Age-related telomere attrition causes aberrant gene expression in sub-telomeric regions. Aging Cell 2020. [Google Scholar] [CrossRef]
- Olivieri, F.; Recchioni, R.; Marcheselli, F.; Marie Abbatecola, A.; Santini, G.; Borghetti, G.; Antonicelli, R.; Domenico Procopio, A. Cellular Senescence in Cardiovascular Diseases: Potential Age-Related Mechanisms and Implications for Treatment. Curr. Pharm. Des. 2013, 19, 1710–1719. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, B.B.; Schneeberger, K.; Vera, M.E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef] [PubMed]
- Dominic, A.; Banerjee, P.; Hamilton, D.J.; Le, N.T.; Abe, J. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020, 37, 101614. [Google Scholar] [CrossRef] [PubMed]
- Hemanthakumar, K.A.; Fang, S.; Anisimov, A.; Mäyränpää, M.I.; Mervaala, E.; Kivelä, R. Cardiovascular disease risk factors induce mesenchymal features and senescence in mouse cardiac endothelial cells. eLife 2021, 10, e62678. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2018, 26, e12487. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Hano, T.; Sawamura, T.; Nishio, I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin. Exp. Pharmacol. Physiol. 2004, 31, 407–413. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 2004, 117, 2417–2426. [Google Scholar] [CrossRef]
- Erdmann, J.; Linsel-Nitschke, P.; Schunkert, H. Genetic Causes of Myocardial Infarction. Dtsch. Arztebl. Int. 2010, 107, 694–699. [Google Scholar] [CrossRef]
- Dai, X.; Wiernek, S.; Evans, J.P.; Runge, M.S. Genetics of coronary artery disease and myocardial infarction. World J. Cardiol. 2016, 8, 1–23. [Google Scholar] [CrossRef]
- Katz, D.H.; Tahir, U.A.; Bick, A.G.; Pampana, A.; Ngo, D.; Benson, M.D.; Yu, Z.; Robbins, J.M.; Chen, Z.-Z.; Cruz, D.E.; et al. Whole Genome Sequence Analysis of the Plasma Proteome in Black Adults Provides Novel Insights Into Cardiovascular Disease. Circulation 2022, 145, 357–370. [Google Scholar] [CrossRef]
- Yun, H.; Noh, N.I.; Lee, E.Y. Genetic risk scores used in cardiovascular disease prediction models: A systematic review. Rev. Cardiovasc. Med. 2022, 23, 8. [Google Scholar] [CrossRef]
- Yegorov, Y.; Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Orekhov, A.N. Role of Telomeres Shortening in Atherogenesis: An Overview. Cells 2021, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2022, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, J.; DePinho, R.A.; Sahin, E. Telomeres and Mitochondria in the Aging Heart. Circ. Res. 2012, 110, 1226–1237. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Wu, C.-M.; Zheng, L.; Wang, Q.; Hu, Y.-W. The emerging role of cell senescence in atherosclerosis. Clin. Chem. Lab. Med. 2020, 59, 27–38. [Google Scholar] [CrossRef]
- Kant, S.; Tran, K.V.; Kvandova, M.; Caliz, A.D.; Yoo, H.J.; Learnard, H.; Dolan, A.C.; Craige, S.M.; Hall, J.D.; Jiménez, J.M.; et al. PGC1α Regulates the Endothelial Response to Fluid Shear Stress via Telomerase Reverse Transcriptase Control of Heme Oxygenase-1. Arterioscler. Thromb. Vasc. Biol. 2021, 42, 19–34. [Google Scholar] [CrossRef]
- Wai, K.M.; Kaori, S.; Itoh, K.; Shinya, O.; Uchikawa, Y.; Hayashi, S.; Shiraki, A.; Murashita, K.; Nakaji, S.; Ihara, K. Telomere Length and Arterial Stiffness Reflected by Brachial-Ankle Pulse Wave Velocity: A Population-Based Cross-Sectional Study. J. Pers. Med. 2021, 11, 1278. [Google Scholar] [CrossRef]
- Matsushita, H.; Chang, E.; Glassford, A.J.; Cooke, J.P.; Chiu, C.P.; Tsao, P.S. eNOS Activity Is Reduced in Senescent Human Endothelial Cells. Preservation by hTERT Immortalization. Circ. Res. 2001, 89, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Mojiri, A.; Walther, B.K.; Jiang, C.; Matrone, G.; Holgate, R.; Xu, Q.; Morales, E.; Wang, G.; Gu, J.; Wang, R.; et al. Telomerase therapy reverses vascular senescence and extends lifespan in progeria mice. Eur. Heart J. 2021, 42, 4352–4369. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial Cell Senescence in Human Atherosclerosis. Role of Telomere in Endothelial Dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Zekavat, S.M.; Chou, E.L.; Zekavat, M.; Pampana, A.; Paruchuri, K.; Lino Cardenas, C.L.; Koyama, S.; Ghazzawi, Y.; Kii, E.; Uddin, M.M.; et al. Fibrillar Collagen Variants in Spontaneous Coronary Artery Dissection. JAMA Cardiol. 2022, 7, 396–406. [Google Scholar] [CrossRef]
- Behjati, M.; Sabri, M.R.; Far, M.E.; Nejati, M. Cardiac complications in inherited mitochondrial diseases. Heart Fail. Rev. 2020, 26, 391–403. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta (BBA) Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.; Tucker, J.; Machin, D.; Liu, Y.; Thomas, T.; Abdeahad, H.; Bramwell, R.; Lesniewski, L.; Donato, A. Aging results in endothelial cell telomere uncapping that induces senescence and physiological dysfunction. FASEB J. 2022, 36 (Suppl. 1). [Google Scholar] [CrossRef]
- Gordon, C.A.; Madamanchi, N.R.; Runge, M.S.; Jarstfer, M.B. Effect of oxidative stress on telomere maintenance in aortic smooth muscle cells. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166397. [Google Scholar] [CrossRef]
- Voghel, G.; Thorin-Trescasesa, N.; Farhata, N.; Nguyen, A.; Villeneuvea, L.; Mamarbachi, A.M.; Fortier, A.; Perrault, L.P.; Carrier, N.; Thorin, E. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech. Ageing Dev. 2007, 128, 662–671. [Google Scholar] [CrossRef]
- Martens, D.S.; Van Der Stukken, C.; Derom, C.; Thiery, E.; Bijnens, E.M.; Nawrot, T.S. Newborn telomere length predicts later life telomere length: Tracking telomere length from birth to child- and adulthood. EBioMedicine 2021, 63, 103164. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Bardeguez, A.; Gardner, J.P.; Rodriguez, P.; Ganesh, V.; Kimura, M.; Skurnick, J.; Awad, G.; Aviv, A. Telomere length in the newborn. Pediatr. Res. 2002, 52, 377–381. [Google Scholar] [CrossRef]
- Lister-Shimauchi, E.H.; McCarthy, B.; Lippincott, M.; Ahmed, S. Genetic and Epigenetic Inheritance at Telomeres. Epigenomes 2022, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Dogan, F.; Forsyth, N.R. Telomerase Regulation: A Role for Epigenetics. Cancers 2021, 13, 1213. [Google Scholar] [CrossRef]
- Jie, M.M.; Chang, X.; Zeng, S.; Liu, C.; Liao, G.B.; Wu, Y.R.; Liu, C.H.; Hu, C.J.; Yang, S.M.; Li, X.Z. Diverse regulatory manners of human telomerase reverse transcriptase. Cell Commun. Signal. 2019, 17, 63. [Google Scholar] [CrossRef]
- Cardenas, A.; Ecker, S.; Fadadu, R.P.; Huen, K.; Orozco, A.; McEwen, L.M.; Engelbrecht, H.R.; Gladish, N.; Kobor, M.S.; Rosero-Bixby, L.; et al. Epigenome-wide association study and epigenetic age acceleration associated with cigarette smoking among Costa Rican adults. Sci. Rep. 2022, 12, 4277. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette Smoke Induces Cellular Senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef]
- Farhat, N.; Thorin-Trescases, N.; Voghel, G.; Villeneuve, L.; Mamarbachi, M.; Perrault, L.P.; Carrier, M.; Thorin, E. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can. J. Physiol. Pharmacol. 2008, 86, 761–769. [Google Scholar] [CrossRef]
- Li, R.; Chen, G.; Liu, X.; Pan, M.; Kang, N.; Hou, X.; Liao, W.; Dong, X.; Yuchi, Y.; Mao, Z.; et al. Aging biomarkers: Potential mediators of association between long-term ozone exposure and risk of atherosclerosis. J. Intern. Med. 2022, 292, 512–522. [Google Scholar] [CrossRef]
- Sharma, K.; Lee, H.H.; Gong, D.S.; Park, S.H.; Yi, E.; Schini-Kerth, V.; Oak, M.H. Fine air pollution particles induce endothelial senescence via redox-sensitive activation of local angiotensin system. Environ. Pollut. 2019, 252 Pt A, 317–329. [Google Scholar] [CrossRef]
- Chang-Chiena, J.; Huang, J.L.; Tsai, H.J.; Wang, S.L.; Kuo, M.L.; Yao, T.C. Particulate matter causes telomere shortening and increase in cellular senescence markers in human lung epithelial cells. Ecotoxicol. Environ. Saf. 2021, 222, 112484. [Google Scholar] [CrossRef] [PubMed]
- Leitão, C.; Mignano, A.; Estrela, M.; Fardilha, M.; Figueiras, A.; Roque, F.; Herdeiro, M.T. The Effect of Nutrition on Aging-A Systematic Review Focusing on Aging-Related Biomarkers. Nutrients 2022, 14, 554. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Bendeck, M.; Gotlieb, A.I. Vascular pathobiology: Atherosclerosis and large vessel disease. In Cardiovascular Pathology; Academic Press: Cambridge, MA, USA, 2016; pp. 85–124. [Google Scholar]
- McGill, H.C., Jr.; McMahan, C.A.; Zieske, A.W.; Tracy, R.E.; Malcom, G.T.; Herderick, E.E.; Strong, J.P. Association of Coronary Heart Disease Risk Factors with microscopic qualities of coronary atherosclerosis in youth. Circulation 2000, 102, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Schade, D.S.; Gonzales, K.; Eaton, R.P. Stop Stenting; Start Reversing Atherosclerosis. Am. J. Med. 2021, 134, 301–303. [Google Scholar] [CrossRef]
- Tuzcu, E.M.; Kapadia, S.R.; Tutar, E.; Ziada, K.M.; Hobbs, R.E.; McCarthy, P.M.; Young, J.B.; Nissen, S.E. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: Evidence from intravascular ultrasound. Circulation 2001, 103, 2705–2710. [Google Scholar] [CrossRef]
- Li, Y.; Gilbert, T.R.; Matsumoto, A.H.; Shi, W. Effect of aging on fatty streak formation in a diet-induced mouse model of atherosclerosis. J. Vasc. Res. 2008, 45, 205–210. [Google Scholar] [CrossRef]
- Vecoli, C.; Borghini, A.; Andreassi, M.G. The Molecular Biomarkers of Vascular Aging and Atherosclerosis: Telomere Length and Mitochondrial DNA 4977 Common Deletion. Mutat. Res. 2020, 784, 108309. [Google Scholar] [CrossRef]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Hägg, S.; Talukdar, H.A.; Asl, H.F.; Jain, R.K.; Cedergren, C.; Shang, M.-M.; Rossignoli, A.; Takolander, R.; Melander, O.; et al. Plasma cholesterol-induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet. 2014, 10, e1004201. [Google Scholar] [CrossRef]
- Parsons, C.; Agasthi, P.; Mookadam, F.; Arsanjani, R. Reversal of coronary atherosclerosis: Role of life style and medical management. Trends Cardiovasc. Med. 2018, 28, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Brown, B.G.; Ganz, P.; Vogel, R.A.; Crowe, T.; Howard, G.; Cooper, C.J.; Brodie, B.; et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: A randomized controlled trial. JAMA 2004, 291, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.J.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M.R. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Ikeda, K.; Urata, R.; Yamazaki, E.; Emoto, N.; Matoba, S. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci. Rep. 2021, 11, 14608. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: Correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef]
- Fossel, M. Accelerated Aging: Progeria. In Genetics; Robinson, R., Ed.; Macmillan: New York, NY, USA, 2003. [Google Scholar]
- Fossel, M. Human Aging and Progeria. J. Ped. Endo. Metab. 2000, 13, 1477–1481. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef]
- Xu, Q.; Mojiri, A.; Boulahouache, L.; Morales, E.; Walther, B.K.; Cooke, J.P. Vascular senescence in progeria: Role of endothelial dysfunction. Eur. Heart J. Open 2022, 2, oeac047. [Google Scholar] [CrossRef]
- Teti, G.; Chiarini, F.; Mazzotti, E.; Ruggeri, A.; Carano, F.; Falconi, M. Cellular senescence in vascular wall mesenchymal stromal cells, a possible contribution to the development of aortic aneurysm. Mech. Ageing Dev. 2021, 197, 111515. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Cellular Senescence in Arterial Diseases. J. Lipid Atheroscler. 2020, 9, 79–91. [Google Scholar] [CrossRef]
- Liao, S.; Curci, J.A.; Kelley, B.J.; Sicard, G.A.; Thompson, R.W. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J. Surg Res. 2000, 92, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The role of cellular senescence in cardiac disease: Basic biology and clinical relevance. Nat. Rev. Cardiol. 2022, 19, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.R.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ. Heart Fail. 2017, 10, e003806. [Google Scholar] [CrossRef]
- Sarig, R.; Rimmer, R.; Bassat, E.; Zhang, L.; Umansky, K.B.; Lendengolts, D.; Perlmoter, G.; Yaniv, K.; Tzahor, E. Transient p53-Mediated Regenerative Senescence in the Injured Heart. Circulation 2019, 139, 2491–2494. [Google Scholar] [CrossRef]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef]
- Guo, G.; Watterson, S.; Zhang, S.-D.; Bjourson, A.; McGilligan, V.; Peace, A.; Rai, T.S. The role of senescence in the pathogenesis of atrial fibrillation: A target process for health improvement and drug development. Ageing Res. Rev. 2021, 69, 101363. [Google Scholar] [CrossRef]
- Ferrari, S.; Pesce, M. Stiffness and Aging in Cardiovascular Diseases: The Dangerous Relationship between Force and Senescence. Int. J. Mol. Sci. 2021, 22, 3404. [Google Scholar] [CrossRef] [PubMed]
- Molnár, A.A.; Nádasy, G.L.; Dörnyei, G.; Patai, B.B.; Delfavero, J.; Fülöp, G.A.; Kirkpatrick, A.C.; Ungvári, Z.; Merkely, B. The aging venous system: From varicosities to vascular cognitive impairment. GeroScience 2021, 43, 2761–2784. [Google Scholar] [CrossRef]
- Vilne, B.; Schunkert, H. Integrating Genes Affecting Coronary Artery Disease in Functional Networks by Multi-OMICs Approach. Front. Cardiovasc. Med. 2018, 5, 89. [Google Scholar] [CrossRef]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schunkert, H. Genetics of coronary artery disease in the post-GWAS era. J. Int. Med. 2021, 290, 980–992. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.P.; Mahajan, B.; Sattar, R.S.A.; Nimisha; Apurva; Kumar, A.; Sharma, A.K.; Ahmad, E.; Ali, A.; Saluja, S.S. Elucidation of Epigenetic Landscape in Coronary Artery Disease: A Review on Basic Concept to Personalized Medicine. Epigenetic Insights 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.V.; Knight, S.; Dogan, T.K.; Knowlton, K.U.; Philibert, R. External validation of integrated genetic-epigenetic biomarkers for predicting incident coronary heart disease. Epigenomics 2021, 13, 1095–1112. [Google Scholar] [CrossRef] [PubMed]
- Garratt, H.; Ashburn, R.; Sopic, M.; Nogara, A.; Caporali, A.; Mitic, T. Long Non-Coding RNA Regulation of Epigenetics in Vascular Cells. Non-Coding RNA 2021, 7, 62. [Google Scholar] [CrossRef]
- Fossel, M. Use of Telomere Length as a Biomarker for Aging and Age-Related Disease. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2012, 1, 121–127. [Google Scholar] [CrossRef][Green Version]
- Semeraro, M.D.; Almer, G.; Renner, W.; Gruber, H.J.; Herrmann, M. Telomere length in leucocytes and solid tissues of young and aged rats. Aging 2022, 14, 1713–1728. [Google Scholar] [CrossRef]
- Bhattacharyya, J.; Mihara, K.; Bhattacharjee, D.; Mukherjee, M. Telomere length as a potential biomarker of coronary artery disease. Indian J. Med. Res. 2017, 145, 730–737. [Google Scholar] [CrossRef]
- Schneider, C.V.; Schneider, K.M.; Teumer, A.; Rudolph, K.L.; Hartmann, D.; Rader, D.J.; Strnad, P. Association of Telomere Length with Risk of Disease and Mortality. JAMA Intern. Med. 2022, 182, 291. [Google Scholar] [CrossRef]
- De Meyer, T.; Van Daele, C.M.; De Buyzere, M.L.; Denil, S.; De Bacquer, D.; Segers, P.; Cooman, L.; De Backer, G.G.; Gillebert, T.C.; Bekaert, S.; et al. No shorter telomeres in subjects with a family history of cardiovascular disease in the Asklepios study. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3076–3081. [Google Scholar] [CrossRef][Green Version]
- Doroschuk, N.A.; Postnov, A.Y.; Doroschuk, A.D.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Khotina, V.A.; Orekhov, A.N.; Sobenin, I.A.; Sazonova, M.A. An original biomarker for the risk of developing cardiovascular diseases and their complications: Telomere length. Tox Rep. 2021, 8, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Codd, V.; Denniff, M.; Swinfield, C.; Warner, S.C.; Papakonstantinou, M.; Sheth, S.; Nanus, D.E.; Budgeon, C.A.; Musicha, C.; Bountziouka, V.; et al. Measurement and initial characterization of leukocyte telomere length in 474,074 participants in UK Biobank. Nat. Aging 2022, 2, 170–179. [Google Scholar] [CrossRef]
- Vecoli, C.; Basta, G.; Borghini, A.; Gaggini, M.; Del Turco, S.; Mercuri, A.; Gastaldelli, A.; Andreassi, M.G. Advanced glycation end products, leukocyte telomere length, and mitochondrial DNA copy number in patients with coronary artery disease and alterations of glucose homeostasis: From the GENOCOR study. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Nordfjäll, K.; Eliasson, M.; Stegmayr, B.; Melander, O.; Nilsson, P.; Roos, G. Telomere Length Is Associated with Obesity Parameters but With a Gender Difference. Obesity 2012, 16, 2682–2689. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Saijonmaa, O.; Strandberg, T. The roles of senescence and telomere shortening in cardiovascular disease. Nat. Rev. Cardiol. 2013, 10, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Zimnitskaya, O.V.; Petrova, M.M.; Lareva, N.V.; Cherniaeva, M.S.; Al-Zamil, M.; Ivanova, A.E.; Shnayder, N.A. Leukocyte Telomere Length as a Molecular Biomarker of Coronary Heart Disease. Genes 2022, 13, 1234. [Google Scholar] [CrossRef] [PubMed]
- Aviv, H.; Khan, M.Y.; Skurnick, J.; Okuda, K.; Kimura, M.; Gardner, J.; Priolo, L.; Aviv, A. Age dependent aneuploidy and telomere length of the human vascular endothelium. Atherosclerosis 2001, 159, 281–287. [Google Scholar] [CrossRef]
- Shi, Q.; Hubbard, G.B.; Kushwaha, R.S.; Rainwater, D.; Thomas, C.A.; Leland, M.M.; VandeBerg, J.L.; Wang, X.L. Endothelial senescence after high-cholesterol, high-fat diet challenge in baboons. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2913–H2920. [Google Scholar] [CrossRef]
- De Meyer, T.; Nawrot, T.; Bekaert, S.; De Buyzere, M.L.; Rietzschel, E.R.; Andrés, V. Telomere Length as Cardiovascular Aging Biomarker. J. Am. Coll. Cardiol. 2018, 72, 805–813. [Google Scholar] [CrossRef]
- Zhang, W.G.; Zhu, S.Y.; Bai, X.J.; Zhao, D.L.; Jiang, S.M.; Li, J.; Li, Z.X.; Fu, B.; Cai, G.Y.; Sun, X.F.; et al. Select aging biomarkers based on telomere length and chronological age to build a biological age equation. Age 2014, 36, 9639. [Google Scholar] [CrossRef]
- Saliha, R.; Syed Tasleem, R.; Farzana, M. Telomere Length Variations in Aging and Age-Related Diseases. Curr. Aging Sci. 2014, 7, 161–167. [Google Scholar]
- Fasching, C.L. Telomere length measurement as a clinical biomarker of aging and disease. Crit. Rev. Clin. Lab. Sci. 2018, 55, 443. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.J.; Constância, M.; Neves, D.; Simm, A. Biomarkers of Aging: From Cellular Senescence to Age-Associated Diseases. Oxidative Med. Cell. Longev. 2017, 55, 443. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, S.; De Meyer, T.; Rietzschel, E.R.; De Buyzere, M.L.; De Bacquer, D.; Langlois, M.; Segers, P.; Cooman, L.; Van Damme, P.; Cassiman, P.; et al. Telomere length and cardiovascular risk factors in a middle-aged population free of overt cardiovascular disease. Aging Cell 2007, 6, 639–647. [Google Scholar] [CrossRef]
- Opstad, T.B.; Berg, T.J.; Bech Holte, K.B.; Arnesen, H.; Solheim, S.; Seljeflot, I. Reduced leukocyte telomere lengths and sirtuin 1 gene expression in long-term survivors of type 1 diabetes: A Dialong substudy. J. Diabetes Investig. 2020, 12, 1183–1192. [Google Scholar] [CrossRef]
- Ali, M.K.; Narayan, K.M.; Tandon, N. Diabetes & coronary heart disease: Current perspectives. Indian J. Med. Res. 2010, 132, 584–597. [Google Scholar]
- Kosmopoulos, M.; Chiriacò, M.; Stamatelopoulos, K.; Tsioufis, C.; Masci, P.G.; Kontogiannis, C.; Mengozzi, A.; Pugliese, N.R.; Taddei, S.; Virdis, A.; et al. The relationship between telomere length and putative markers of vascular ageing: A Systematic Review and Meta-analysis. Mech. Ageing Dev. 2021, 201, 111604. [Google Scholar] [CrossRef]
- Willeit, P.; Willeit, J.; Brandstätter, A.; Ehrlenbach, S.; Mayr, A.; Gasperi, A.; Weger, S.; Oberhollenzer, F.; Reindl, M.; Kronenberg, F.; et al. Cellular Aging Reflected by Leukocyte Telomere Length Predicts Advanced Atherosclerosis and Cardiovascular Disease Risk. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1649–1656. [Google Scholar] [CrossRef]
- Pusceddu, I.; Kleber, M.; Delgado, G.; Herrmann, W.; Marz, W.; Herrmann, M. Telomere length and mortality in the Ludwigshafen Risk and Cardiovascular Health study. PLoS ONE 2018, 13, e0198373. [Google Scholar] [CrossRef]
- Sun, X.; Feinberg, M.W. Vascular Endothelial Senescence: Pathobiological Insights, Emerging Long Noncoding RNA Targets, Challenges and Therapeutic Opportunities. Front. Physiol. 2021, 12, 693067. [Google Scholar] [CrossRef]
- Khan, S.; Naidoo, D.P.; Chuturgoon, A.A. Telomeres and atherosclerosis: Review. S. Afr. J. Diabetes Vasc. Dis. 2015, 12. Available online: https://hdl.handle.net/10520/EJC181963 (accessed on 15 October 2022).
- den Buijs, J.O.; Musters, M.; Verrips, T.; Post, J.P.; Braam, B.; van Riel, N. Mathematical modeling of vascular endothelial layer maintenance: The role of endothelial cell division, progenitor cell homing, and telomere shortening. Am. J. Physiol. 2004, 287, H2651–H2658. [Google Scholar] [CrossRef]
- Xie, Y.; Lou, D.; Zhang, D. Melatonin Alleviates Age-Associated Endothelial Injury of Atherosclerosis via Regulating Telomere Function. J. Inflamm. Res. 2021, 14, 6799–6812. [Google Scholar] [CrossRef]
- Zglinicki, T.; Martin-Ruiz, C.M. Telomeres as Biomarkers for Ageing and Age-Related Diseases. Curr. Mol. Med. 2005, 5, 197–203. [Google Scholar] [CrossRef]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere Shortening in Human Coronary Artery Diseases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Andres, V. Telomeres and Cardiovascular Disease. Does Size Matter? Circ. Res. 2004, 94, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Aviv, A.; Aviv, H. Reflections on Telomeres, Growth, Aging, and Essential Hypertension. Hypertension 1997, 29, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Aviv, A.; Aviv, H. Telomeres and essential hypertension. Am. J. Hypertens. 1999, 12, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Khan, M.Y.; Skurnick, J.; Kimura, M.; Aviv, H.; Aviv, A. Telomere attrition of the human abdominal aorta: Relationships with age and atherosclerosis. Atherosclerosis 2000, 152, 391–398. [Google Scholar] [CrossRef]
- Edo, M.D.; Andres, V. Aging, telomeres, and atherosclerosis. Cardiovascular. Res. 2005, 66, 213–221. [Google Scholar] [CrossRef]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent High Glucose Implements Stress-Induced Senescence in Human Vascular Endothelial Cells: Role of Superoxide Production by NADPH Oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Milara, J.; Belhadj, N.; Cortijo, J. Senescence Alterations in Pulmonary Hypertension. Cells 2021, 10, 3456. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.G.; Wenceslau, C.F.; Webb, R.C.; Joe, B. Novel Contributors and Mechanisms of Cellular Senescence in Hypertension-Associated Premature Vascular Aging. Am. J. Hypertens. 2019, 32, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, J.H.; Hilgers, K.F.; Steinbach, M.P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. Hypertension Induces Somatic Cellular Senescence in Rats and Humans by Induction of Cell Cycle Inhibitor p16INK4a. Hypertension 2008, 52, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.J.L.; White, S.; Hassan, O.; Zhu, L.; Scott, A.M.; Clark, J.; Glasziou, P. Evaluation of the Incremental Value of a Coronary Artery Calcium Score Beyond Traditional Cardiovascular Risk Assessment: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2022, 182, 634. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis revisited from a metabolic perspective: Role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017, 7, 170219. [Google Scholar] [CrossRef]
- Graves, S.I.; Baker, D.J. Implicating endothelial cell senescence to dysfunction in the ageing and diseased brain. Basic Clin. Pharmacol. Toxicol. 2020, 127, 102–110. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Mattson, M.P. Telomere shortening in neurological disorders: An abundance of unanswered questions. Trends Neurosci. 2014, 37, 256–263. [Google Scholar] [CrossRef]
- Kota, L.N.; Bharath, S.; Purushottam, M.; Moily, N.S.; Sivakumar, P.T.; Varghes, M.; Pal, P.K.; Jain, S. Reduced telomere length in neurodegenerative disorders may suggest shared biology. J. Neuropsychiatry Clin. Neurosci. 2015, 27, e92–e96. [Google Scholar] [CrossRef]
- Liu, M.Y.; Neme, A.; Zhou, Q.G. The Emerging Roles for Telomerase in the Central Nervous System. Front. Mol. Neurosci. 2018, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.; Emoto, N. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat. Commun. 2020, 11, 481. [Google Scholar] [CrossRef] [PubMed]
- Regulsk, M. Understanding Diabetic Induction of Cellular Senescence: A Concise Review. Wounds 2018, 30, 96–101. [Google Scholar]
- Liu, J.; Huang, K.; Cai, G.-Y.; Chen, X.-M.; Yang, J.-R.; Lin, L.-R.; Yang, J.; Huo, B.-G.; Zhan, J.; He, Y.-N. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal. 2014, 26, 110–121. [Google Scholar] [CrossRef]
- Narasimhan, A.; Flores, R.R.; Robbins, P.D.; Niedernhofer, L.J. Role of Cellular Senescence in Type II Diabetes. Endocrinology 2021, 162, bqab136. [Google Scholar] [CrossRef] [PubMed]
- Kuki, S.; Imanishi, T.; Kobayashi, K.; Matsuo, Y.; Obana, M.; Akasaka, T. Hyperglycemia accelerated endothelial progenitor cell senescence via the activation of p38 mitogen-activated protein kinase. Circ. J. 2006, 70, 1076–1081. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef]
- Senthil, K.K.J.; Gokila, V.M.; Wang, S.Y. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-induced senescence and apoptosis in human endothelial cells. Oncotarget 2017, 8, 96568–96587. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.G.; Tang, Q.Z. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis. 2022, 13, 103–128. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 154. [Google Scholar] [CrossRef]
- Behmoaras, J.; Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 2021, 220, e202010162. [Google Scholar] [CrossRef] [PubMed]
- Pamukcu, B.; Lip, G.Y.; Devitt, A.; Griffiths, H.; Shantsila, E. The role of monocytes in atherosclerotic coronary artery disease. Ann. Med. 2010, 42, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Daquinag, A.C.; Fussell, C.; Zhao, Z.; Dai, Y.; Rivera, A.; Snyder, B.E.; Eckel-Mahan, K.L.; Kolonin, M.G. Age-associated telomere attrition in adipocyte progenitors predisposes to metabolic disease. Nat. Metab. 2020, 2, 1482–1497. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.; Latre, A.R.; Kolonin, M.G. Adipose Stromal Cell Expansion and Exhaustion: Mechanisms and Consequences. Cells 2020, 9, 863. [Google Scholar] [CrossRef]
- Ribas-Latre, A.; Santos, R.B.; Fekry, B.; Tamim, Y.M.; Shivshankar, S.; Mohamed, A.M.T.; Baumgartner, C.; Kwok, C.; Gebhardt, C.; Rivera, A.; et al. Cellular and physiological circadian mechanisms drive diurnal cell proliferation and expansion of white adipose tissue. Nat. Commun. 2021, 12, 3482. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nature reviews. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Kim, S.M.; Lun, M.; Wang, M.; Senyo, S.E.; Guillermier, C.; Patwari, P.; Steinhauser, M.L. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014, 20, 1049–1058. [Google Scholar] [CrossRef]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Hammarstedt, A.; Gogg, S.; Hedjazifa, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.; Jeffery, E.; Rodeheffe, M.S. Weighing in on adipocyte precursors. Cell Metab. 2014, 19, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Lakowa, N.; Trieu, N.; Flehmig, G.; Lohmann, T.; Schon, M.R.; Dietrich, A.; Zeplin, P.H.; Langer, S.; Stumvoll, M.; Bluher, M.; et al. Telomere length differences between subcutaneous and visceral adipose tissue in humans. Biochem. Biophys. Res. Commun. 2015, 457, 426–432. [Google Scholar] [CrossRef]
- Lee, Y.H.; Petkova, A.P.; Mottillo, E.P.; Granneman, J.G. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012, 15, 480–491. [Google Scholar] [CrossRef]
- Liu, W.Q.; Zhang, Y.Z.; Wu, Y.; Zhang, J.J.; Li, T.B.; Jiang, T.; Xiong, X.-M.; Luo, X.-J.; Ma, Q.-L.; Peng, J. Myeloperoxidase-derived hypochlorous acid promotes ox-LDL-induced senescence of endothelial cells through a mechanism involving β-catenin signaling in hyperlipidemia. Biochem. Biophys. Res. Commun. 2015, 467, 859–865. [Google Scholar] [CrossRef]
- Oh, S.T.; Park, H.; Yoon, H.J.; Yang, S.Y. Long-Term Treatment of Native LDL Induces Senescence of Cultured Human Endothelial Cells. Oxid Med. Cell Longev. 2017, 2017, 6487825. [Google Scholar] [CrossRef]
- Conley, S.M.; Hickson, L.J.; Kellogg, T.A.; McKenzie, T.; Heimbach, J.K.; Taner, T.; Tang, H.; Jordan, K.L.; Saadiq, I.M.; Woollard, J.R.; et al. Human Obesity Induces Dysfunction and Early Senescence in Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells. Front. Cell Dev. Biol. 2020, 8, 197. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077. [Google Scholar] [CrossRef]
- Rabhi, N.; Desevin, K.; Belkina, A.C.; Tilston-Lunel, A.; Varelas, X.; Layne, M.D.; Farmer, S.R. Obesity-induced senescent macrophages activate a fibrotic transcriptional program in adipocyte progenitors. Life Sci. Alliance 2022, 5, e202101286. [Google Scholar] [CrossRef]
- Ahima, R. Connecting obesity, aging and diabetes. Nat. Med. 2009, 15, 996–997. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Li, Q.; Rydén, M.; Spalding, K.L. Cellular senescence and its role in white adipose tissue. Int. J. Obes. 2021, 45, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.A.; Faragher, R.G.A. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology 2018, 19, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-Induced Hypertension—Interaction of Neurohumoral and Renal Mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Cercato, C.; Fonseca, F.A. Cardiovascular risk and obesity. Diabetol. Metab. Syndr. 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells. Front. Endocrinol 2022, 13, 869414. [Google Scholar] [CrossRef]
- Stone, N.J. Treating Severe Hypercholesterolemia—If Not Now, When? JAMA Cardiol. 2022, 7, 128–129. [Google Scholar] [CrossRef]
- Navar, A.M.; Fonarow, G.C. Transforming the Paradigm for Lipid Lowering. JAMA Cardiol. 2021, 6, 1414. [Google Scholar] [CrossRef]
- Newton, S.L.; Hoffmann, A.P.; Zhi Yu, Z.; Haidermota, S.; Pradeep Natarajan, P.; Michael CHonigberg, M.C. Management of Severe and Moderate Hypercholesterolemia in Young Women and Men. JAMA Cardiol. 2022, 7, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679. [Google Scholar] [CrossRef]
- Michos, E.D.; McEvoy, J.W.; Blumenthal, R.S. Lipid Management for the Prevention of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2019, 381, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.A.; Chen, Y.Y. T cells to fix a broken heart. Science 2022, 375, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Fernández, P.O.M.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, W.; Lu, Z.; Wang, B.; Li, Y.; Yang, J.; Li, P.; Zhao, M. Functional gene module-base identification of phyllyrin as an anticardiac fibrosis agent. Front. Pharmacol. 2020, 11, 1077. [Google Scholar] [CrossRef]
- Orecchioni, M.; Kobiyama, K.; Winkels, H.; Ghosheh, Y.; McArdle, S.; Mikulski, Z.; Kiosses, W.B.; Fan, Z.; Wen, L.; Jung, Y.; et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science 2022, 375, 214–221. [Google Scholar] [CrossRef]
- Berenson, G.S.; Srinivasan, S.R.; Bao, W.; Newman, W.P.; Tracy, R.E.; Wattigney, W.A.; M.S. for the Bogalusa Heart Study. Association between Multiple Cardiovascular Risk Factors and Atherosclerosis in Children and Young Adults. N. Engl. J. Med. 1998, 338, 1650–1656. [Google Scholar] [CrossRef]
- Lee, Y.T.H.; Fang, J.; Schieb, L.; Park, S.; Casper, M.; Gillespie, C. Prevalence and Trends of Coronary Heart Disease in the United States 2011 to 2018. JAMA Cardiol. 2022, 7, 459–462. [Google Scholar] [CrossRef]
- Brinks, J.; Fowler, A.; Franklin, B.A.; Dulai, J. Lifestyle Modification in Secondary Prevention. Am. J. Lifestyle Med. 2016, 11, 137–152. [Google Scholar] [CrossRef]
- Robinson, M.M.; Dasari, S.; Konopka, A.R.; Johnson, M.L.; Manjunatha, S.; Esponda, R.R.; Carter, R.E.; Lanza, I.R.; Nair, K.S. Enhanced Protein Translation Underlies Improved Metabolic and Physical Adaptations to Different Exercise Training Modes in Young and Old Humans. Clin. Transl. Rep. 2017, 25, 581–592. [Google Scholar] [CrossRef]
- Rubio-Tomás, T.; Rueda-Robles, A.; Plaza-Díaz, J.; Álvarez-Mercado, A.I. (). Nutrition and cellular senescence in obesity-related disorders. J. Nutr. Biochem. 2022, 99, 108861. [Google Scholar] [CrossRef]
- Werner, C.; Fürster, T.; Widmann, T.; Pöss, J.; Roggia, C.; Hanhoun, M.; Scharhag, J.; Büchner, N.; Meyer, T.; Kindermann, W.; et al. Physical Exercise Prevents Cellular Senescence in Circulating Leukocytes and in the Vessel Wall. Circulation 2009, 120, 2438–2447. [Google Scholar] [CrossRef]
- Lagoumtzia, S.M.; Chondrogiannia, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free. Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef]
- Stojanović, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef] [PubMed]
- Fossel, M. Cell Senescence, Telomerase, and Senolytic Therapy. OBM Geriatr. 2019, 3, 1–14. [Google Scholar] [CrossRef]
- Sun, Q.; Shi, L.; Prescott, J.; Chiuve, S.; Hu, F.B.; De Vivo, I.; Stampfer, M.J.; Franks, P.; Manson, J.E.; Rexrode, K. Healthy Lifestyle and Leukocyte Telomere Length in U.S. Women. PLoS ONE 2012, 7, e38374. [Google Scholar] [CrossRef]
- Funovic, P.; Korda, M.; Kubant, R.; Barlag, R.E.; Jacob, R.F.; Mason, R.P.; Malinski, T. Effect of beta-blockers on endothelial function during biological aging: A nanotechnological approach. J. Cardiovasc. Pharmacol. 2008, 51, 208–215. [Google Scholar] [CrossRef]
- Khemais-Benkhiat, S.; Idris-Khoja, N.; Ribeiro, T.P.; Silva, G.C.; Abbas, M.; Kheloufi, M.; Lee, J.O.; Toti, F.; Auger, C.; Schini-Kert, V.B. The Redox-sensitive Induction of the Local Angiotensin System Promotes Both Premature and Replicative Endothelial Senescence: Preventive Effect of a Standardized Crataegus Extract. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1581–1590. [Google Scholar] [CrossRef]
- Bode-Böger, A.M.; Martens-Lobenhoffer, J.; Täger, M.; Schröder, H.; Scalera, F. Aspirin reduces endothelial cell senescence. Biochem. Biophys. Res. Commun. 2005, 334, 1226–1232. [Google Scholar] [CrossRef]
- Martinez, P.; Blasco, M.A. An enzyme to cure age-related diseases. Nat. Catal. 2021, 4, 738–739. [Google Scholar] [CrossRef]
- Chiodi, I.; Mondello, C. Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front. Oncol. 2012, 2, 133. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The non-canonical functions of telomerase: To turn off or not to turn off. Mol. Biol. Rep. 2019, 46, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.K.; Lin, M.H.; Wang, C.Y. Telomeres as Therapeutic Targets in Heart Disease. JACC 2019, 4, 855–865. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Funk, W.D.; Wang, C.K.; Shelton, D.N.; Harley, C.B.; Pagon, G.B.; Hoeffler, W.K. Telomerase Expression Restores Dermal Integrity to in Vitro-Aged Fibroblasts in a Reconstituted Skin Model. Exp. Cell Res. 2000, 258, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; Matsuno, H.; Nakazawa, F.; Katayama, R.; Kimura, T. Reconstituting Telomerase Activity Using the Telomerase Catalytic Subunit Prevents the Telomere Shorting and Replicative Senescence in Human Osteoblasts. J. Bone Miner. Res. 2001, 16, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.S.; Horner, J.W.; Wu, C.J.; Li, J.; Lan, Z.D.; Jiang, S.; Xu, X.; Hsu, W.H.; Zal, T.; Flores, I.I.; et al. Telomerase Reverse Transcriptase Preserves Neuron Survival and Cognition in Alzheimer’s Disease Models. Nat. Aging 2021, 1, 1162–1174. [Google Scholar] [CrossRef]
- Harley, C.B.; Liu, W.; Flom, P.L.; Raffaele, J.M. A natural product telomerase activator as part of a health maintenance program: Metabolic and cardiovascular response. Rejuv. Res. 2013, 16, 386–395. [Google Scholar] [CrossRef]
- Salvador, L.; Singaravelu, G.; Harley, C.B.; Flom, P.; Suram, A.; Raffaele, J.M. A Natural Product Telomerase Activator Lengthens Telomeres in Humans: A Randomized, Double Blind, and Placebo Controlled Study. Rejuv. Res. 2016, 19, 478–484. [Google Scholar] [CrossRef]
- Harley, C.B.; Liu, W.; Blasco, M.; Vera, E.; Andrews, W.H.; Briggs, L.A.; Raffaele, J.M. A natural product telomerase activator as part of a health maintenance program. Rejuv. Res. 2011, 14, 45–56. [Google Scholar] [CrossRef]
- Ale-Agha, N.; Jakobs, P.; Goy, C.; Zurek, M.; Rosen, J.; Dyballa-Rukes, N.; Metzger, S.; Greulich, J.; von Ameln, F.; Eckermann, O.; et al. Mitochondrial Telomerase Reverse Transcriptase Protects from Myocardial Ischemia/reperfusion Injury by Improving Complex I Composition and Function. Circulation 2021, 144, 1876–1890. [Google Scholar] [CrossRef] [PubMed]
- Guinobert, I.; Blondeau, C.; Colicchio, B.; Oudrhiri, N.; Dieterlen, A.; Jeandidier, E.; Deschenes, G.; Bardot, V.; Cotte, C.; Ripoche, I.; et al. The Use of Natural Agents to Counteract Telomere Shortening: Effects of a Multi-Component Extract of Astragalus mongholicus Bunge and Danazol. Biomedicines 2020, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.; Bawamia, B.; Bennaceur, K.; Dunn, S.; Marsay, L.; Amoah, R.; Kasim, A.; Filby, A.; Austin, D.; Hancock, H.; et al. Telomerase Activation to Reverse Immunosenescence in Elderly Patients With Acute Coronary Syndrome: Protocol for a Randomized Pilot Trial. JMIR Res. Protoc. 2020, 9, e19456. [Google Scholar] [CrossRef]
- Hoffmann, J.; Richardson, G.; Haendeler, J.; Altschmied, J.; Andrés, V.; Spyridopoulos, I. Telomerase as a Therapeutic Target in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1047–1061. [Google Scholar] [CrossRef]
- Townsley, D.M.; Dumitriu, B.; Liu, D.; Biancotto, A.; Weinstein, B.; Chen, C.; Hardy, N.; Mihalek, A.D.; Lingala, S.; Kim, Y.J.; et al. Danazol Treatment for Telomere Diseases. N. Engl. J. Med. 2016, 374, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Kobayashi, K.; Kuroi, A.; Ikejima, H.; Akasaka, T. Pioglitazone Inhibits Angiotensin II–Induced Senescence of Endothelial Progenitor Cell. Hypertens. Res. 2008, 31, 757–765. [Google Scholar] [CrossRef][Green Version]
- de Jesus, B.B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef]
- Realizing Gene Therapy and Cell Regeneration in Heart Disease|BioSpace. Available online: https://www.biospace.com/article/imagining-the-possibilities-of-gene-therapy-and-cell-regeneration-in-heart-disease-/ (accessed on 15 October 2022).
- Lombardi, L.; Greer-Short, A.; Leon, E.C.; Qureshi, T.N.; Greenwood, A.; Reid, C.A.; Yang, J.; Hunsdorfer, N.; Elmojahid, S.; Mandegar, M.; et al. Reversal of Cardiac Hypertrophy with an Optimized MYBPC3 Gene Therapy. Mol. Ther. 2021, 29, 256. [Google Scholar]
- Arze, C.A.; Springer, S.; Dudas, G.; Patel, S.; Bhattacharyya, A.; Swaminathan, H.; Brugnara, C.; Delagrave, S.; Ong, T.; Kahvejian, A.; et al. Global genome analysis reveals a vast and dynamic anellovirus landscape within the human virome. Cell Host Microbe 2021, 29, 1305–1315. [Google Scholar] [CrossRef]
- Chanda, P.K.; Sukhovershin, R.; Cooke, J.P. mRNA-Enhanced Cell Therapy and Cardiovascular Regeneration. Cells 2021, 10, 187. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, H.; Huang, X.; Chaurasiya, B.; Dong, D.; Shanley, T.P.; Zhao, Y.-Y. Robust genome editing in adult vascular endothelium by nanoparticle delivery of CRISPR-Cas9 plasmid DNA. Cell Rep. 2022, 38, 110196. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Derevyanko, A.; Whittemore, K.; Schneidere, R.P.; Jimenez, V.; Bosch, F.; Blasco, M.A. Gene therapy with the TRF1 telomere gene rescues decreased TRF1 levels with aging and prolongs mouse health span. Aging Cell 2017, 16, 1353–1368. [Google Scholar] [CrossRef] [PubMed]
- Ramunas, J.; Yakubov, E.; Brady, J.J.; Corbel, S.Y.; Holbrook, C.; Brandt, M.; Stein, J.; Santiago, J.G.; Cooke, J.P.; Blau, H.M. Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 2015, 29, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, G.; Bruno, I.G.; Zhang, N.; Sho, S.; Tedone, E.; Lai, T.; Cooke, J.P.; Shay, J.W. Transient introduction of human telomerase mRNA improves hallmarks of progeria cells. Aging Cell 2019, 18, e12979. [Google Scholar] [CrossRef] [PubMed]
- Jaijyan, D.K.; Selariu, A.; Cruz-Cosme, R.; Tong, M.; Yang, S.; Stefa, A.; Kekich, D.; Sadoshima, J.; Herbig, U.; Tang, Q.; et al. New intranasal and injectable gene therapy for healthy life extension. Proc. Natl. Acad. Sci. USA 2022, 119, e2121499119. [Google Scholar] [CrossRef]
- Bär, C.; de Jesus, B.B.; Serrano, R.M.; Tejera, A.M.; Ayuso, E.; Jimenez, V.; Formentini, I.; Bobadilla, M.; Mizrahi, J.; De Martino, A.; et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat. Commun. 2014, 5, 5863. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Muñoz-Lorente, M.A.; Cano-Martin, A.C.; Blasco, M.A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019, 10, 4723. [Google Scholar] [CrossRef]
- Fossel, M.; Whittemore, K. Telomerase and Cancer: A Complex Relationship. OBM Geriatr. 2021, 5, 18. [Google Scholar] [CrossRef]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomerase is not an oncogene. Oncogene 2002, 21, 494–502. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fossel, M.; Bean, J.; Khera, N.; Kolonin, M.G. A Unified Model of Age-Related Cardiovascular Disease. Biology 2022, 11, 1768. https://doi.org/10.3390/biology11121768
Fossel M, Bean J, Khera N, Kolonin MG. A Unified Model of Age-Related Cardiovascular Disease. Biology. 2022; 11(12):1768. https://doi.org/10.3390/biology11121768
Chicago/Turabian StyleFossel, Michael, Joe Bean, Nina Khera, and Mikhail G. Kolonin. 2022. "A Unified Model of Age-Related Cardiovascular Disease" Biology 11, no. 12: 1768. https://doi.org/10.3390/biology11121768
APA StyleFossel, M., Bean, J., Khera, N., & Kolonin, M. G. (2022). A Unified Model of Age-Related Cardiovascular Disease. Biology, 11(12), 1768. https://doi.org/10.3390/biology11121768