
Fecal Microbiota Dynamics Reveal the Feasibility of Early Weaning of Yak Calves under Conventional Grazing System

 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
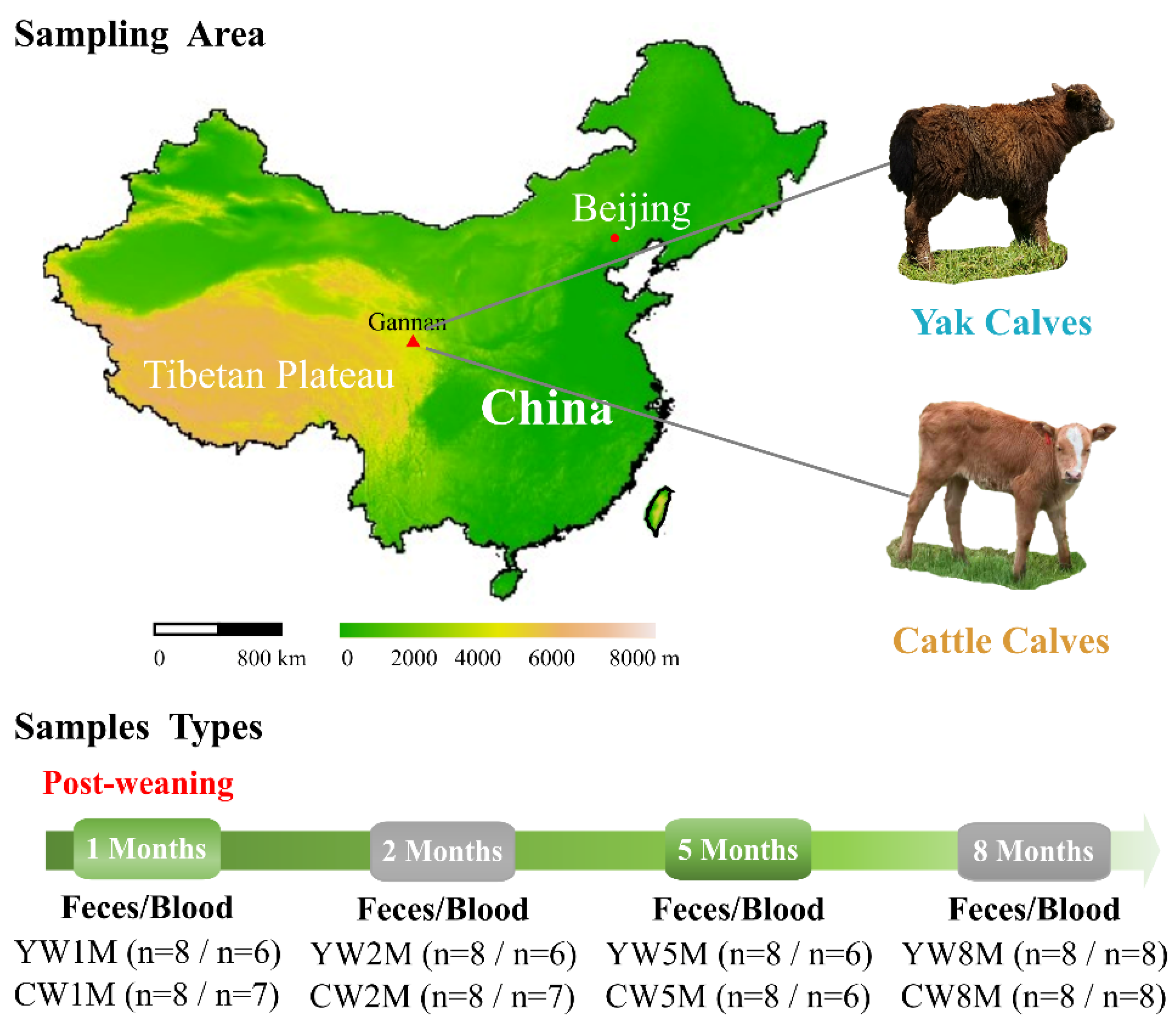
2.1. Animal and Sampling
2.2. Detection of Blood Biochemical Indexes
2.3. DNA Extraction and Illumina Sequencing of 16S rRNA Genes
2.4. Bioinformatics and Statistical Analysis
3. Results
3.1. Comparison of Blood Biochemical Indexes in Post-Weaning Calve
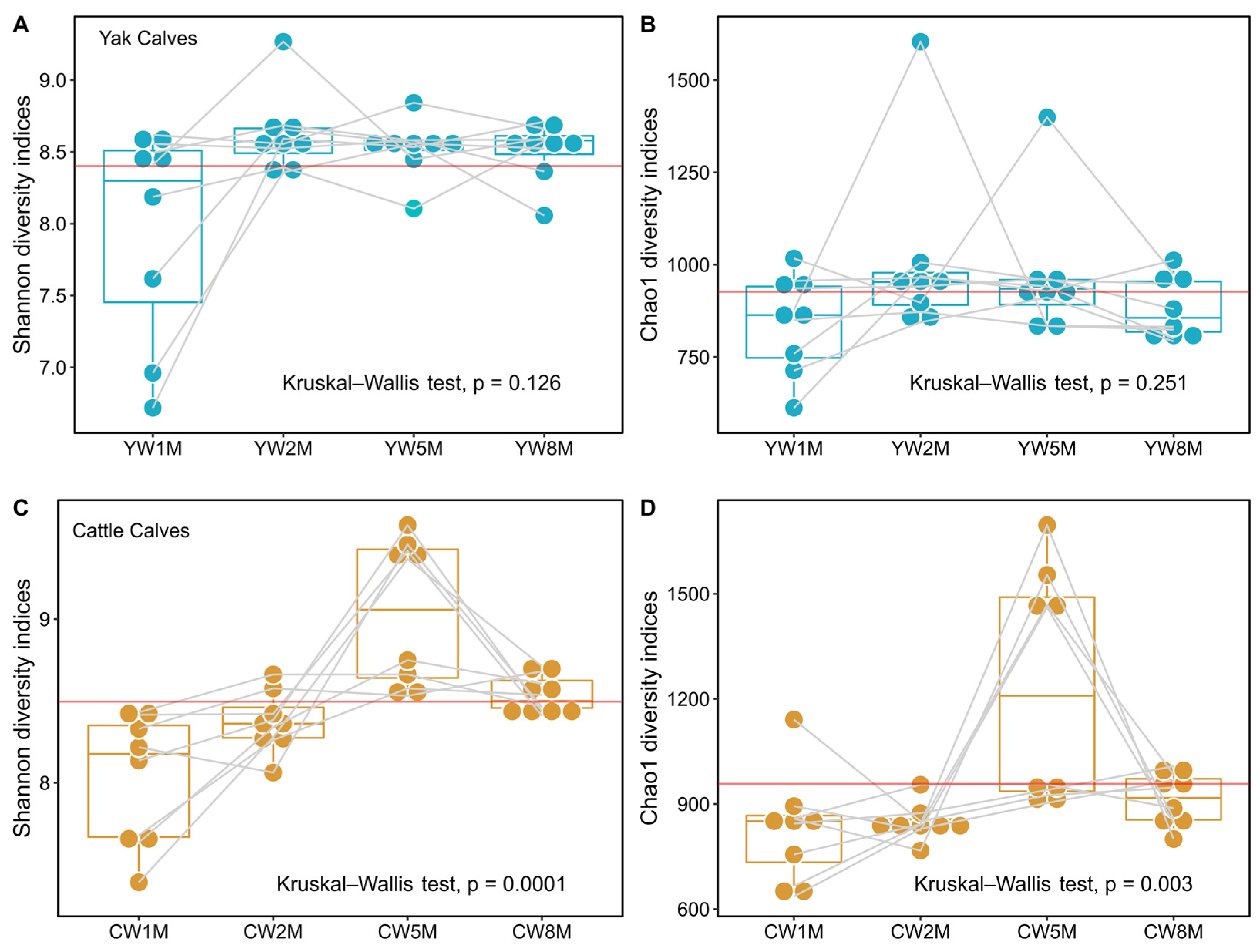
3.2. Diversity of Fecal Bacterial Microbiota in Post-Weaning Calve
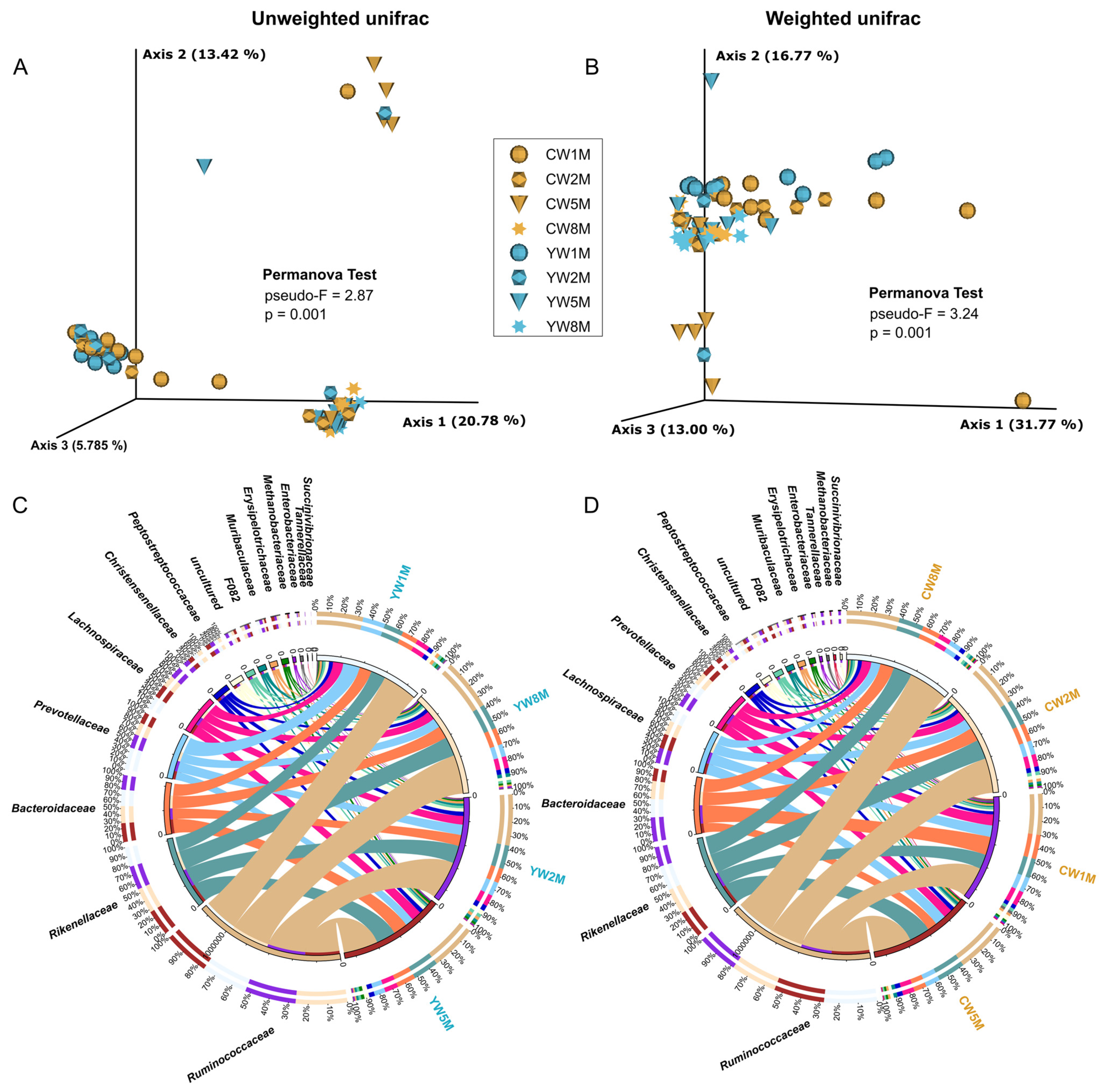
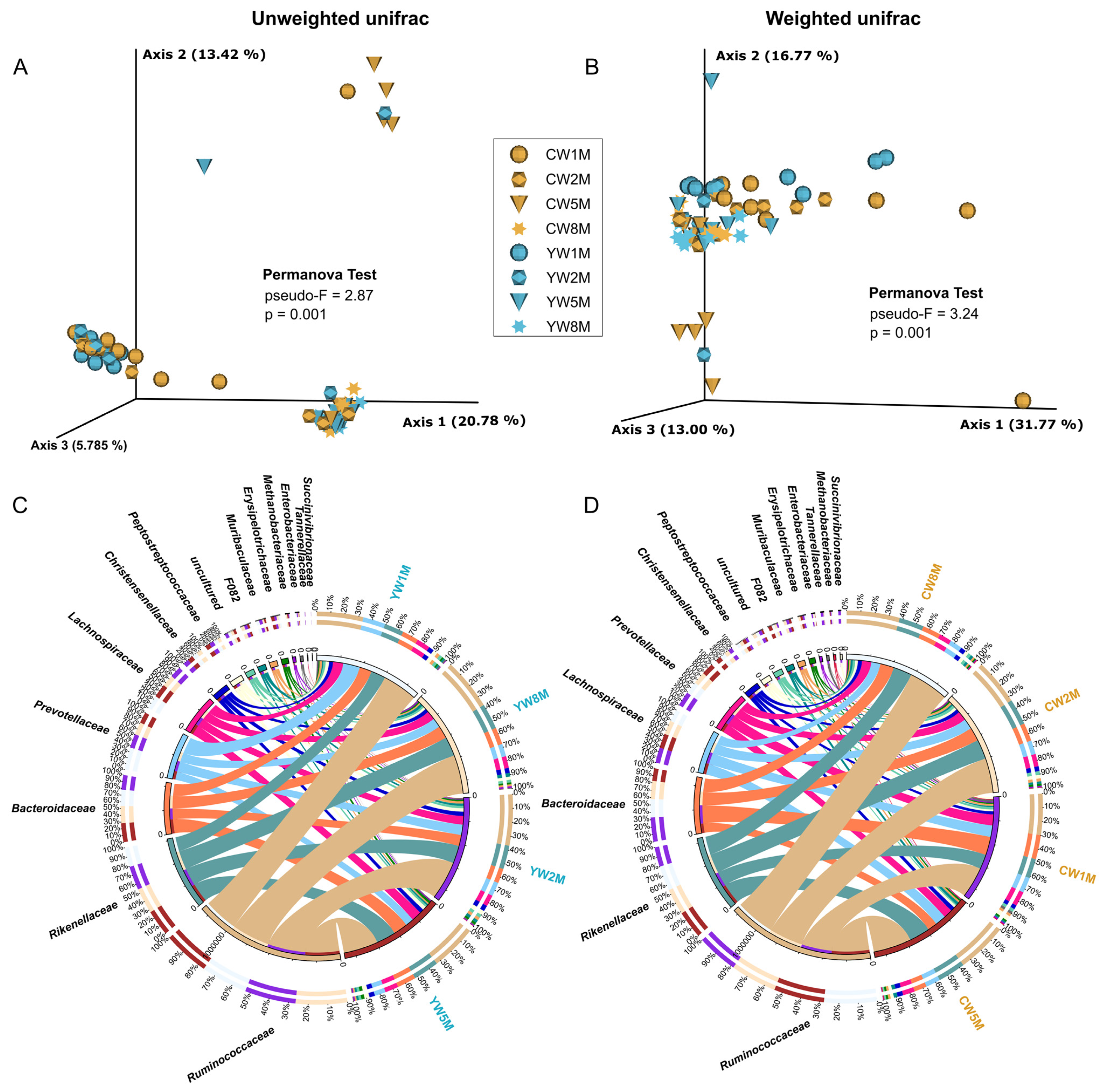
3.3. Comparison and Structure of Fecal Bacterial Communities of Weaned Calves
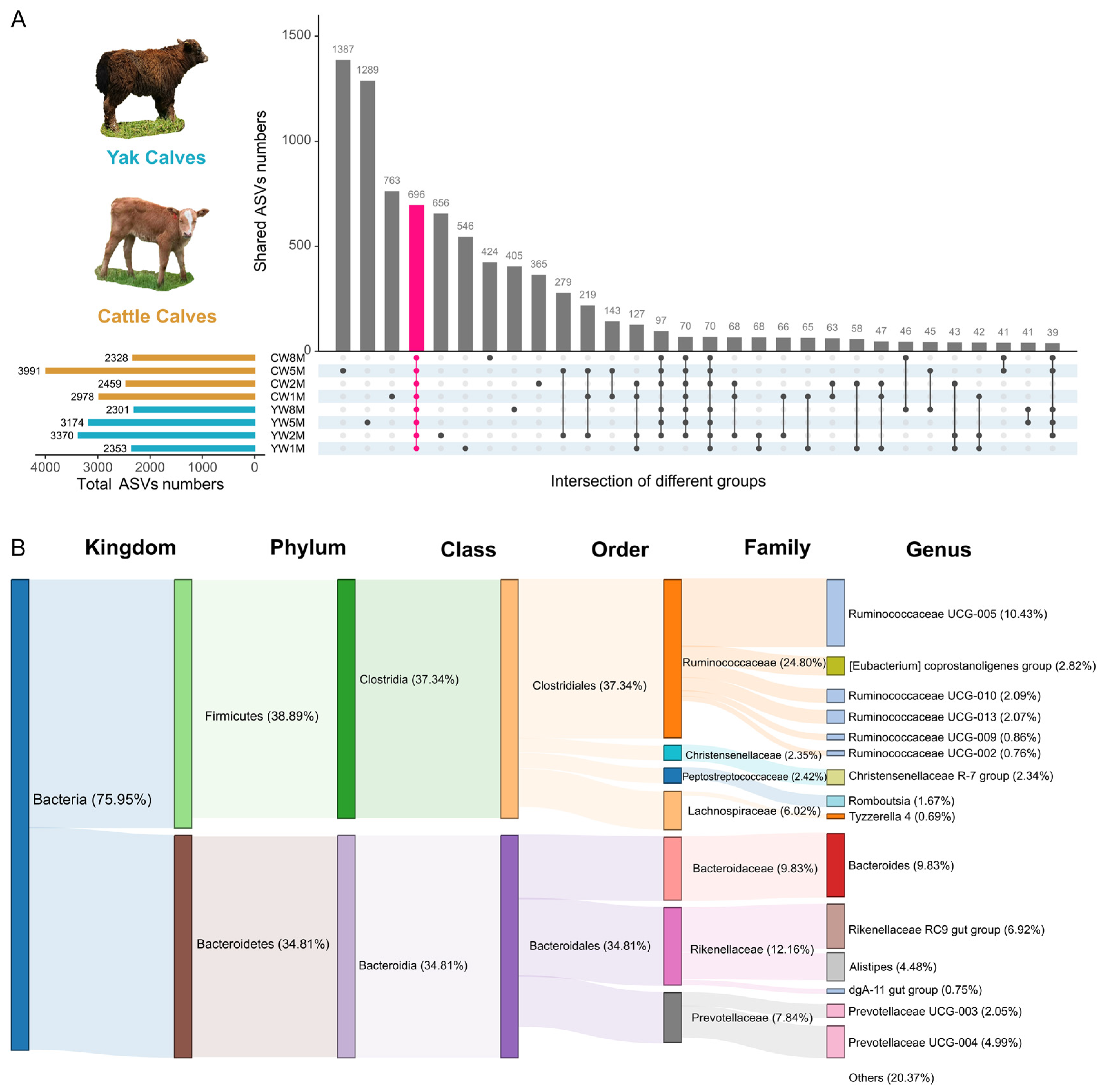
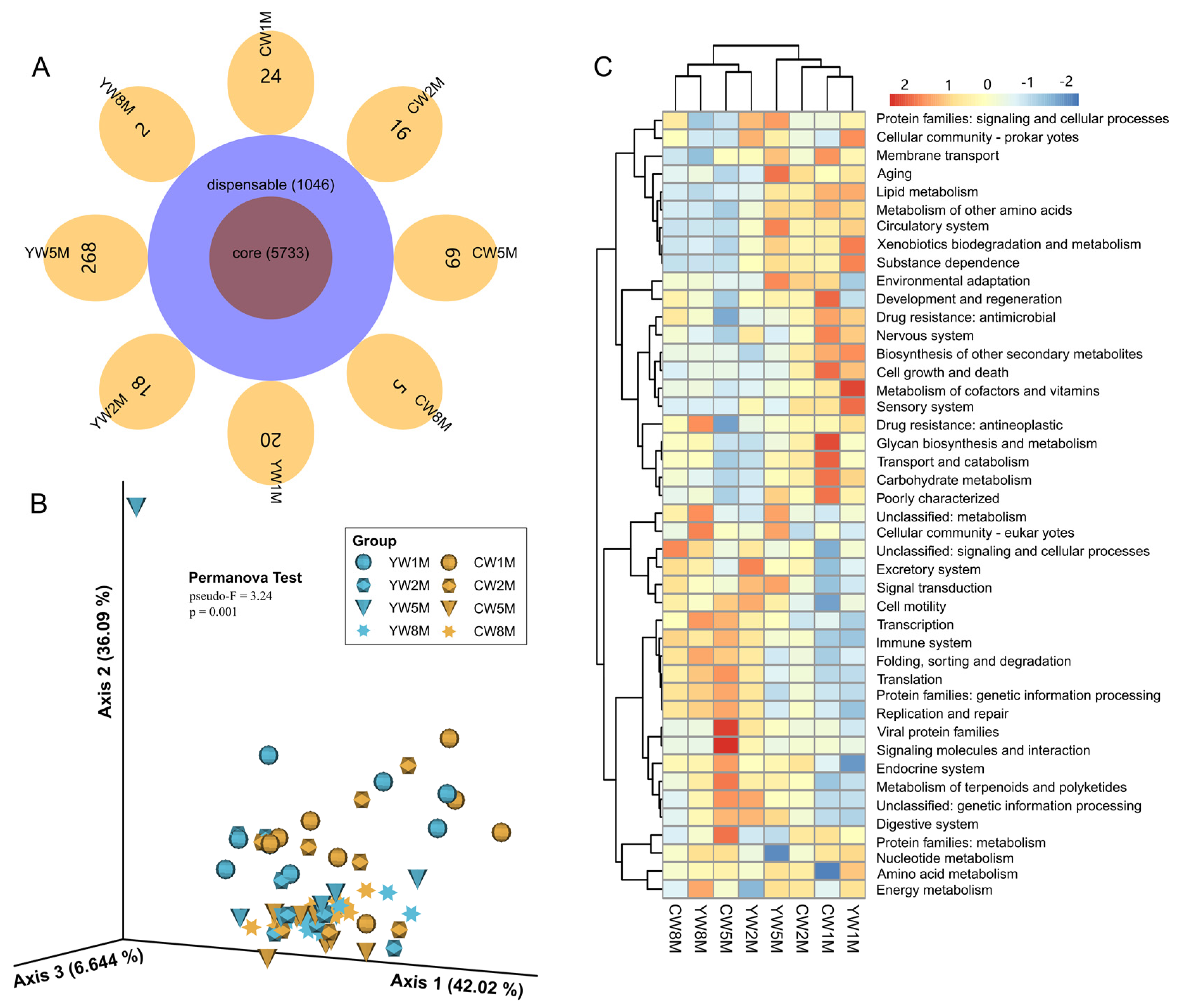
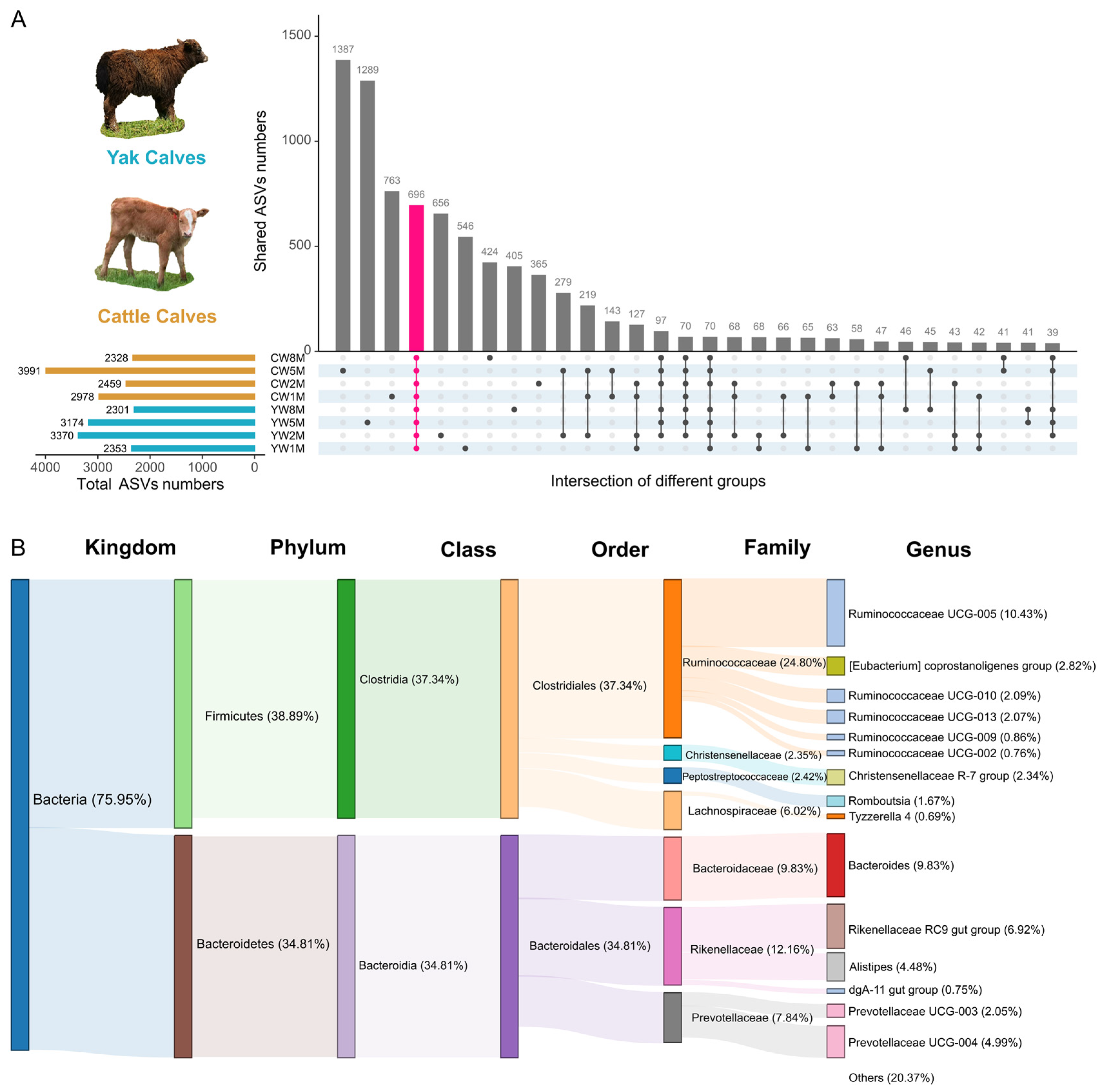
3.4. Core Fecal Microbiota in Weaned Yak and Cattle Calves
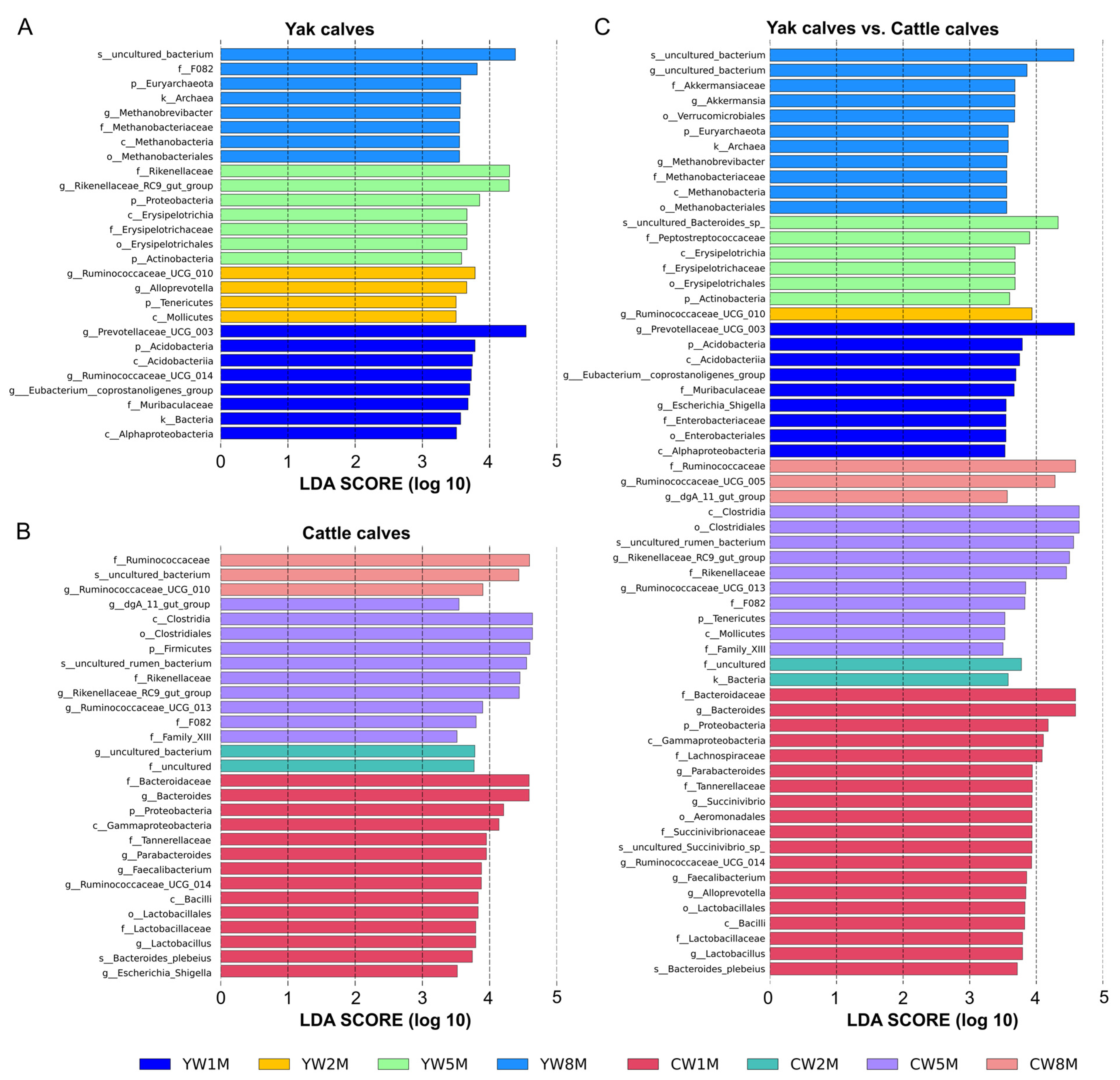
3.5. Differential Taxa between Weaned Yak and Cattle Calves
3.6. Potential Function of the Microbial Community in Weaned Calves
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, R.J.; Ding, L.M.; Shang, Z.H.; Guo, X.H. The yak grazing system on the Qinghai-Tibetan plateau and its status. Rangeland J. 2008, 30, 241–246. [Google Scholar] [CrossRef]
- Gu, Z.; Zhao, X.; Li, N.; Wu, C. Complete sequence of the yak (Bos grunniens) mitochondrial genome and its evolutionary relationship with other ruminants. Mol. Phylogenet. Evol. 2007, 42, 248–255. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, D.; Wang, L.; Hao, J.; Wang, J.; Zhou, X.; Wang, W.; Qiu, Q.; Huang, X.; Zhou, J.; et al. Convergent Evolution of Rumen Microbiomes in High-Altitude Mammals. Curr. Biol. 2016, 26, 1873–1879. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Long, R.; Shang, Z.; Wang, C.; Yang, Y.; Xu, S. Feeding behaviour of yaks on spring, transitional, summer and winter pasture in the alpine region of the Qinghai–Tibetan plateau. Appl. Anim. Behav. Sci. 2008, 111, 373–390. [Google Scholar]
- Shao, B.; Long, R.; Ding, Y.; Wang, J.; Ding, L.; Wang, H. Morphological adaptations of yak (Bos grunniens) tongue to the foraging environment of the Qinghai-Tibetan Plateau. J. Anim. Sci. 2010, 88, 2594–2603. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Long, K.; Ma, J.; Zhang, J.; He, D.; Jin, L.; Tang, Q.; Jiang, A.; Wang, X.; Hu, Y.; et al. Comparative analysis of the microRNA transcriptome between yak and cattle provides insight into high-altitude adaptation. PeerJ 2017, 5, e3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.W.; Liu, H.; Zhong, C.L.; Degen, A.A.; Yang, G.; Zhang, Y.; Qian, J.L.; Wang, W.W.; Hao, L.Z.; Qiu, Q.; et al. Apparent digestibility, rumen fermentation, digestive enzymes and urinary purine derivatives in yaks and Qaidam cattle offered forage-concentrate diets differing in nitrogen concentration. Livest. Sci. 2018, 208, 14–21. [Google Scholar] [CrossRef]
- Ding, X.Z.; Long, R.J.; Kreuzer, M.; Mi, J.D.; Yang, B. Methane emissions from yak (Bos grunniens) steers grazing or kept indoors and fed diets with varying forage concentrate ratio during the cold season on the Qinghai-Tibetan Plateau. Anim. Feed Sci. Technol. 2010, 162, 91–98. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Yang, C.; Zhang, J.B.; Kalwar, Q.; Liang, Z.Y.; Li, C.; Du, M.; Yan, P.; Long, R.J.; Han, J.L.; et al. Effects of dietary energy levels on rumen fermentation, microbial diversity, and feed efficiency of yaks (Bos grunniens). Front. Microbiol. 2020, 11, 625. [Google Scholar] [CrossRef]
- Yang, C.; Ahmad, A.A.; Bao, P.J.; Guo, X.; Wu, X.Y.; Liu, J.B.; Chu, M.; Liang, C.N.; Pei, J.; Long, R.J.; et al. Increasing dietary energy level improves growth performance and lipid metabolism through up-regulating lipogenic gene expression in yak (Bos grunniens). Anim. Feed Sci. Technol. 2020, 263, 114455. [Google Scholar] [CrossRef]
- An, D.; Dong, X.; Dong, Z. Prokaryote diversity in the rumen of yak (Bos grunniens) and Jinnan cattle (Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe 2005, 11, 207–215. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, M.; Ma, T.; Bi, S.; Wang, W.; Zhang, Y.; Huang, X.; Guan, L.; Long, R. Survey of rumen microbiota of domestic grazing yak during different growth stages revealed novel maturation patterns of four key microbial groups and their dynamic interactions. Anim. Microbiome 2020, 2, 23. [Google Scholar] [CrossRef]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Yeoman, C.J.; White, B.A. Gastrointestinal tract microbiota and probiotics in production animals. Annu. Rev. Anim. Biosci. 2014, 2, 469–486. [Google Scholar] [CrossRef]
- Parker, A.; Lawson, M.A.E.; Vaux, L.; Pin, C. Host-microbe interaction in the gastrointestinal tract. Environ. Microbiol. 2018, 20, 2337–2353. [Google Scholar] [CrossRef] [Green Version]
- Hills, R.D., Jr.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, G.; Teixeira, A.G.; Foditsch, C.; Bicalho, M.L.; Machado, V.S.; Bicalho, R.C. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS ONE 2013, 8, e63157. [Google Scholar]
- Malmuthuge, N.; Griebel, P.J.; Guan le, L. The gut microbiome and its potential role in the development and function of newborn calf gastrointestinal tract. Front. Vet. Sci. 2015, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Uetake, K. Newborn calf welfare: A review focusing on mortality rates. Anim. Sci. J. 2013, 84, 101–105. [Google Scholar] [CrossRef]
- Hoover, W.H. Digestion and absorption in the hindgut of ruminants. J. Anim. Sci. 1978, 46, 1789–1799. [Google Scholar] [CrossRef]
- Gressley, T.F.; Hall, M.B.; Armentano, L.E. Ruminant Nutrition Symposium: Productivity, digestion, and health responses to hindgut acidosis in ruminants. J. Anim. Sci. 2011, 89, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Rey, M.; Enjalbert, F.; Combes, S.; Cauquil, L.; Bouchez, O.; Monteils, V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 2014, 116, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Ruiz, D.R.; Abecia, L.; Newbold, C.J. Manipulating rumen microbiome and fermentation through interventions during early life: A review. Front. Microbiol. 2015, 6, 1133. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (capra hircus) during preweaning development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill-McFarland, K.A.; Breaker, J.D.; Suen, G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef] [Green Version]
- Meale, S.J.; Li, S.; Azevedo, P.; Derakhshani, H.; Plaizier, J.C.; Khafipour, E.; Steele, M.A. Development of ruminal and fecal microbiomes are affected by weaning but not weaning strategy in dairy calves. Front. Microbiol. 2016, 7, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honoré-Bouakline, S.; Vincensini, J.P.; Giacuzzo, V.; Lagrange, P.H.; Herrmann, J.L. Rapid diagnosis of extrapulmonary tuberculosis by PCR: Impact of sample preparation and DNA extraction. J. Clin. Microbiol. 2003, 41, 2323–2329. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Zhang, L.; Fan, C.; Li, W.; Liu, C.; Zhang, H.; Cheng, Q.; Zhang, Y. Sympatric yaks and plateau pikas promote microbial diversity and similarity by the mutual utilization of gut microbiota. Microorganisms 2021, 9, 1890. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Author Correction: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Agrawal, S.; Thi Kinh, C.; Schwartz, T.; Hosomi, M.; Terada, A.; Lackner, S. Determining uncertainties in PICRUSt analysis—An easy approach for autotrophic nitrogen removal. Biochem. Eng. J. 2019, 152, 107328. [Google Scholar] [CrossRef]
- Xue, D.; Chen, H.; Zhao, X.; Xu, S.; Hu, L.; Xu, T.; Jiang, L.; Zhan, W. Rumen prokaryotic communities of ruminants under different feeding paradigms on the Qinghai-Tibetan Plateau. Syst. Appl. Microbiol. 2017, 40, 227–236. [Google Scholar] [CrossRef]
- Cui, Z.; Wu, S.; Liu, S.; Sun, L.; Feng, Y.; Cao, Y.; Chai, S.; Zhang, G.; Yao, J. From maternal grazing to barn feeding during pre-weaning period: Altered gastrointestinal microbiota contributes to change the development and function of the rumen and intestine of yak calves. Front. Microbiol. 2020, 11, 485. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Malmuthuge, N.; Guan, L.L. Understanding host-microbial interactions in rumen: Searching the best opportunity for microbiota manipulation. J. Anim. Sci. Biotechnol. 2017, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Abecia, L.; Martín-García, A.I.; Martínez, G.; Newbold, C.J.; Yáñez-Ruiz, D.R. Nutritional intervention in early life to manipulate rumen microbial colonization and methane output by kid goats postweaning. J. Anim. Sci. 2013, 91, 4832–4840. [Google Scholar] [CrossRef] [Green Version]
- Abecia, L.; Waddams, K.E.; Martínez-Fernandez, G.; Martín-García, A.I.; Ramos-Morales, E.; Newbold, C.J.; Yáñez-Ruiz, D.R. An antimethanogenic nutritional intervention in early life of ruminants modifies ruminal colonization by Archaea. Archaea 2014, 2014, 841463. [Google Scholar] [CrossRef] [Green Version]
- Malmuthuge, N.; Griebel, P.J.; le Guan, L. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl. Environ. Microbiol. 2014, 80, 2021–2028. [Google Scholar] [CrossRef] [Green Version]
- Yeoman, C.J.; Ishaq, S.L.; Bichi, E.; Olivo, S.K.; Lowe, J.; Aldridge, B.M. Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci. Rep. 2018, 8, 3197. [Google Scholar] [CrossRef]
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.L.; Huse, S.M.; Sogin, M.L. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Nuriel-Ohayon, M.; Neuman, H.; Koren, O. Microbial Changes during Pregnancy, Birth, and Infancy. Front. Microbiol. 2016, 7, 1031. [Google Scholar] [CrossRef] [Green Version]
- Fernando, S.C.; Purvis, H.T., 2nd; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G.; Roe, B.A.; Desilva, U. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Liu, G.; Shafer, A.B.A.; Wei, Y.; Zhou, J.; Lin, S.; Wu, H.; Zhou, M.; Hu, D.; Liu, S. Comparative analysis of the gut microbial communities in forest and alpine musk deer using high-throughput sequencing. Front. Microbiol. 2017, 8, 572. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Zhao, Y.; Liu, Z.; Liu, G.; Du, M.; Wu, J.; Bai, D.; Li, B.; Bou, G.; Zhang, X.; et al. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments of Mongolian horses. Microbiol. Open 2020, 9, 1085–1101. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Kelly, W.J.; Ganesh, S.; Rakonjac, J.; Attwood, G.T. Butyrivibrio hungatei MB2003 competes effectively for soluble sugars released by butyrivibrio proteoclasticus B316(T) during growth on xylan or pectin. Appl. Environ. Microbiol. 2019, 85, e02056-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Makino, H.; Ishizuka, N.; Iwamoto, E.; Masaki, T.; Ikuta, K.; Kim, Y.H.; Sato, S. Long-term high-grain diet altered the ruminal pH, fermentation, and composition and functions of the rumen bacterial community, leading to enhanced lactic acid production in Japanese Black beef cattle during fattening. PLoS ONE 2019, 14, e0225448. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Pinchak, E.; Dowd, S.E.; Osterstock, J.; Gontcharova, V.; Youn, E.; Dorton, K.; Yoon, I.; Min, B.R.; Fulford, J.D.; et al. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb. Ecol. 2010, 59, 511–522. [Google Scholar] [CrossRef] [PubMed]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K. Diet, microbiota, and metabolic health: Trade-off between saccharolytic and proteolytic fermentation. Annu. Rev. Food Sci. Technol. 2018, 9, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, F.; Nguyen, T.L.; Brinkman, B.; Yunta, R.G.; Cauwe, B.; Vandenabeele, P.; Liston, A.; Raes, J. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013, 14, R4. [Google Scholar] [CrossRef] [Green Version]
- Martens, E.C.; Lowe, E.C.; Chiang, H.; Pudlo, N.A.; Wu, M.; McNulty, N.P.; Abbott, D.W.; Henrissat, B.; Gilbert, H.J.; Bolam, D.N.; et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011, 9, e1001221. [Google Scholar] [CrossRef]
- Castro, J.J.; Gomez, A.; White, B.A.; Mangian, H.J.; Loften, J.R.; Drackley, J.K. Changes in the intestinal bacterial community, short-chain fatty acid profile, and intestinal development of preweaned Holstein calves. 1. Effects of prebiotic supplementation depend on site and age. J. Dairy Sci. 2016, 99, 9682–9702. [Google Scholar] [CrossRef]
- Ma, T.; Villot, C.; Renaud, D.; Skidmore, A.; Chevaux, E.; Steele, M.; Guan, L.L. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: Prediction of diarrhea. ISME J. 2020, 14, 2223–2235. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 59–64. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, C.K.; Chen, Y.Y.; Fang, A.; Shaw, G.T.; Hung, C.M.; Wang, D. Maternal gut microbes shape the early-life assembly of gut microbiota in passerine chicks via nests. Microbiome 2020, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Taft, D.H.; Maldonado-Gomez, M.X.; Johnson, D.; Treiber, M.L.; Lemay, D.G.; DePeters, E.J.; Mills, D.A. The fecal resistome of dairy cattle is associated with diet during nursing. Nat. Commun. 2019, 10, 4406. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Wu, Q.; Shi, F.; Niu, J.; Zhang, T.; Degen, A.A.; Fang, Q.; Ding, L.; Shang, Z.; Zhang, Z.; et al. Seasonal dynamics of diet-gut microbiota interaction in adaptation of yaks to life at high altitude. NPJ Biofilms Microbiomes 2021, 7, 38. [Google Scholar] [CrossRef]
- De Paula Vieira, A.; von Keyserling, M.A.G.; Weary, D.M. Presence of an older weaned companion influences feeding behavior and improves performance of dairy calves before and after weaning from milk. J. Dairy Sci. 2012, 95, 3218–3224. [Google Scholar] [CrossRef]
- Phillips, C.J. The effects of forage provision and group size on the behavior of calves. J. Dairy Sci. 2004, 87, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Galef, B.G., Jr.; Giraldeau, L.A. Social influences on foraging in vertebrates: Causal mechanisms and adaptive functions. Anim. Behav. 2001, 61, 3–15. [Google Scholar] [CrossRef]
- Saro, C.; Hohenester, U.M.; Bernard, M.; Lagrée, M.; Martin, C.; Doreau, M.; Boudra, H.; Popova, M.; Morgavi, D.P. Effectiveness of interventions to modulate the rumen microbiota composition and function in pre-ruminant and ruminant lambs. Front. Microbiol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Montiel-Castro, A.J.; Gonzalez-Cervantes, R.M.; Bravo-Ruiseco, G.; Pacheco-Lopez, G. The microbiota-gut-brain axis: Neurobehavioral correlates, health and sociality. Front. Integr. Neurosci. 2013, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- McNeil, N.I. The contribution of the large intestine to energy supplies in man. Am. J. Clin. Nutr. 1984, 39, 338–342. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Species | Different Months | SEM | p-Value | ||||
|---|---|---|---|---|---|---|---|---|
| 1 M | 2 M | 5 M | 8 M | Inter-Species | Months | |||
| GLU (mmol/L) | Yak | 3.85 | 3.30 | 3.62 | 4.20 | 0.15 | 0.99 | 0.19 |
| Cattle | 3.61 | 3.82 | 3.34 | 4.22 | 0.19 | 0.43 | ||
| TC (mmol/L) | Yak | 2.06 | 2.71 | 1.97 | 2.59 | 0.18 | 0.16 | 0.41 |
| Cattle | 2.72 | 3.20 | 1.89 | 3.03 | 0.21 | 0.16 | ||
| TG (mmol/L) | Yak | 0.29 | 0.27 | 0.22 | 0.36 | 0.04 | 0.09 | 0.62 |
| Cattle | 0.43 | 0.41 | 0.24 | 0.42 | 0.04 | 0.26 | ||
| LDL-C (mmol/L) | Yak | 0.44 | 0.46 | 0.47 | 0.51 | 0.06 | 0.21 | 0.98 |
| Cattle | 0.63 | 0.83 | 0.31 | 0.57 | 0.08 | 0.14 | ||
| HDL-C (mmol/L) | Yak | 1.47 | 1.53 | 1.34 | 1.84 | 0.12 | 0.03 | 0.50 |
| Cattle | 1.84 | 2.03 | 1.58 | 2.12 | 0.10 | 0.26 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Wang, P.; Dingkao, R.; Du, M.; Ahmad, A.A.; Liang, Z.; Zheng, J.; Shen, J.; Yan, P.; Ding, X. Fecal Microbiota Dynamics Reveal the Feasibility of Early Weaning of Yak Calves under Conventional Grazing System. Biology 2022, 11, 31. https://doi.org/10.3390/biology11010031
Zhang J, Wang P, Dingkao R, Du M, Ahmad AA, Liang Z, Zheng J, Shen J, Yan P, Ding X. Fecal Microbiota Dynamics Reveal the Feasibility of Early Weaning of Yak Calves under Conventional Grazing System. Biology. 2022; 11(1):31. https://doi.org/10.3390/biology11010031
Chicago/Turabian StyleZhang, Jianbo, Peng Wang, Renqing Dingkao, Mei Du, Anum Ali Ahmad, Zeyi Liang, Juanshan Zheng, Jiahao Shen, Ping Yan, and Xuezhi Ding. 2022. "Fecal Microbiota Dynamics Reveal the Feasibility of Early Weaning of Yak Calves under Conventional Grazing System" Biology 11, no. 1: 31. https://doi.org/10.3390/biology11010031
APA StyleZhang, J., Wang, P., Dingkao, R., Du, M., Ahmad, A. A., Liang, Z., Zheng, J., Shen, J., Yan, P., & Ding, X. (2022). Fecal Microbiota Dynamics Reveal the Feasibility of Early Weaning of Yak Calves under Conventional Grazing System. Biology, 11(1), 31. https://doi.org/10.3390/biology11010031