
Lysosomes and Their Role in Regulating the Metabolism of Hematopoietic Stem Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
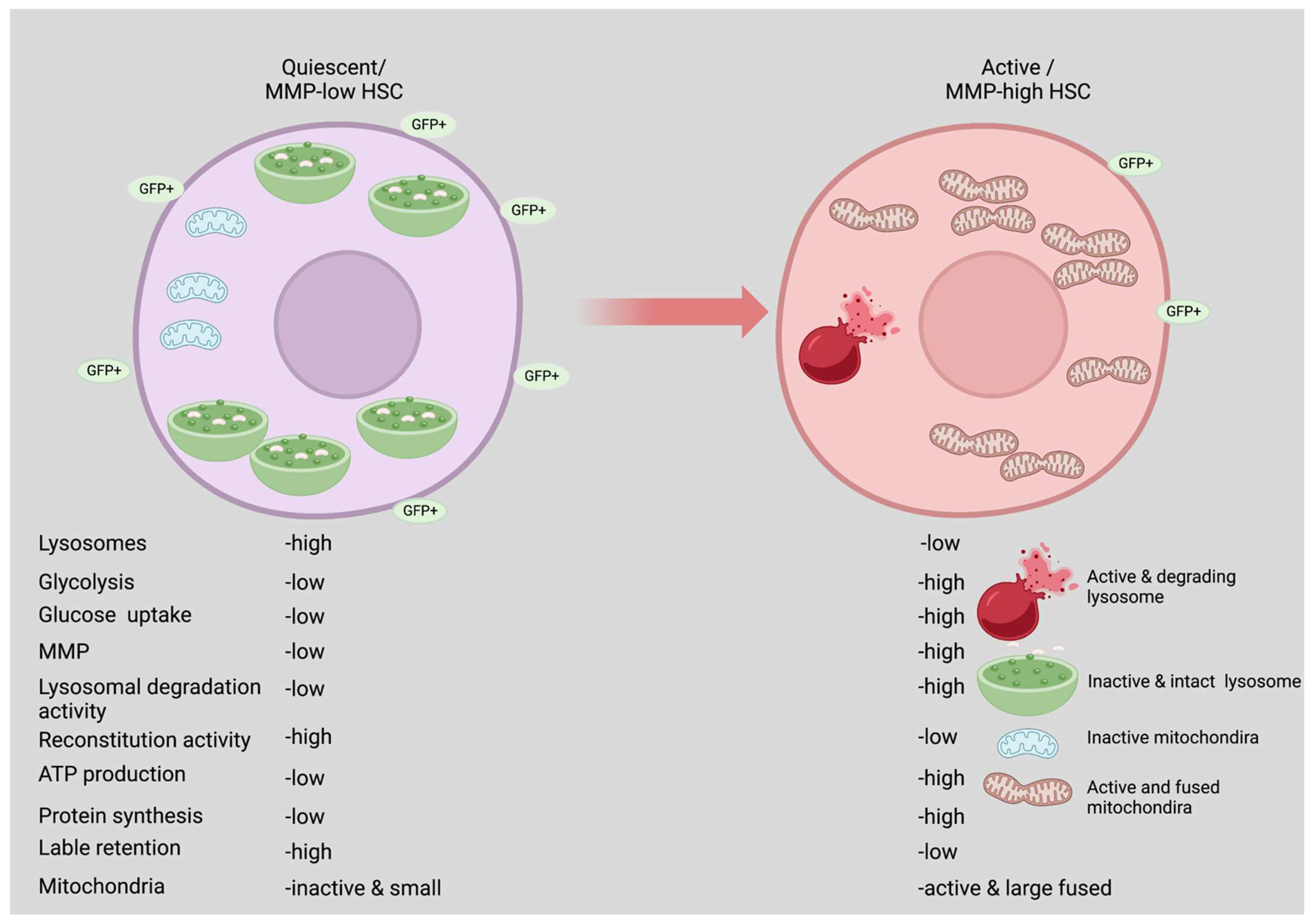
2. Quiescence vs. Active State
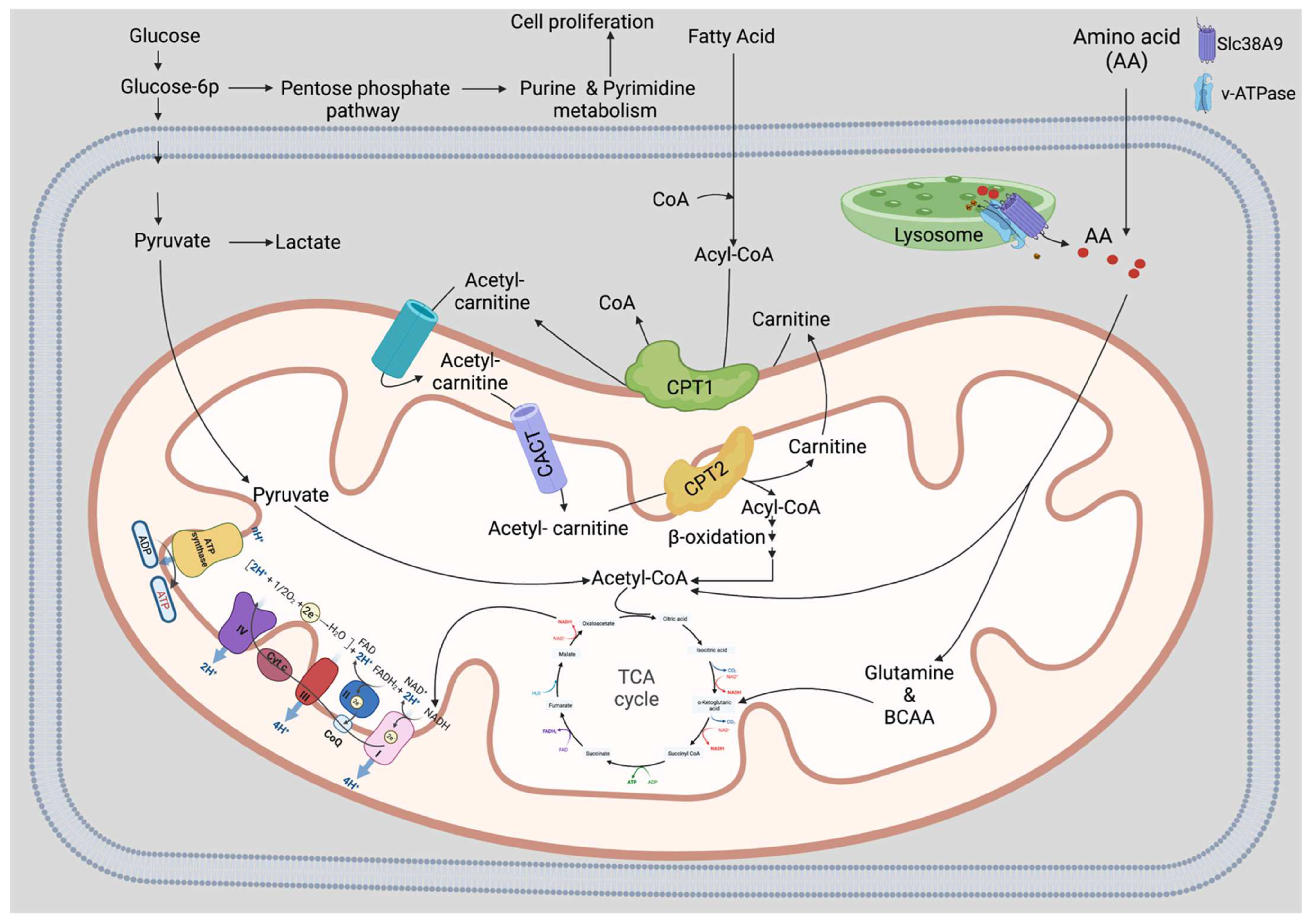
3. Factors Regulating Glycolysis in HSCs
4. Glycolysis Is Required Mainly by Activated HSCs
5. Lysosomal Metabolism
Lysosomal Disorders
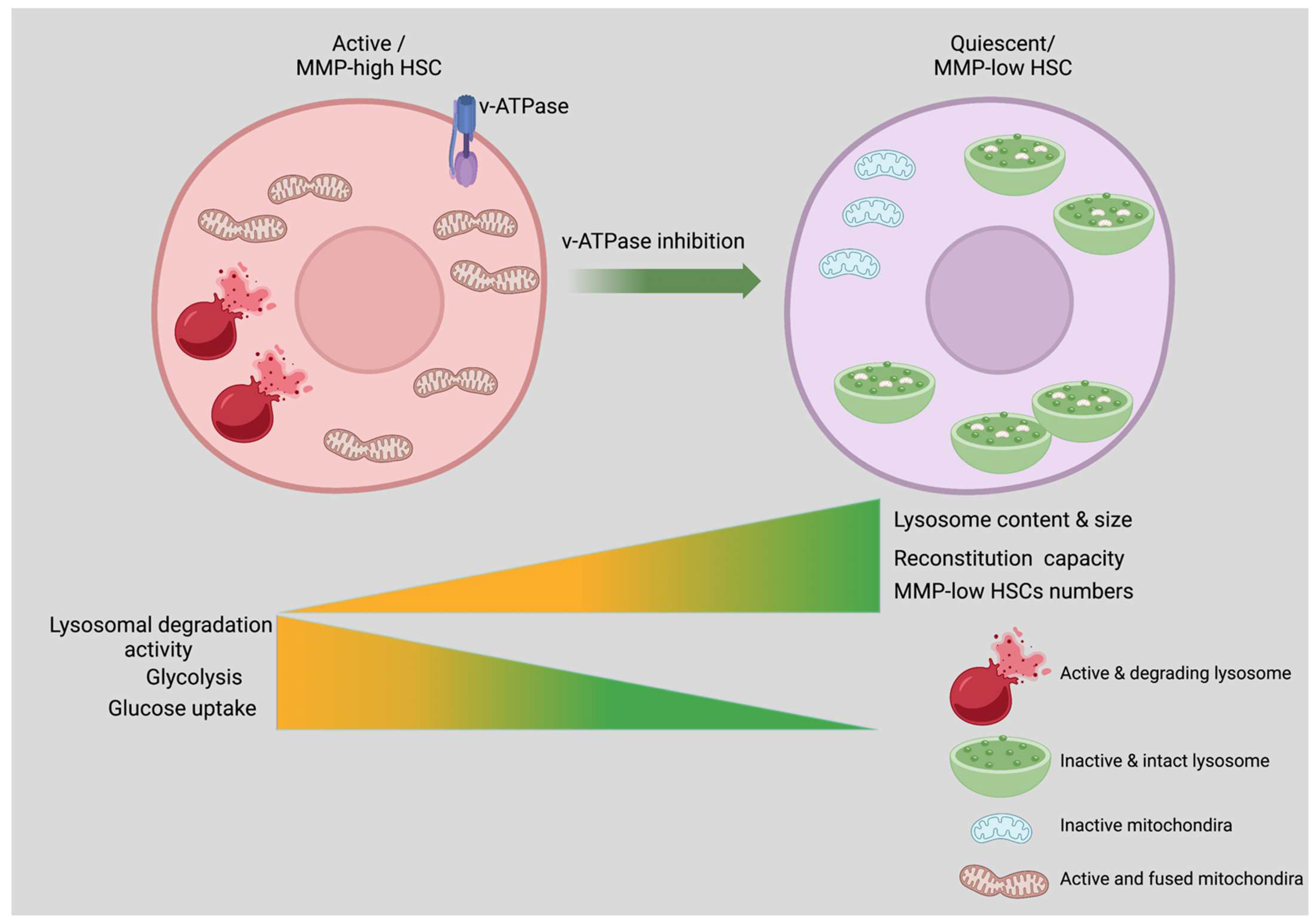
6. Role of Lysosomes in Maintaining HSCs Quiescence
6.1. Glycolysis and Mitochondrial Biogenesis
6.2. How HSCs Switch from Catabolic to Anabolic Activity?
6.3. Role of Anabolic Regulators in HSCs
7. Nutrients in the Maintenance of HSCs
7.1. Amino Acids (AAs)
7.2. Lipids
7.3. Metabolites
7.4. Other Nutrients
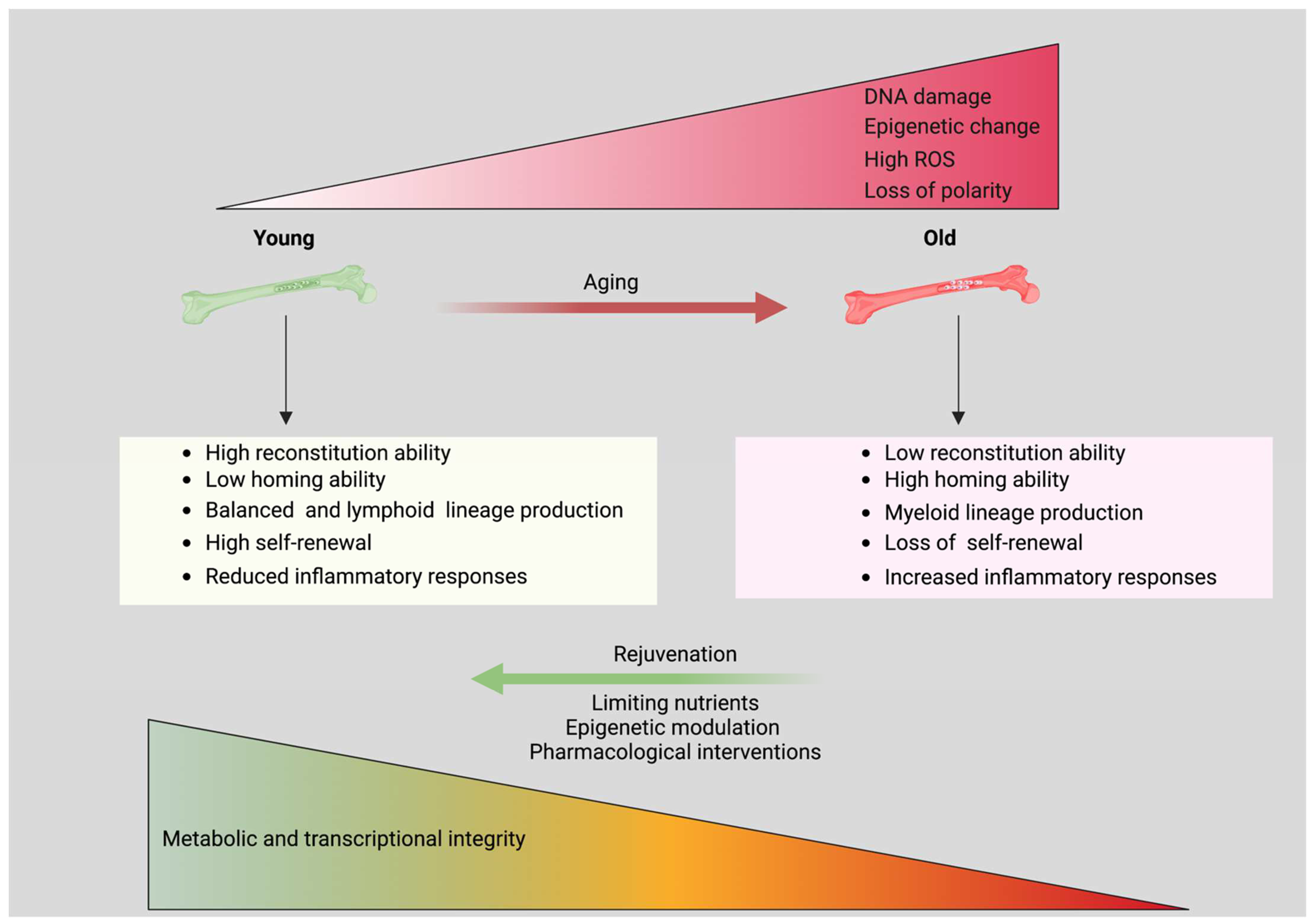
8. Aging HSCs Metabolism
9. HSC Aging
10. Conclusions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mann, Z.; Sengar, M.; Verma, Y.K.; Rajalingam, R.; Raghav, P.K. Hematopoietic Stem Cell Factors: Their Functional Role in Self-Renewal and Clinical Aspects. Front. Cell Dev. Biol. 2022, 10, 664261. [Google Scholar] [CrossRef] [PubMed]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. WIREs Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.T.; Passegué, E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 2013, 41, 915–923. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, M.J.; Luchsinger, L.L.; Corrigan, D.J.; Williams, L.J.; Snoeck, H.-W. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 2017, 21, 725–729.e4. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, K. Hematopoietic stem cell fate through metabolic control. Exp. Hematol. 2018, 64, 1–11. [Google Scholar] [CrossRef]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430.e6. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Hashimoto, M.; Matsumura, T.; Nakamura-Ishizu, A.; Suda, T. Ca2+–mitochondria axis drives cell division in hematopoietic stem cells. J. Exp. Med. 2018, 215, 2097–2113. [Google Scholar] [CrossRef]
- Hsu, P.; Qu, C.-K. Metabolic plasticity and hematopoietic stem cell biology. Curr. Opin. Hematol. 2013, 20, 289–294. [Google Scholar] [CrossRef]
- Baba, M.; Takeshige, K.; Baba, N.; Ohsumi, Y. Ultrastructural analysis of the autophagic process in yeast: Detection of autophagosomes and their characterization. J. Cell Biol. 1994, 124, 903–913. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef]
- De Araujo, M.E.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal size matters. Traffic 2019, 21, 60–75. [Google Scholar] [CrossRef]
- Jain, V.; Bose, S.; Arya, A.K.; Arif, T. Lysosomes in Stem Cell Quiescence: A Potential Therapeutic Target in Acute Myeloid Leukemia. Cancers 2022, 14, 1618. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376.e7. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; D’Azzo, A.; Davidson, B.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Mejia-Ramirez, E.; Florian, M.C. Understanding intrinsic hematopoietic stem cell aging. Haematologica 2019, 105, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Clevers, H. Coexistence of Quiescent and Active Adult Stem Cells in Mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef]
- Mantel, C.; Messina-Graham, S.; Broxmeyer, H.E. Upregulation of nascent mitochondrial biogenesis in mouse hematopoietic stem cells parallels upregulation of CD34 and loss of pluripotency: A potential strategy for reducing oxidative risk in stem cells. Cell Cycle 2010, 9, 2008–2017. [Google Scholar] [CrossRef]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating Mitochondrial DNA Mutations Drive Premature Hematopoietic Aging Phenotypes Distinct from Physiological Stem Cell Aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef]
- Piccoli, C.; Ria, R.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilio, A.; Capitanio, N. Characterization of Mitochondrial and Extra-mitochondrial Oxygen Consuming Reactions in Human Hematopoietic Stem Cells. J. Biol. Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef]
- Inoue, S.-I.; Noda, S.; Kashima, K.; Nakada, K.; Hayashi, J.-I.; Miyoshi, H. Mitochondrial respiration defects modulate differentiation but not proliferation of hematopoietic stem and progenitor cells. FEBS Lett. 2010, 584, 3402–3409. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal Transduction by Mitochondrial Oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.-Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Mauch, P.; Vergilio, J.-A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, L.; Chen, J.; Song, S.; Lee, I.H.; Quijano, C.; Liu, H.; Keyvanfar, K.; Chen, H.; Cao, L.-Y.; et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009, 459, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, F.; Xie, L.; Xie, J.; Yu, Z.; DeBerardinis, R.J.; Kimura, W.; Thet, S.; Elshamy, A.F.; Abouellail, H.; Muralidhar, S.; et al. Hypoxic metabolism in human hematopoietic stem cells. Cell Biosci. 2015, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Messing, E.M.; Wu, G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1α. EMBO Rep. 2005, 6, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lu, Z.; Kocabas, F.; Böttcher, R.T.; Costell, M.; Kang, X.; Liu, X.; DeBerardinis, R.J.; Wang, Q.; Chen, G.-Q.; et al. Profilin 1 is essential for retention and metabolism of mouse hematopoietic stem cells in bone marrow. Blood 2014, 123, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-H.; Kang, J.H.; Hwang, S.-L.; Cho, H.-J.; Park, K.-K.; Park, Y.-Y.; Chung, I.-K.; Chang, H.-W.; Kim, C.-H.; Min, K.-S.; et al. 4-O-methylascochlorin, methylated derivative of ascochlorin, stabilizes HIF-1α via AMPK activation. Biochem. Biophys. Res. Commun. 2011, 406, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, S.; Yamada, Y.; Gonzalez, F.J.; Nakajima, T.; Murohara, T.; Ichihara, G. Inhibition of ischemia-induced angiogenesis by benzo[a]pyrene in a manner dependent on the aryl hydrocarbon receptor. Biochem. Biophys. Res. Commun. 2009, 381, 44–49. [Google Scholar] [CrossRef]
- Zhou, F.; Li, X.; Wang, W.; Zhu, P.; Zhou, J.; He, W.; Ding, M.; Xiong, F.; Zheng, X.; Li, Z.; et al. Tracing haematopoietic stem cell formation at single-cell resolution. Nature 2016, 533, 487–492. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, F.; Zhang, Y.; Li, X.; Chen, C.; Zhou, M.; Yu, Z.; Liu, Y.; Zhao, Y.; Hao, X.; et al. PPM1K Regulates Hematopoiesis and Leukemogenesis through CDC20-Mediated Ubiquitination of MEIS1 and p21. Cell Rep. 2018, 23, 1461–1475. [Google Scholar] [CrossRef]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef]
- Piccoli, C.; D’Aprile, A.; Ripoli, M.; Scrima, R.; Boffoli, D.; Tabilio, A.; Capitanio, N. The hypoxia-inducible factor is stabilized in circulating hematopoietic stem cells under normoxic conditions. FEBS Lett. 2007, 581, 3111–3119. [Google Scholar] [CrossRef]
- Rehn, M.; Olsson, A.; Reckzeh, K.; Diffner, E.; Carmeliet, P.; Landberg, G.; Cammenga, J. Hypoxic induction of vascular endothelial growth factor regulates murine hematopoietic stem cell function in the low-oxygenic niche. Blood 2011, 118, 1534–1543. [Google Scholar] [CrossRef]
- Vukovic, M.; Sepulveda, C.; Subramani, C.; Guitart, A.; Mohr, J.; Allen, L.; Panagopoulou, T.I.; Paris, J.; Lawson, H.; Villacreces, A.; et al. Adult hematopoietic stem cells lacking Hif-1α self-renew normally. Blood 2016, 127, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Kaidi, A.; Williams, A.; Paraskeva, C. Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 2007, 9, 210–217. [Google Scholar] [CrossRef]
- Kim, C.G.; Lee, J.J.; Jung, D.Y.; Jeon, J.; Heo, H.S.; Kang, H.C.; Shin, J.H.; Cho, Y.S.; Cha, K.J.; Gil Kim, C.; et al. Profiling of differentially expressed genes in human stem cells by cDNA microarray. Mol. Cells 2006, 21, 343–355. [Google Scholar]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef]
- Chandel, N.S.; Jasper, H.; Ho, T.T.; Passegue, E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol. 2016, 18, 823–832. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef]
- Balazs, A.B.; Fabian, A.J.; Esmon, C.T.; Mulligan, R.C. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood 2006, 107, 2317–2321. [Google Scholar] [CrossRef]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef]
- Garg, S.; Sharma, M.; Ung, C.; Tuli, A.; Barral, D.C.; Hava, D.L.; Veerapen, N.; Besra, G.S.; Hacohen, N.; Brenner, M.B. Lysosomal Trafficking, Antigen Presentation, and Microbial Killing Are Controlled by the Arf-like GTPase Arl8b. Immunity 2011, 35, 182–193. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma Membrane Repair Is Mediated by Ca2+-Regulated Exocytosis of Lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Tancini, B.; Buratta, S.; Delo, F.; Sagini, K.; Chiaradia, E.; Pellegrino, R.M.; Emiliani, C.; Urbanelli, L. Lysosomal Exocytosis: The Extracellular Role of an Intracellular Organelle. Membranes 2020, 10, 406. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Hesketh, G.G.; Wartosch, L.; Davis, L.J.; Bright, N.A.; Luzio, J.P. The Lysosome and Intracellular Signalling. Molluscs 2018, 57, 151–180. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2018, 218, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Kondo, K.; Itoh, T.; Ikari, S.; Nada, S.; Okada, M.; Noda, T. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 2017, 131, 8017. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Yagi, M.; Toshima, T.; Amamoto, R.; Do, Y.; Hirai, H.; Setoyama, D.; Kang, D.; Uchiumi, T. Mitochondrial translation deficiency impairs NAD + -mediated lysosomal acidification. EMBO J. 2021, 40, e105268. [Google Scholar] [CrossRef]
- Bautista, S.J.; Boras, I.; Vissa, A.; Mecica, N.; Yip, C.M.; Kim, P.K.; Antonescu, C.N. mTOR complex 1 controls the nuclear localization and function of glycogen synthase kinase 3β. J. Biol. Chem. 2018, 293, 14723–14739. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal v-ATPase-Ragulator Complex Is a Common Activator for AMPK and mTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Inpanathan, S.; Botelho, R.J. The Lysosome Signaling Platform: Adapting with the Times. Front. Cell Dev. Biol. 2019, 7, 113. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Li, H.; Puertollano, R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Experientia 2014, 71, 2483–2497. [Google Scholar] [CrossRef]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 Is a Master Transcriptional Repressor of Autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Barbato, D.L.; Tatulli, G.; Aquilano, K.; Ciriolo, M.R. FoxO1 controls lysosomal acid lipase in adipocytes: Implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 2013, 4, e861. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.-I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e9. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; Van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Ther. Nucleic Acids 2019, 17, 24–37. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro-Oncology 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wu, Q.; Zhou, F. Targeting acute myeloid leukemia stem cells: Current therapies in development and potential strategies with new dimensions. Crit. Rev. Oncol. 2020, 152, 102993. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Hu, D.; Shilatifard, A. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 2016, 30, 2021–2041. [Google Scholar] [CrossRef]
- Passegue, E.; Jamieson, C.H.M.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. S1), 11842–11849. [Google Scholar] [CrossRef]
- Ianniciello, A.; Rattigan, K.M.; Helgason, G.V. The Ins and Outs of Autophagy and Metabolism in Hematopoietic and Leukemic Stem Cells: Food for Thought. Front. Cell Dev. Biol. 2018, 6, 120. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2019, 21, 101–118. [Google Scholar] [CrossRef]
- Fujimaki, K.; Li, R.; Chen, H.; Della Croce, K.; Zhang, H.H.; Xing, J.; Bai, F.; Yao, G. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. USA 2019, 116, 22624–22634. [Google Scholar] [CrossRef]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018, 359, 1277–1283. [Google Scholar] [CrossRef]
- García-Prat, L.; Kaufmann, K.B.; Schneiter, F.; Voisin, V.; Murison, A.; Chen, J.; Chan-Seng-Yue, M.; Gan, O.I.; McLeod, J.L.; Smith, S.A.; et al. TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell 2021, 28, 1838–1850.e10. [Google Scholar] [CrossRef] [PubMed]
- Ansó, E.; Weinberg, S.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef]
- Filippi, M.-D.; Ghaffari, S. Mitochondria in the maintenance of hematopoietic stem cells: New perspectives and opportunities. Blood 2019, 133, 1943–1952. [Google Scholar] [CrossRef]
- Jun, S.; Mahesula, S.; Mathews, T.P.; Martin-Sandoval, M.S.; Zhao, Z.; Piskounova, E.; Agathocleous, M. The requirement for pyruvate dehydrogenase in leukemogenesis depends on cell lineage. Cell Metab. 2021, 33, 1777–1792.e8. [Google Scholar] [CrossRef] [PubMed]
- Karigane, D.; Kobayashi, H.; Morikawa, T.; Ootomo, Y.; Sakai, M.; Nagamatsu, G.; Kubota, Y.; Goda, N.; Matsumoto, M.; Nishimura, E.K.; et al. p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 2016, 19, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.; Schneiter, F.; Wang, W.; Wehling, A.; Kull, T.; Lengerke, C.; Manz, M.G.; Schroeder, T. Asymmetric organelle inheritance predicts human blood stem cell fate. Blood 2022, 139, 2011–2023. [Google Scholar] [CrossRef]
- Loeffler, D.; Wehling, A.; Schneiter, F.; Zhang, Y.; Müller-Bötticher, N.; Hoppe, P.S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Endele, M.; Schroeder, T. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 2019, 573, 426–429. [Google Scholar] [CrossRef]
- DeVilbiss, A.W.; Zhao, Z.; Martin-Sandoval, M.S.; Ubellacker, J.M.; Tasdogan, A.; Agathocleous, M.; Mathews, T.P.; Morrison, S.J. Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. eLife 2021, 10, e61980. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.-R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Ghaffari, S. Lysosomal Regulation of Metabolism in Quiescent Hematopoietic Stem Cells: More than Just Autophagy. Cell Stem Cell 2021, 28, 374–377. [Google Scholar] [CrossRef]
- Signer, R.A.; Qi, L.; Zhao, Z.; Thompson, D.; Sigova, A.A.; Fan, Z.P.; DeMartino, G.N.; Young, R.A.; Sonenberg, N.; Morrison, S.J. The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev. 2016, 30, 1698–1703. [Google Scholar] [CrossRef]
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T.; Sugiyama, N.; Soga, T.; Araki, K.; Yamamura, K.-I.; Hirao, A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Investig. 2012, 122, 2114–2129. [Google Scholar] [CrossRef] [PubMed]
- Hoshii, T.; Matsuda, S.; Hirao, A. Pleiotropic roles of mTOR complexes in haemato-lymphopoiesis and leukemogenesis. J. Biochem. 2014, 156, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.-L.; Liu, Y.; Zheng, P. TSC–mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.; Gilliland, D.G.; Morrison, S.J. mTOR Activation Induces Tumor Suppressors that Inhibit Leukemogenesis and Deplete Hematopoietic Stem Cells after Pten Deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzidis, D.; Sykes, S.M.; Wang, Z.; Punt, N.; Tang, Y.; Ragu, C.; Sinha, A.U.; Lane, S.W.; Souza, A.L.; Clish, C.B.; et al. mTOR Complex 1 Plays Critical Roles in Hematopoiesis and Pten-Loss-Evoked Leukemogenesis. Cell Stem Cell 2012, 11, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef]
- Peng, H.; Kasada, A.; Ueno, M.; Hoshii, T.; Tadokoro, Y.; Nomura, N.; Ito, C.; Takase, Y.; Vu, H.T.; Kobayashi, M.; et al. Distinct roles of Rheb and Raptor in activating mTOR complex 1 for the self-renewal of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 2017, 495, 1129–1135. [Google Scholar] [CrossRef]
- Luo, Y.; Li, L.; Zou, P.; Wang, J.; Shao, L.; Zhou, D.; Liu, L. Rapamycin Enhances Long-Term Hematopoietic Reconstitution of Ex Vivo Expanded Mouse Hematopoietic Stem Cells by Inhibiting Senescence. Transplantation 2014, 97, 20–29. [Google Scholar] [CrossRef]
- Rohrabaugh, S.L.; Campbell, T.B.; Hangoc, G.; Broxmeyer, H.E. Ex vivo rapamycin treatment of human cord blood CD34+ cells enhances their engraftment of NSG mice. Blood Cells Mol. Dis. 2011, 46, 318–320. [Google Scholar] [CrossRef]
- Huang, J.; Nguyen-McCarty, M.; Hexner, E.O.; Danet-Desnoyers, G.; Klein, P.S. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat. Med. 2012, 18, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Zheng, P. mTOR Regulation and Therapeutic Rejuvenation of Aging Hematopoietic Stem Cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.E. Amino Acid Balance and Imbalance. J. Nutr. 1959, 68, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Taya, Y.; Ota, Y.; Wilkinson, A.C.; Kanazawa, A.; Watarai, H.; Kasai, M.; Nakauchi, H.; Yamazaki, S. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 2016, 354, 1152–1155. [Google Scholar] [CrossRef]
- Li, Z.; Takubo, K.; Qian, P.; Suda, T.; Li, L. Amino Acid Transporter X Is Required for Hematopoietic Stem Cell Maintenance through Regulating Specific Amino Acids Level. Blood 2015, 126, 1166. [Google Scholar] [CrossRef]
- Buszczak, M.; Signer, R.; Morrison, S.J. Cellular Differences in Protein Synthesis Regulate Tissue Homeostasis. Cell 2014, 159, 242–251. [Google Scholar] [CrossRef]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef]
- Lu, Z.; Xie, J.; Wu, G.; Shen, J.; Collins, R.; Chen, W.; Kang, X.; Luo, M.; Zou, Y.; Huang, L.J.-S.; et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat. Med. 2016, 23, 79–90. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef]
- van Galen, P.; Kreso, A.; Mbong, N.; Kent, D.G.; Fitzmaurice, T.; Chambers, J.E.; Xie, S.; Laurenti, E.; Hermans, K.G.; Eppert, K.; et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 2014, 510, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Morita, M.; Nakauchi, H.; Yamazaki, S. Branched-chain amino acid depletion conditions bone marrow for hematopoietic stem cell transplantation avoiding amino acid imbalance-associated toxicity. Exp. Hematol. 2018, 63, 12–16.e1. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Han, X.; Wang, J.; Wang, C.; Sun, X.; Xie, J.; Wu, G.; Phan, H.; Liu, Z.; Yeh, E.T.; et al. SHP-1 regulates hematopoietic stem cell quiescence by coordinating TGF-β signaling. J. Exp. Med. 2018, 215, 1337–1347. [Google Scholar] [CrossRef]
- Hu, L.; Cheng, H.; Gao, Y.; Shi, M.; Liu, Y.; Hu, Z.; Xu, J.; Qiu, L.; Yuan, W.; Leung, A.Y.-H.; et al. Antioxidant N-acetyl-l-cysteine increases engraftment of human hematopoietic stem cells in immune-deficient mice. Blood 2014, 124, e45–e48. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, B.; Li, Y.; Chen, J.; Ye, Z.; Tian, X.; Wang, J.; Xu, X.; Pan, S.; Zheng, Y.; et al. Amino acid catabolism regulates hematopoietic stem cell proteostasis via a GCN2-eIF2α axis. Cell Stem Cell 2022, 29, 1119–1134.e7. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Baragetti, A.; Bonacina, F.; Catapano, A.L.; Norata, G.D. Effect of Lipids and Lipoproteins on Hematopoietic Cell Metabolism and Commitment in Atherosclerosis. Immunometabolism 2021, 3, 1–20. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Li, X.; Chavakis, T. Immunometabolic control of hematopoiesis. Mol. Asp. Med. 2020, 77, 100923. [Google Scholar] [CrossRef]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.-L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [PubMed]
- Gomaraschi, M.; Bonacina, F.; Norata, G. Lysosomal Acid Lipase: From Cellular Lipid Handler to Immunometabolic Target. Trends Pharmacol. Sci. 2019, 40, 104–115. [Google Scholar] [CrossRef]
- Qu, P.; Shelley, W.C.; Yoder, M.C.; Wu, L.; Du, H.; Yan, C. Critical Roles of Lysosomal Acid Lipase in Myelopoiesis. Am. J. Pathol. 2010, 176, 2394–2404. [Google Scholar] [CrossRef]
- Aryal, B.; Rotllan, N.; Araldi, E.; Ramírez, C.M.; He, S.; Chousterman, B.G.; Fenn, A.M.; Wanschel, A.; Madrigal-Matute, J.; Warrier, N.; et al. ANGPTL4 deficiency in haematopoietic cells promotes monocyte expansion and atherosclerosis progression. Nat. Commun. 2016, 7, 12313. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Denecke, B.; Braunschweig, T.; Stahlschmidt, J.; Ziegler, S.; Brandenburg, L.-O.; Stope, M.B.; Martincuks, A.; Vogt, M.; Görtz, D.; et al. Angptl4 is upregulated under inflammatory conditions in the bone marrow of mice, expands myeloid progenitors, and accelerates reconstitution of platelets after myelosuppressive therapy. J. Hematol. Oncol. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Orsini, M.; Chateauvieux, S.; Rhim, J.; Gaigneaux, A.; Cheillan, D.; Christov, C.; Dicato, M.; Morceau, F.; Diederich, M. Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells. Cell Death Differ. 2018, 26, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Kinder, M.; Wei, C.; Shelat, S.G.; Kundu, M.; Zhao, L.; Blair, I.A.; Puré, E. Hematopoietic stem cell function requires 12/15-lipoxygenase–dependent fatty acid metabolism. Blood 2010, 115, 5012–5022. [Google Scholar] [CrossRef] [PubMed]
- Schönberger, K.; Obier, N.; Romero-Mulero, M.C.; Cauchy, P.; Mess, J.; Pavlovich, P.V.; Zhang, Y.W.; Mitterer, M.; Rettkowski, J.; Lalioti, M.-E.; et al. Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell 2021, 29, 131–148.e10. [Google Scholar] [CrossRef]
- Pernes, G.; Flynn, M.C.; Lancaster, G.I.; Murphy, A.J. Fat for fuel: Lipid metabolism in haematopoiesis. Clin. Transl. Immunol. 2019, 8, e1098. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Guo, B.; Huang, X.; Lee, M.R.; Lee, S.A.; Broxmeyer, H.E. Antagonism of PPAR-γ signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat. Med. 2018, 24, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.J.; Hellmich, C.; Moore, J.A.; Jibril, A.; Macaulay, I.; Moreno-Gonzalez, M.; Di Palma, F.; Beraza, N.; Bowles, K.M.; Rushworth, S.A. Free fatty-acid transport via CD36 drives β-oxidation-mediated hematopoietic stem cell response to infection. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e19. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Martin-Sandoval, M.S.; Merchant, S.; Gu, W.; Eckhardt, M.; Mathews, T.P.; Zhao, Z.; Agathocleous, M.; Morrison, S.J. Aspartate availability limits hematopoietic stem cell function during hematopoietic regeneration. Cell Stem Cell 2021, 28, 1982–1999.e8. [Google Scholar] [CrossRef]
- Alkan, H.F.; Bogner-Strauss, J.G. Maintaining cytosolic aspartate levels is a major function of the TCA cycle in proliferating cells. Mol. Cell. Oncol. 2019, 6, e1536843. [Google Scholar] [CrossRef]
- Cortes, M.; Chen, M.J.; Stachura, D.L.; Liu, S.Y.; Kwan, W.; Wright, F.; Vo, L.T.; Theodore, L.N.; Esain, V.; Frost, I.M.; et al. Developmental Vitamin D Availability Impacts Hematopoietic Stem Cell Production. Cell Rep. 2016, 17, 458–468. [Google Scholar] [CrossRef]
- Paubelle, E.; Zylbersztejn, F.; Maciel, T.T.; Carvalho, C.; Mupo, A.; Cheok, M.; Lieben, L.; Sujobert, P.; Decroocq, J.; Yokoyama, A.; et al. Vitamin D Receptor Controls Cell Stemness in Acute Myeloid Leukemia and in Normal Bone Marrow. Cell Rep. 2020, 30, 739–754.e4. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Girotra, M.; Naveiras, O.; Vannini, N. Targeting mitochondria to stimulate hematopoiesis. Aging 2020, 12, 1042–1043. [Google Scholar] [CrossRef]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 2019, 24, 405–418.e7. [Google Scholar] [CrossRef]
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.-J.; Klein, A.M.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal Analysis Unveils Self-Renewing Lineage-Restricted Progenitors Generated Directly from Hematopoietic Stem Cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef]
- Singh, R.P.; Grinenko, T.; Ramasz, B.; Franke, K.; Lesche, M.; Dahl, A.; Gassmann, M.; Chavakis, T.; Henry, I.; Wielockx, B. Hematopoietic Stem Cells but Not Multipotent Progenitors Drive Erythropoiesis during Chronic Erythroid Stress in EPO Transgenic Mice. Stem Cell Rep. 2018, 10, 1908–1919. [Google Scholar] [CrossRef]
- Singh, S.K.; Singh, S.; Gadomski, S.; Sun, L.; Pfannenstein, A.; Magidson, V.; Chen, X.; Kozlov, S.; Tessarollo, L.; Klarmann, K.D.; et al. Id1 Ablation Protects Hematopoietic Stem Cells from Stress-Induced Exhaustion and Aging. Cell Stem Cell 2018, 23, 252–265.e8. [Google Scholar] [CrossRef]
- Nakamura-Ishizu, A.; Matsumura, T.; Stumpf, P.; Umemoto, T.; Takizawa, H.; Takihara, Y.; O’Neil, A.; Majeed, A.B.B.A.; MacArthur, B.D.; Suda, T. Thrombopoietin Metabolically Primes Hematopoietic Stem Cells to Megakaryocyte-Lineage Differentiation. Cell Rep. 2018, 25, 1772–1785.e6. [Google Scholar] [CrossRef]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and Cancer Risk: A Potentially Modifiable Relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- De Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-H.; del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; García-García, A.; Macías, D.; González-Gómez, C.; del Monte, A.; Wittner, M.; et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 2019, 25, 407–418.e6. [Google Scholar] [CrossRef]
- Ho, Y.-H.; Méndez-Ferrer, S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica 2019, 105, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 Reverses Aging-Associated Degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged Fasting Reduces IGF-1/PKA to Promote Hematopoietic-Stem-Cell-Based Regeneration and Reverse Immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.-D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 Activity Regulates Hematopoietic Stem Cell Aging and Rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef]
- Satoh, Y.; Yokota, T.; Sudo, T.; Kondo, M.; Lai, A.; Kincade, P.W.; Kouro, T.; Iida, R.; Kokame, K.; Miyata, T.; et al. The Satb1 Protein Directs Hematopoietic Stem Cell Differentiation toward Lymphoid Lineages. Immunity 2013, 38, 1105–1115. [Google Scholar] [CrossRef]
- Wang, J.; Morita, Y.; Han, B.; Niemann, S.; Löffler, B.; Rudolph, K.L. Per2 induction limits lymphoid-biased haematopoietic stem cells and lymphopoiesis in the context of DNA damage and ageing. Nature 2016, 18, 480–490. [Google Scholar] [CrossRef]
- Geiger, H.; Zheng, Y. Cdc42 and aging of hematopoietic stem cells. Curr. Opin. Hematol. 2013, 20, 295–300. [Google Scholar] [CrossRef]
- Mansell, E.; Sigurdsson, V.; Deltcheva, E.; Brown, J.; James, C.; Miharada, K.; Soneji, S.; Larsson, J.; Enver, T. Mitochondrial Potentiation Ameliorates Age-Related Heterogeneity in Hematopoietic Stem Cell Function. Cell Stem Cell 2020, 28, 241–256.e6. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.; Domann, F.; Van Houten, B.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.; et al. Mitochondria, Energetics, Epigenetics, and Cellular Responses to Stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Sperling, A.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Cancer 2016, 17, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef]
- Hennrich, M.L.; Romanov, N.; Horn, P.; Jaeger, S.; Eckstein, V.; Steeples, V.; Ye, F.; Ding, X.; Poisa-Beiro, L.; Lai, M.C.; et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat. Commun. 2018, 9, 4004. [Google Scholar] [CrossRef]
- Poisa-Beiro, L.; Landry, J.J.M.; Raffel, S.; Tanaka, M.; Zaugg, J.; Gavin, A.-C.; Ho, A.D. Glucose Metabolism and Aging of Hematopoietic Stem and Progenitor Cells. Int. J. Mol. Sci. 2022, 23, 3028. [Google Scholar] [CrossRef]
- Suo, M.; Rommelfanger, M.K.; Chen, Y.; Amro, E.M.; Han, B.; Chen, Z.; Szafranski, K.; Chakkarappan, S.R.; Boehm, B.O.; MacLean, A.L.; et al. Age-dependent effects of Igf2bp2 on gene regulation, function, and aging of hematopoietic stem cells in mice. Blood 2022, 139, 2653–2665. [Google Scholar] [CrossRef]
- Hinge, A.; Xu, J.; Javier, J.; Mose, E.; Kumar, S.; Kapur, R.; Srour, E.F.; Malik, P.; Aronow, B.J.; Filippi, M.-D. p190-B RhoGAP and intracellular cytokine signals balance hematopoietic stem and progenitor cell self-renewal and differentiation. Nat. Commun. 2017, 8, 14382. [Google Scholar] [CrossRef] [PubMed][Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arif, T. Lysosomes and Their Role in Regulating the Metabolism of Hematopoietic Stem Cells. Biology 2022, 11, 1410. https://doi.org/10.3390/biology11101410
Arif T. Lysosomes and Their Role in Regulating the Metabolism of Hematopoietic Stem Cells. Biology. 2022; 11(10):1410. https://doi.org/10.3390/biology11101410
Chicago/Turabian StyleArif, Tasleem. 2022. "Lysosomes and Their Role in Regulating the Metabolism of Hematopoietic Stem Cells" Biology 11, no. 10: 1410. https://doi.org/10.3390/biology11101410
APA StyleArif, T. (2022). Lysosomes and Their Role in Regulating the Metabolism of Hematopoietic Stem Cells. Biology, 11(10), 1410. https://doi.org/10.3390/biology11101410