
m6A Methylation Analysis Reveals Networks and Key Genes Underlying the Coarse and Fine Wool Traits in a Full-sib Merino Family

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
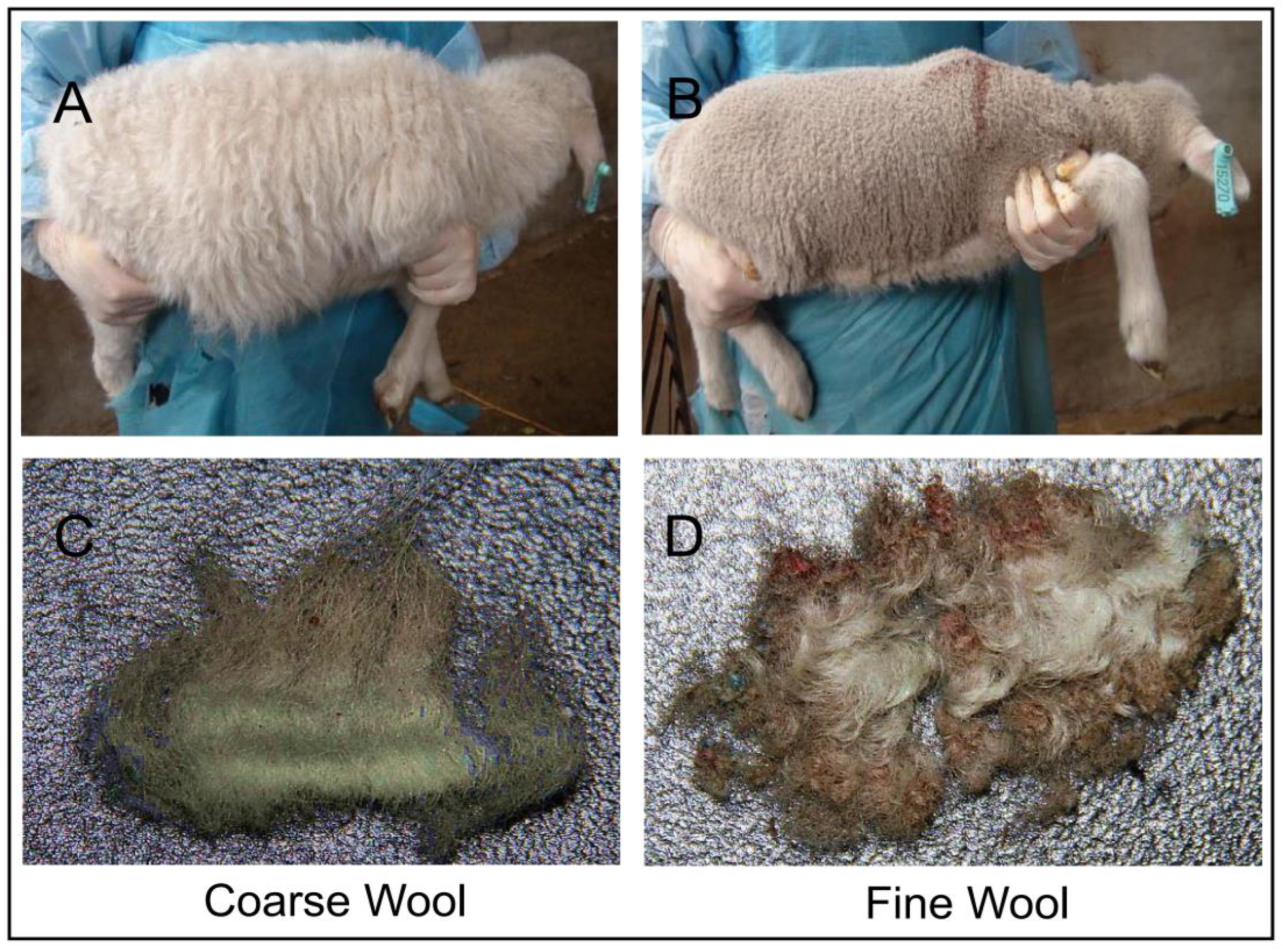
2.1. Sample Collection
2.2. Experimental Procedure
2.3. Bioinformatic Analysis Process
2.4. Quantitative Real-Time PCR Validation
3. Results
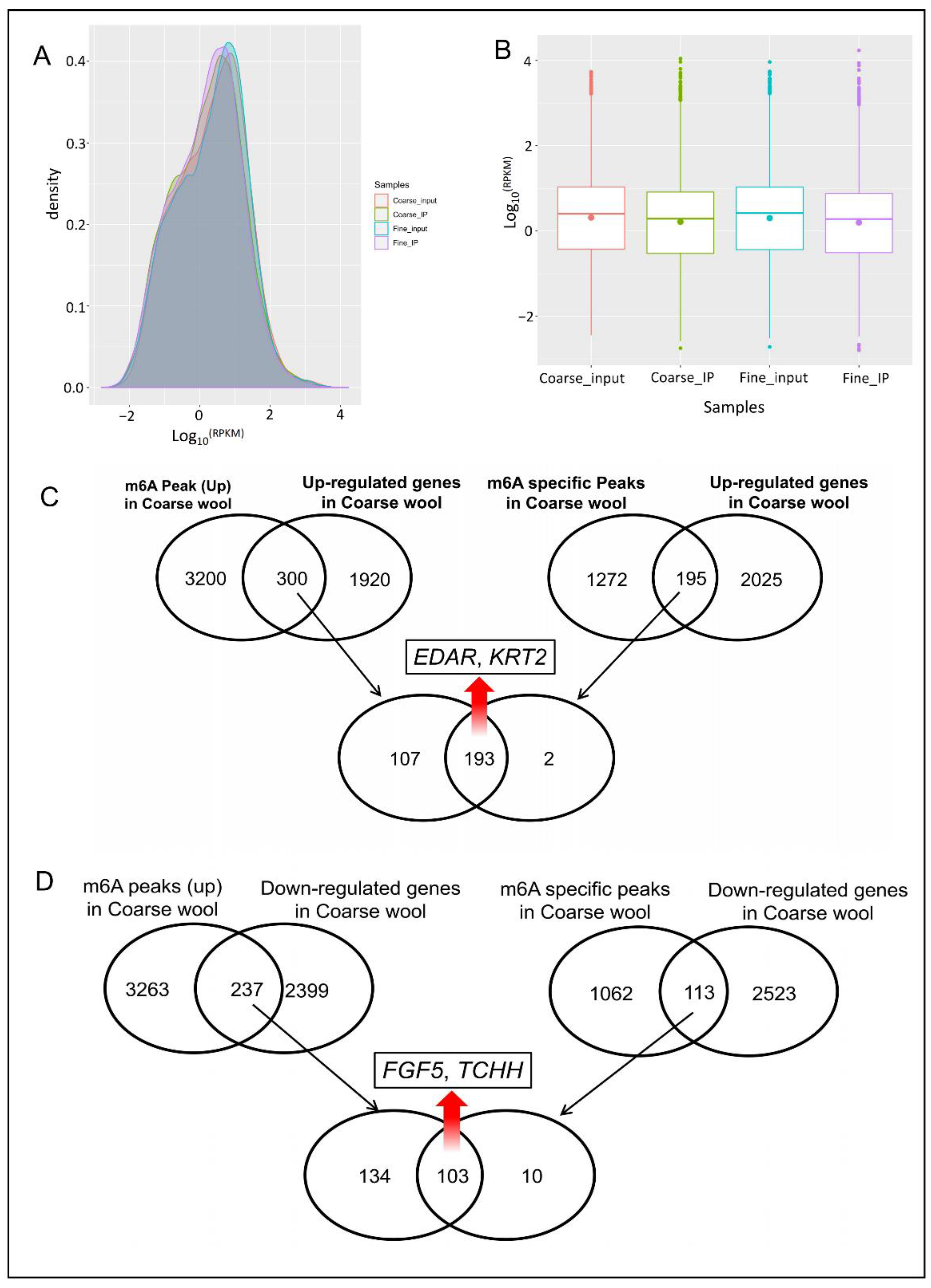
3.1. DATA Quality Control
3.2. Mapping Reads to the Reference Genome
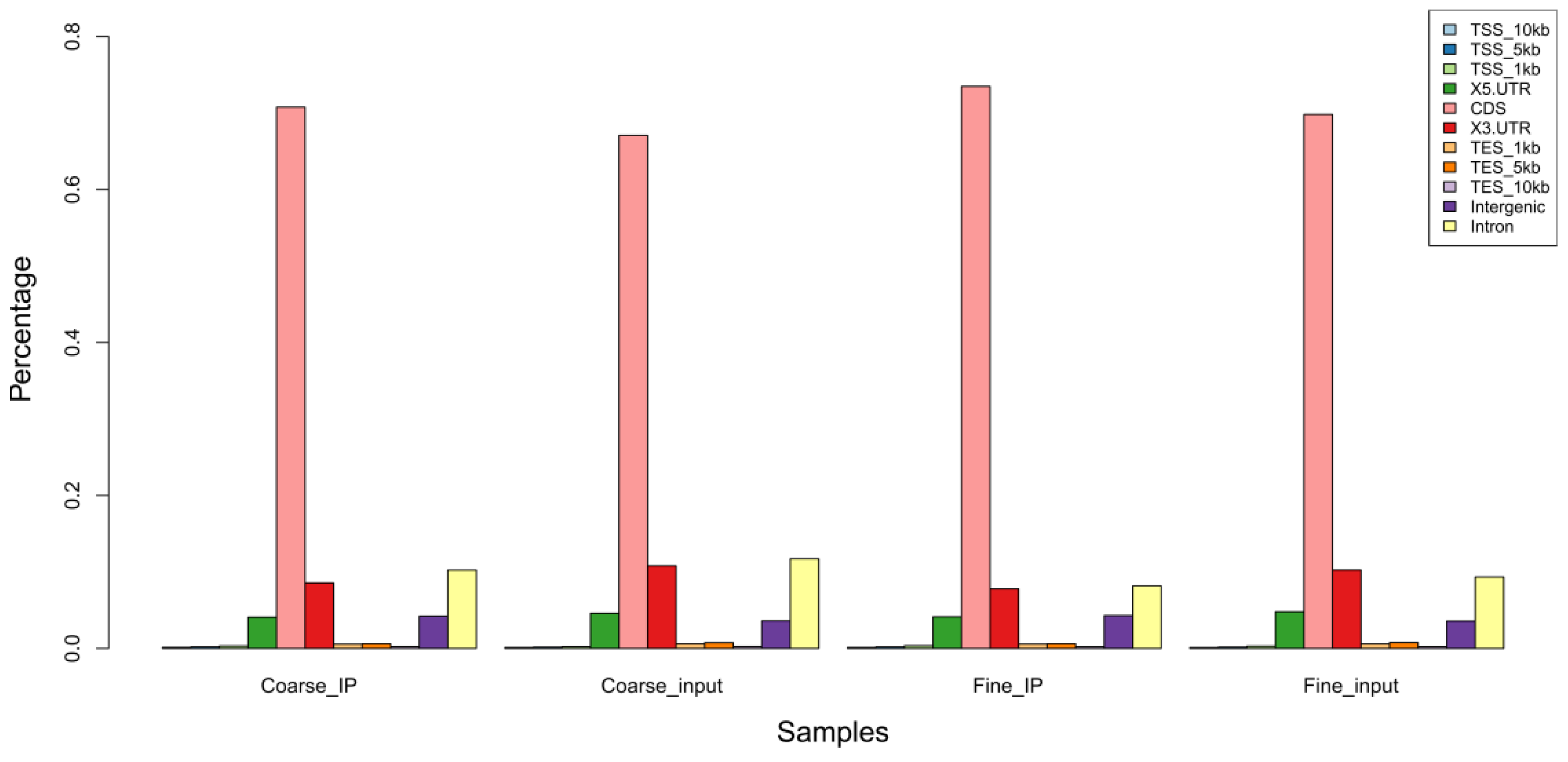
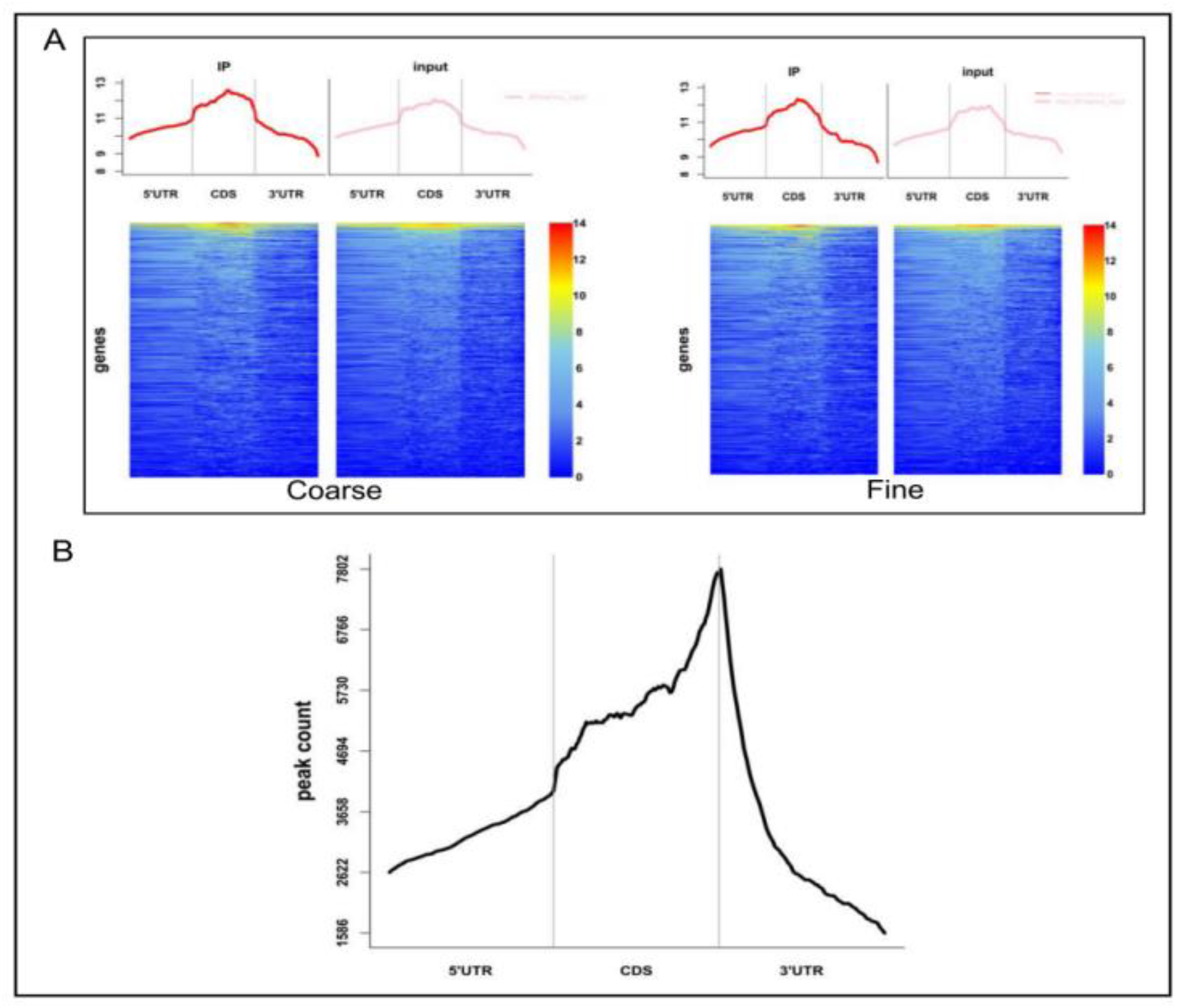
3.3. m6A Peak Calling and Differential Methylation Analysis
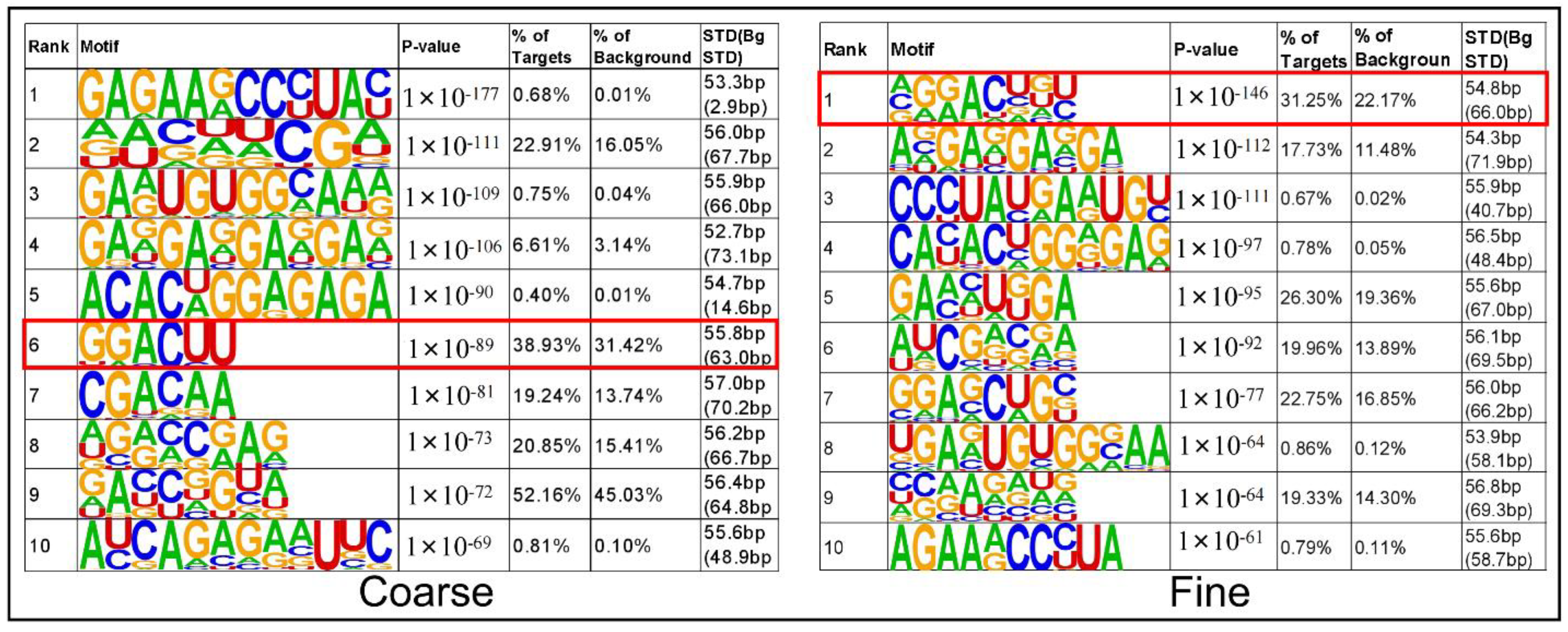
3.4. Motif Analysis
3.5. Association Analysis between m6A Modification and Genes Expression
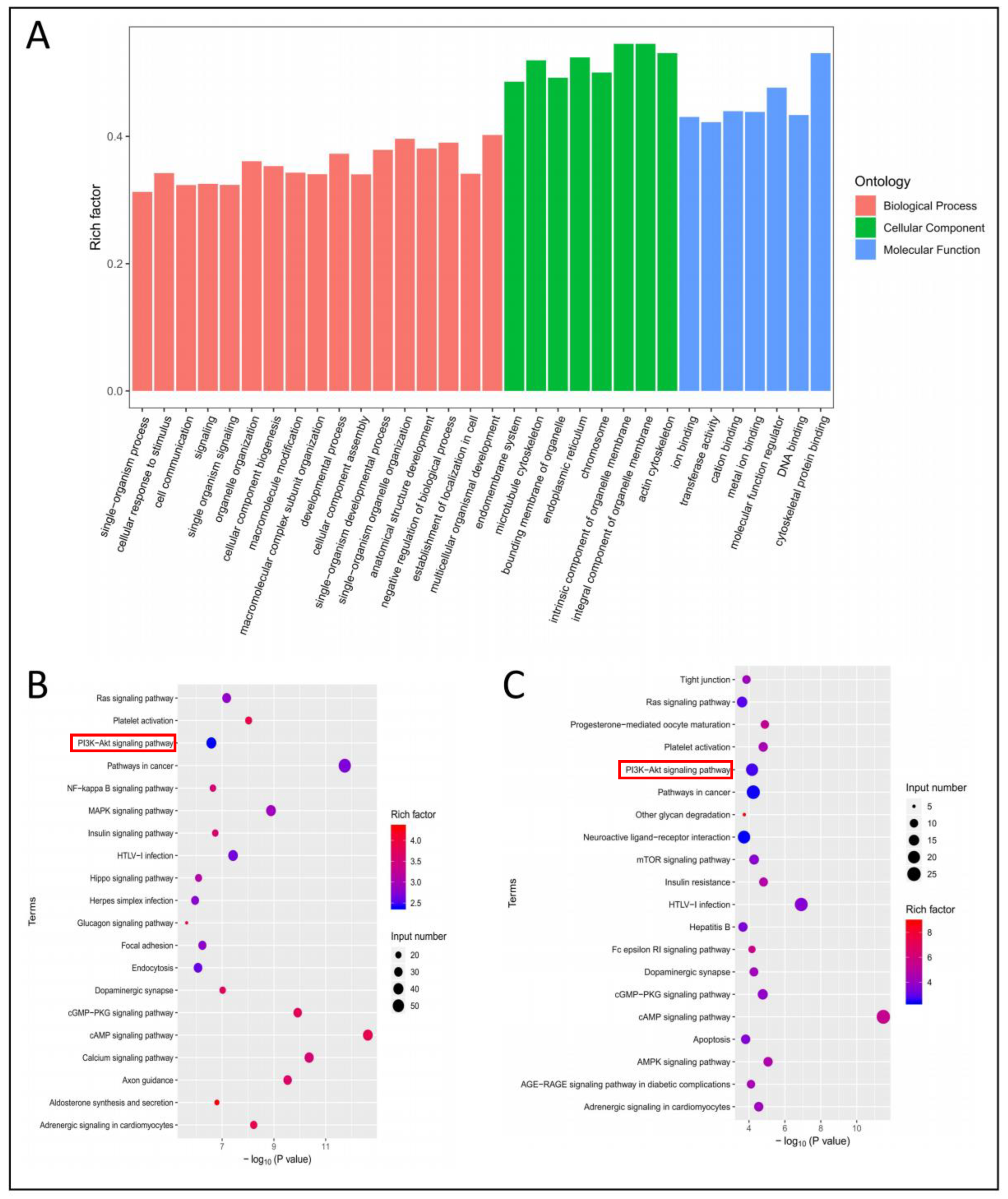
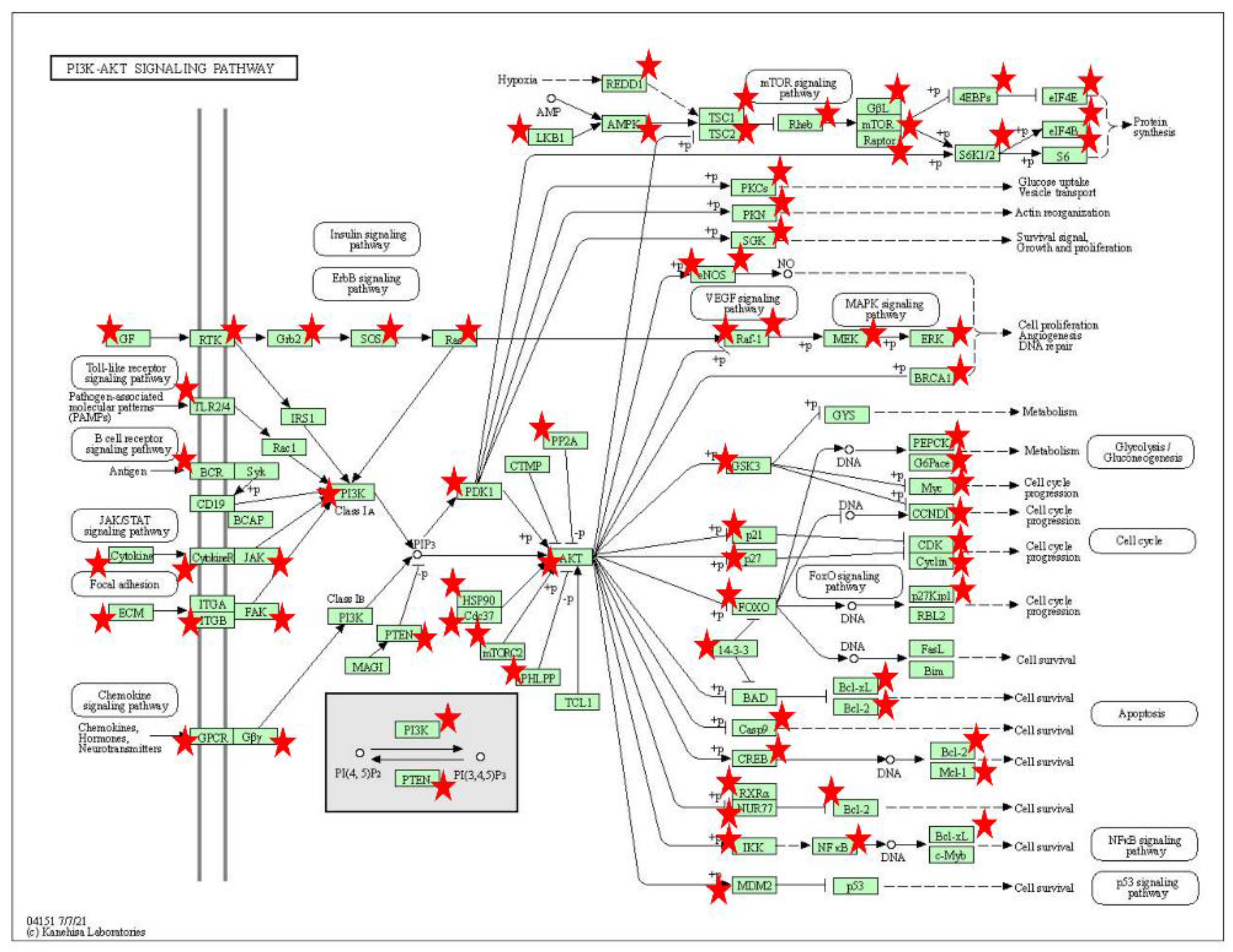
3.6. GO Analysis and KEGG Pathway Analysis of Differentially Methylated Genes
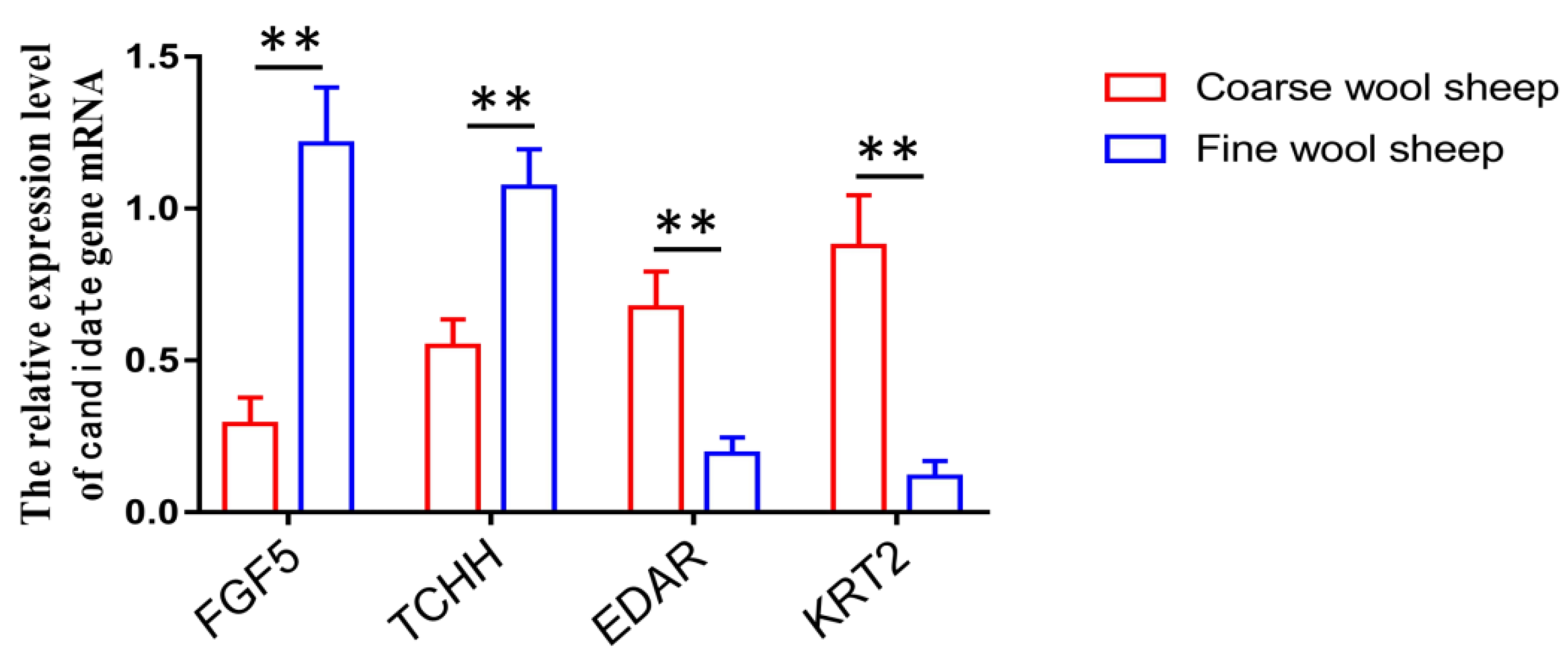
3.7. Validation of Candidate Genes by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.P.; Kelley, D.E.; Friderici, K.; Rottman, F. The methylated constituents of L cell messenger RNA: Evidence for an unusual cluster at the 5′ terminus. Cell 1975, 4, 387–394. [Google Scholar] [CrossRef]
- Cui, X.; Meng, J.; Zhang, S.; Chen, Y.; Huang, Y. A novel algorithm for calling mRNA m6A peaks by modeling biological variances in MeRIP-seq data. Bioinformatics 2016, 32, i378–i385. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.; Li, H.B.; Yin, Z.; Flavell, R.A. Recent advances in dynamic m6A RNA modification. Open Biol. 2016, 6, 160003. [Google Scholar] [CrossRef] [Green Version]
- Scobie, D.R.; Grosvenor, A.J.; Bray, A.R.; Tandon, S.K.; Meade, W.J.; Cooper, A.M.B. A review of wool fibre variation across the body of sheep and the effects on wool processing. Small Ruminant Res. 2015, 133, 43–53. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Yang, J.; Lv, F.H.; Hu, X.J.; Xie, X.L.; Zhang, M.; Li, W.R.; Liu, M.J.; Wang, Y.T.; Li, J.Q.; et al. Genomic Reconstruction of the History of Native Sheep Reveals the Peopling Patterns of Nomads and the Expansion of Early Pastoralism in East Asia. Mol. Biol. Evol. 2017, 34, 2380–2395. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M.; et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef] [Green Version]
- Rong, E.G.; Yang, H.; Zhang, Z.W.; Wang, Z.P.; Yan, X.H.; Li, H.; Wang, N. Association of methionine synthase gene polymorphisms with wool production and quality traits in Chinese Merino population. J. Anim. Sci. 2015, 93, 4601–4609. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Getmantseva, L.; Shirockova, N. Sheep breeding resources in Rostov region, Russia. World Appl. Sci. J. 2013, 23, 1322–1324. [Google Scholar]
- Ponzoni, R.W. A profit equation for the definition of the breeding objective of australian merino sheep. J. Anim. Breed. Genet. Z. Tierz. Zucht. 1986, 103, 342–357. [Google Scholar] [CrossRef]
- Ciani, E.; Lasagna, E.; D’Andrea, M.; Alloggio, I.; Marroni, F.; Ceccobelli, S.; Delgado Bermejo, J.V.; Sarti, F.M.; Kijas, J.; Lenstra, J.A.; et al. Merino and Merino-derived sheep breeds: A genome-wide intercontinental study. Genet. Sel. Evol. 2015, 47, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.; Birtles, M.J.; Harris, P.M.; Parry, A.L.; Paterson, E.; Wickham, G.A.; McCutcheon, S.N. Pre- and post-natal wool follicle development and density in sheep of five genotypes. J. Agr. Sci. 1996, 126, 363–370. [Google Scholar] [CrossRef]
- Burns, M. Merino birthcoat fibre types and their follicular origin. J. Agr. Sci. 1966, 66, 155. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Meng, J.; Lu, Z.; Liu, H.; Zhang, L.; Zhang, S.; Chen, Y.; Rao, M.K.; Huang, Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 2014, 69, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [Green Version]
- Ribo-Perez, D.; Herraiz-Canete, A.; Alfonso-Solar, D.; Vargas-Salgado, C.; Gomez-Navarro, T. Modelling biomass gasifiers in hybrid renewable energy microgrids; a complete procedure for enabling gasifiers simulation in HOMER. Renew Energ. 2021, 174, 501–512. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Peng, J.; Yi, C. The m6A Consensus Motif Provides a Paradigm of Epitranscriptomic Studies. Biochemistry 2021, 60, 3410–3412. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, K.; Quan, W.; Yu, L.; Chen, S.; Cheng, C.; Wu, Q.; Zhao, S.; Zhang, Y.; Zhou, L. The dynamics of FTO binding and demethylation from the m(6)A motifs. RNA Biol. 2019, 16, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Pan, T. Structures of the m(6)A Methyltransferase Complex: Two Subunits with Distinct but Coordinated Roles. Mol Cell 2016, 63, 183–185. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theor. Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Mou, C.; Jackson, B.; Schneider, P.; Overbeek, P.A.; Headon, D.J. Generation of the primary hair follicle pattern. Proc. Natl. Acad. Sci. USA 2006, 103, 9075–9080. [Google Scholar] [CrossRef] [Green Version]
- Westgate, G.E.; Ginger, R.S.; Green, M.R. The biology and genetics of curly hair. Exp. Dermatol. 2017, 26, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Jiang, M.; Chen, L.; Niu, B.; Cai, Y. Prediction of Gene Phenotypes Based on GO and KEGG Pathway Enrichment Scores. Biomed Res. Int. 2013, 2013, 870795. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.E. Biology of the wool follicle: An excursion into a unique tissue interaction system waiting to be re-discovered. Exp. Dermatol. 2006, 15, 931–949. [Google Scholar] [CrossRef]
- Higgins, C.A.; Petukhova, L.; Harel, S.; Ho, Y.Y.; Drill, E.; Shapiro, L.; Wajid, M.; Christiano, A.M. FGF5 is a crucial regulator of hair length in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 10648–10653. [Google Scholar] [CrossRef] [Green Version]
- Drogemuller, C.; Rufenacht, S.; Wichert, B.; Leeb, T. Mutations within the FGF5 gene are associated with hair length in cats. Anim. Genet. 2007, 38, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Li, W.R.; Liu, C.X.; Zhang, X.M.; Chen, L.; Peng, X.R.; He, S.G.; Lin, J.P.; Han, B.; Wang, L.Q.; Huang, J.C.; et al. CRISPR/Cas9-mediated loss of FGF5 function increases wool staple length in sheep. Febs. J. 2017, 284, 2764–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirgis, R.A. Study of variability in some wool traits in a coarse wool breed of sheep. J. Agr. Sci. 1973, 80, 233–238. [Google Scholar] [CrossRef]
- Kang, X.; Liu, G.; Liu, Y.; Xu, Q.; Zhang, M.; Fang, M. Transcriptome profile at different physiological stages reveals potential mode for curly fleece in Chinese tan sheep. PLoS ONE 2013, 8, e71763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Tan, J.; Yang, Y.; Peng, Q.; Zhang, M.; Li, J.; Lu, D.; Liu, Y.; Lou, H.; Feng, Q.; et al. Genome-wide scans reveal variants at EDAR predominantly affecting hair straightness in Han Chinese and Uyghur populations. Hum. Genet. 2016, 135, 1279–1286. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Z.; Wang, X.; Mo, M.; Zeng, S.B.; Xu, R.H.; Wang, X.; Wu, Y. PI3K/Akt signaling pathway is essential for de novo hair follicle regeneration. Stem Cell Res. Ther. 2020, 11, 144. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw_Reads | Clean_Reads | Clean% | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|
| Coarse_IP | 65,836,752 | 60,538,940 | 66.83 | 100 | 98.95 | 54.28 |
| Coarse_input | 59,483,600 | 52,447,544 | 75.24 | 100 | 99.25 | 53.92 |
| Fine_IP | 65,814,440 | 61,402,346 | 65.06 | 100 | 98.9 | 54.76 |
| Fine_input | 66,350,948 | 59,261,076 | 74.39 | 100 | 99.1 | 54.45 |
| Sample | Total Reads | Total Mapped (%) | Unique (%) | Non-Unique (%) |
|---|---|---|---|---|
| Coarse_IP | 52,622,230 | 50,790,053 (96.52) | 47,142,268 (92.82) | 3,647,785 (7.18) |
| Coarse_Input | 45,974,696 | 44,149,378 (96.03) | 40,487,767 (91.71) | 3,661,611 (8.29) |
| Fine_IP | 53,580,892 | 51,594,011 (96.29) | 48,063,658 (93.16) | 3,530,353 (6.84) |
| Fine_Input | 51,915,058 | 49,857,573 (96.04) | 45,846,904 (91.96) | 4,010,669 (8.04) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, G.; Yang, X.; Ma, Y.; Li, T.; Wang, J.; Deng, X. m6A Methylation Analysis Reveals Networks and Key Genes Underlying the Coarse and Fine Wool Traits in a Full-sib Merino Family. Biology 2022, 11, 1637. https://doi.org/10.3390/biology11111637
Hua G, Yang X, Ma Y, Li T, Wang J, Deng X. m6A Methylation Analysis Reveals Networks and Key Genes Underlying the Coarse and Fine Wool Traits in a Full-sib Merino Family. Biology. 2022; 11(11):1637. https://doi.org/10.3390/biology11111637
Chicago/Turabian StyleHua, Guoying, Xue Yang, Yuhao Ma, Tun Li, Jiankui Wang, and Xuemei Deng. 2022. "m6A Methylation Analysis Reveals Networks and Key Genes Underlying the Coarse and Fine Wool Traits in a Full-sib Merino Family" Biology 11, no. 11: 1637. https://doi.org/10.3390/biology11111637
APA StyleHua, G., Yang, X., Ma, Y., Li, T., Wang, J., & Deng, X. (2022). m6A Methylation Analysis Reveals Networks and Key Genes Underlying the Coarse and Fine Wool Traits in a Full-sib Merino Family. Biology, 11(11), 1637. https://doi.org/10.3390/biology11111637