
Multiple Introductions of Rabbit Hemorrhagic Disease Virus Lagovirus europaeus/GI.2 in Africa

 ,
,  , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Samples and Genome Amplification
2.2. Phylogenetic Analysis
2.3. Genetic Characterization and Subspecies Identification of the European Rabbits
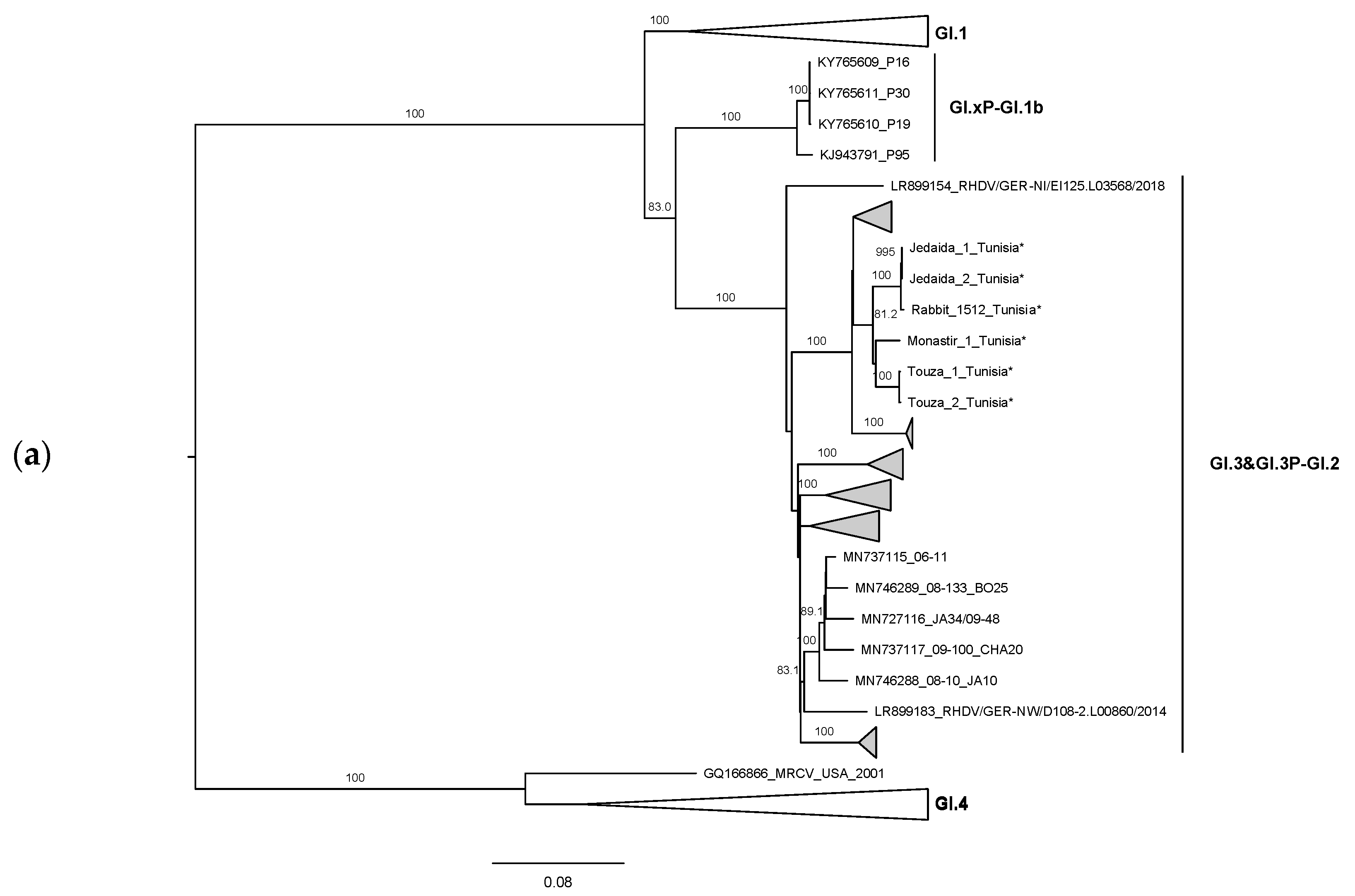
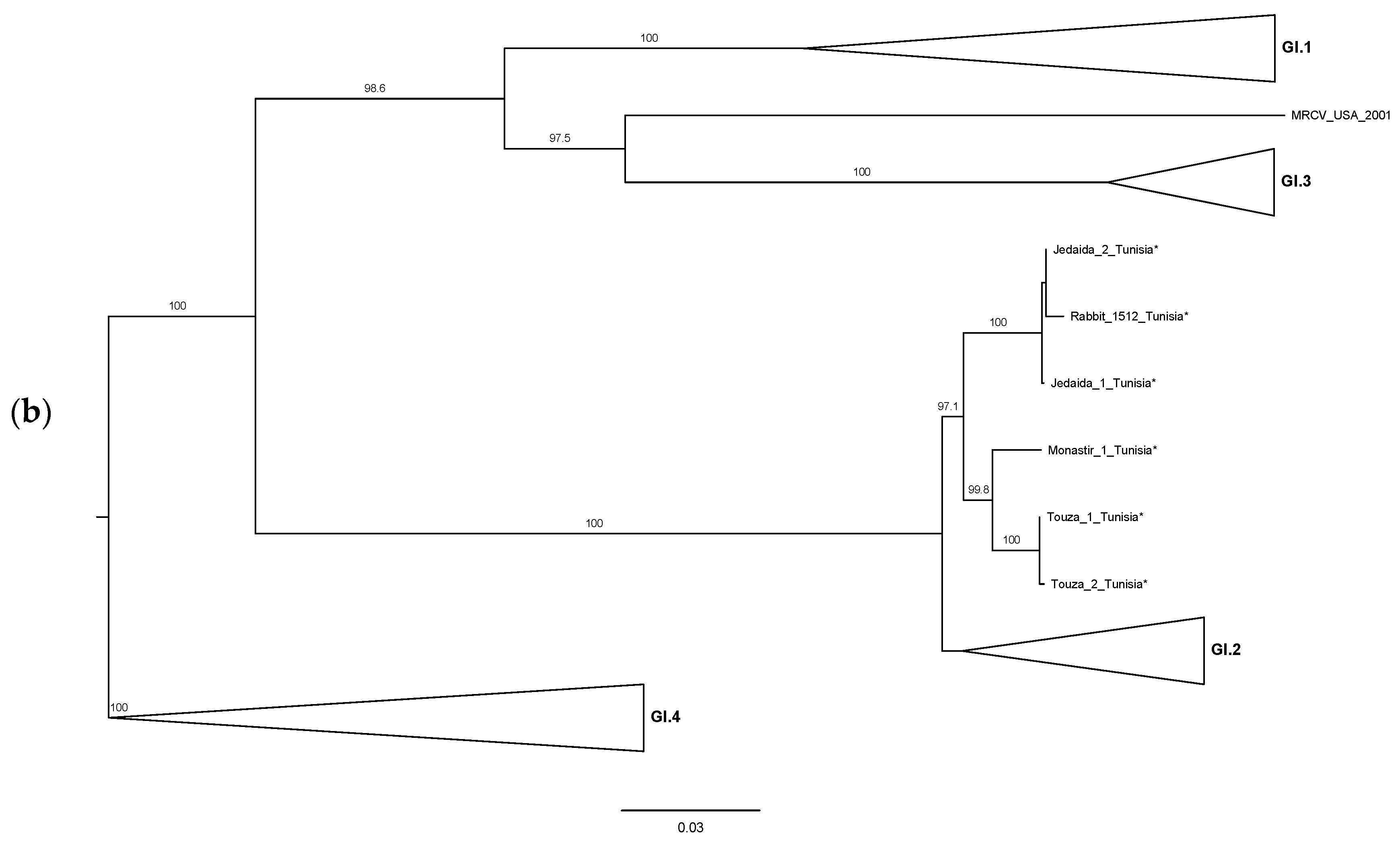
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abrantes, J.; van der Loo, W.; Le Pendu, J.; Esteves, P.J. Rabbit haemorrhagic disease (RHD) and rabbit haemorrhagic disease virus (RHDV): A review. Vet. Res. 2012, 43, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Pendu, J.; Abrantes, J.; Bertagnoli, S.; Guitton, J.S.; Le Gall-Recule, G.; Lopes, A.M.; Marchandeau, S.; Alda, F.; Almeida, T.; Celio, A.P.; et al. Proposal for a unified classification system and nomenclature of lagoviruses. J. Gen. Virol. 2017, 98, 1658–1666. [Google Scholar] [CrossRef]
- Morisse, J.P.; Le Gall, G.; Boilletot, E. Hepatitis of viral origin in Leporidae: Introduction and aetiological hypotheses. Rev. Sci. Tech. 1991, 10, 263–310. [Google Scholar]
- Bouslama, A.; De Mia, G.M.; Hammami, S.; Aouina, T.; Soussi, H.; Frescura, T. Identification of the virus of rabbit haemorrhagic disease in Tunisia. Vet. Rec. 1996, 138, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, C.; Hamouda, B.; Kaboudi, K.; Sghaier, S. La nouvelle forme de la maladie hémorragique virale (VHD) en Tunisie due au virus variant. Bull. d’Inf. Avic. Cunic. (GIPAC) 2015, 56, 23–26. [Google Scholar]
- OIE. Available online: https://wahis.oie.int (accessed on 14 July 2021).
- Lopes, A.M.; Rouco, C.; Esteves, P.J.; Abrantes, J. GI.1b/GI.1b/GI.2 recombinant rabbit hemorrhagic disease virus 2 (Lagovirus europaeus/GI.2) in Morocco, Africa. Arch. Virol. 2019, 164, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Erfan, A.M.; Shalaby, A.G. Genotyping of rabbit hemorrhagic disease virus detected in diseased rabbits in Egyptian Provinces by VP60 sequencing. Vet. World 2020, 13, 1098–1107. [Google Scholar] [CrossRef]
- Daodu, O.B.; Shaibu, J.O.; Richards, A.B.; Folaranmi, E.B.; Adegoke, S.; Ajadi, A.; Olorunshola, I.D.; Akanbi, O.B.; Afolabi, A.A.; Daodu, O.C.; et al. Detection and molecular characterization of a first isolate of rabbit haemorrhagic disease virus in Nigeria. Trop. Anim. Health Prod. 2021, 53, 185. [Google Scholar] [CrossRef]
- Happi, A.N.; Ogunsanya, O.A.; Oguzie, J.U.; Oluniyi, P.E.; Olono, A.S.; Heeney, J.L.; Happi, C.T. Microbial Metagenomic Approach Uncovers the First Rabbit Haemorrhagic Disease Virus genome in Sub-Saharan Africa. Sci. Rep. 2020, 11, 13689. [Google Scholar] [CrossRef]
- Ambagala, A.; Ababio, P.; Lamboo, L.; Goolia, M.; Lung, O.; Berhane, Y.; Odoom, T. Outbreak of Rabbit Hemorrhagic Disease Virus 2, Ghana. Emerg. Infect. Dis. 2021, 27, 1999. [Google Scholar] [CrossRef]
- Abrantes, J.; Droillard, C.; Lopes, A.M.; Lemaitre, E.; Lucas, P.; Blanchard, Y.; Marchandeau, S.; Esteves, P.J.; Le Gall-Reculé, G. Recombination at the emergence of the pathogenic rabbit haemorrhagic disease virus Lagovirus europaeus/GI.2. Sci. Rep. 2020, 10, 14502. [Google Scholar] [CrossRef]
- Silvério, D.; Lopes, A.M.; Melo-Ferreira, J.; Magalhães, M.J.; Monterroso, P.; Serronha, A.; Maio, E.; Alves, P.C.; Esteves, P.J.; Abrantes, J. Insights into the evolution of the new variant rabbit haemorrhagic disease virus (GI.2) and the identification of novel recombinant strains. Transbound. Emerg. Dis. 2018, 65, 983–992. [Google Scholar] [CrossRef]
- Szillat, K.P.; Höper, D.; Beer, M.; König, P. Full-genome sequencing of German rabbit haemorrhagic disease virus uncovers recombination between RHDV (GI.2) and EBHSV (GII.1). Virus Evol. 2020, 6, veaa080. [Google Scholar] [CrossRef]
- Hall, R.N.; Mahar, J.E.; Read, A.J.; Mourant, R.; Piper, M.; Huang, N.; Strive, T. A strain-specific multiplex RT-PCR for Australian rabbit haemorrhagic disease viruses uncovers a new recombinant virus variant in rabbits and hares. Transbound. Emerg. Dis. 2018, 65, e444–e456. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Dalton, K.P.; Magalhaes, M.J.; Parra, F.; Esteves, P.J.; Holmes, E.C.; Abrantes, J. Full genomic analysis of new variant rabbit hemorrhagic disease virus revealed multiple recombination events. J. Gen. Virol. 2015, 96, 1309–1319. [Google Scholar] [CrossRef]
- Mahar, J.; Jenckel, M.; Huang, N.; Smertina, E.; Holmes, E.; Strive, T.; Hall, R.J.B. Frequent intergenotypic recombination between the non-structural and structural genes is a major driver of epidemiological fitness in caliciviruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rahali, N.; Sghaier, S.; Kbaier, H.; Zanati, A.; Bahloul, C. Genetic characterization and phylogenetic analysis of rabbit hemorrhagic disease virus isolated in Tunisia from 2015 to 2018. Arch. Virol. 2019, 164, 2327–2332. [Google Scholar] [CrossRef]
- Le Gall-Recule, G.; Lavazza, A.; Marchandeau, S.; Bertagnoli, S.; Zwingelstein, F.; Cavadini, P.; Martinelli, N.; Lombardi, G.; Guerin, J.L.; Lemaitre, E.; et al. Emergence of a new lagovirus related to Rabbit Haemorrhagic Disease Virus. Vet. Res. 2013, 44, 81. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Queirós, J.; Carneiro, M.; Lopes, S.; Alves, P.C. Wild or domestic? Algirus or cuniculus? A novel genetic approach to infer the genetic integrity of European rabbit subspecies. In Proceedings of the First Iberian Congress of Applied Science on Game Resources (CICARC), Ciudad Real, Spain, 1–4 July 2019. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945. [Google Scholar] [CrossRef]
- Rouco, C.; Aguayo-Adán, J.A.; Santoro, S.; Abrantes, J.; Delibes-Mateos, M. Worldwide rapid spread of the novel rabbit haemorrhagic disease virus (GI.2/RHDV2/b). Transbound. Emerg. Dis. 2019, 66, 1762–1764. [Google Scholar] [CrossRef] [PubMed]
- Le Gall-Recule, G.; Zwingelstein, F.; Boucher, S.; Le Normand, B.; Plassiart, G.; Portejoie, Y.; Decors, A.; Bertagnoli, S.; Guerin, J.L.; Marchandeau, S. Detection of a new variant of rabbit haemorrhagic disease virus in France. Vet. Rec. 2011, 168, 137–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villafuerte, R.; Delibes-Mateos, M. Oryctolagus Cuniculus. Available online: https://www.iucnredlist.org/species/41291/170619657#text-fields (accessed on 24 May 2021).

{kind=link}
{kind=link}
{kind=link}
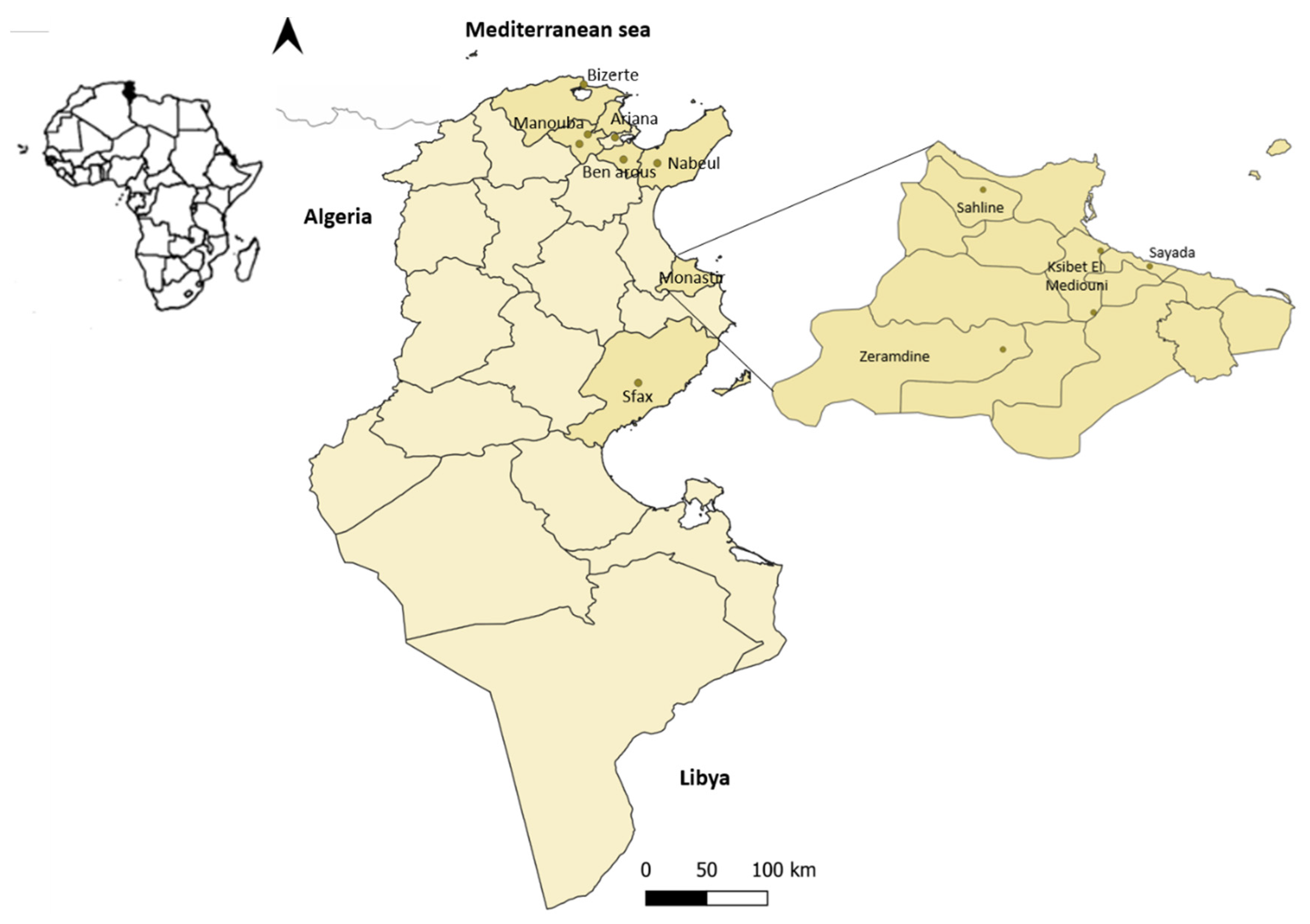
| Governorate | Region | Collection Date (Month/Year) | Sequence ID 1 |
|---|---|---|---|
| Monastir | Sahline | 10/2018 | - |
| Zeramdine | 10/2018 | Monastir 1 | |
| Ksibet el Mediouni (north) Ksibet el Mediouni (south) | 10/2018 12/2019 | - Touza 1; Touza 2 | |
| Sayada | 10/2018 | - | |
| Manouba | Jedaida | 12/2019 | Jedaida 1; Jedaida 2 |
| Manouba (center) | 06/2020 | Rabbit 1512 | |
| Bizerte | Bizerte (south) | 01/2020 | - |
| Ben arous | Ben arous | 2020 | - |
| Ariana | Bassatine | 11/2020 | - |
| Sfax | Sfax | 11/2020 | - |
| Nabeul | Grombalia | 2020 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Chehida, F.; Lopes, A.M.; Côrte-Real, J.V.; Sghaier, S.; Aouini, R.; Messadi, L.; Abrantes, J. Multiple Introductions of Rabbit Hemorrhagic Disease Virus Lagovirus europaeus/GI.2 in Africa. Biology 2021, 10, 883. https://doi.org/10.3390/biology10090883
Ben Chehida F, Lopes AM, Côrte-Real JV, Sghaier S, Aouini R, Messadi L, Abrantes J. Multiple Introductions of Rabbit Hemorrhagic Disease Virus Lagovirus europaeus/GI.2 in Africa. Biology. 2021; 10(9):883. https://doi.org/10.3390/biology10090883
Chicago/Turabian StyleBen Chehida, Faten, Ana M. Lopes, João V. Côrte-Real, Soufien Sghaier, Rim Aouini, Lilia Messadi, and Joana Abrantes. 2021. "Multiple Introductions of Rabbit Hemorrhagic Disease Virus Lagovirus europaeus/GI.2 in Africa" Biology 10, no. 9: 883. https://doi.org/10.3390/biology10090883
APA StyleBen Chehida, F., Lopes, A. M., Côrte-Real, J. V., Sghaier, S., Aouini, R., Messadi, L., & Abrantes, J. (2021). Multiple Introductions of Rabbit Hemorrhagic Disease Virus Lagovirus europaeus/GI.2 in Africa. Biology, 10(9), 883. https://doi.org/10.3390/biology10090883