Simple Summary
The search for genomic regions of putative selective signaling is instrumental in obtaining information about selection history in various species and populations. Domestic animals are subject to long-term artificial selection that leaves certain footprints in their genomes one can explore using genome-wide SNP screen. We examined here genomes of two contrasting chicken breeds, the native egg-type Russian White and meat-type White Cornish. Using three statistics, we identified genomic regions under putative selection, both breed-specific and shared between two breeds, that harbor key candidate genes for economically important traits. Our findings will be useful in further understanding selection history and genomic diversity in domestic chickens that would be pivotal in their productive breeding.
Abstract
Comparison of genomic footprints in chicken breeds with different selection history is a powerful tool in elucidating genomic regions that have been targeted by recent and more ancient selection. In the present work, we aimed at examining and comparing the trajectories of artificial selection in the genomes of the native egg-type Russian White (RW) and meat-type White Cornish (WC) breeds. Combining three different statistics (top 0.1% SNP by FST value at pairwise breed comparison, hapFLK analysis, and identification of ROH island shared by more than 50% of individuals), we detected 45 genomic regions under putative selection including 11 selective sweep regions, which were detected by at least two different methods. Four of such regions were breed-specific for each of RW breed (on GGA1, GGA5, GGA8, and GGA9) and WC breed (on GGA1, GGA5, GGA8, and GGA28), while three remaining regions on GGA2 (two sweeps) and GGA3 were common for both breeds. Most of identified genomic regions overlapped with known QTLs and/or candidate genes including those for body temperatures, egg productivity, and feed intake in RW chickens and those for growth, meat and carcass traits, and feed efficiency in WC chickens. These findings were concordant with the breed origin and history of their artificial selection. We determined a set of 188 prioritized candidate genes retrieved from the 11 overlapped regions of putative selection and reviewed their functions relative to phenotypic traits of interest in the two breeds. One of the RW-specific sweep regions harbored the known domestication gene, TSHR. Gene ontology and functional annotation analysis provided additional insight into a functional coherence of genes in the sweep regions. We also showed a greater candidate gene richness on microchromosomes relative to macrochromosomes in these genomic areas. Our results on the selection history of RW and WC chickens and their key candidate genes under selection serve as a profound information for further conservation of their genomic diversity and efficient breeding.
1. Introduction
Since the middle of the last century, the poultry industry has focused on the exploitation of a few highly specialized, productive lines selected for egg or meat (broiler) production traits. In the chicken (Gallus gallus; GGA), this has led to a drastic decline in the number and population size of native breeds that were extensively used for agricultural production in the past. Many native breeds are now at the risk of being lost, while potentially harboring significant historical footprints of selection for economically important phenotypes in their genomes. Conservation and molecular genetic characterization of biodiversity among domestic animal and poultry species used for food production are two of the key objectives to ensure the sustainability of local and regional agricultural production systems [1,2,3,4,5,6,7]. Genomic tools and resources for poultry-related research have advanced and been immensely enriched since the time of the first chicken reference genome sequence publication [8].
Two main evolutionary lineages of domestic chickens are represented by egg-type and meat-type breeds [9]. One of the distinctive native egg-type chicken breeds is the Russian White (RW), developed for egg production in the former USSR in 1929–1953 by crossing local low-productive hens with White Leghorn roosters of Danish, British, and American origins [10]. Before 1965, the RW was a major chicken breed employed for egg production in the USSR. In 1975, the number of RW chickens was 29.73 million heads; however, there was a dramatic drop by 1980 to 4.4 million [10], with a further essential population reduction in later years.
Historically, RW chickens were brought to the Russian Research Institute of Farm Animal Genetics and Breeding (RRIFAGB), Pushkin, from the Leningradskaya egg production plant in 1952. Along with the selective breeding for high egg performance in 1952–1999, the RW chickens were selected for tolerance to low temperatures. This gave rise to establishing in 1957 and developing an inbred RS subpopulation selected for cold tolerance of chicks [11,12]. The breeding RS stock was kept at 15–22 °C during the first five days after hatch, with a gradual temperature lowering to 14 and 11 °C by 21–30 days. Adult individuals were kept in winter at a temperature below 0 °C, while safeguarding the laying performance at a high level [13]. As a result of the long-term selection for critically low temperatures, the RS phenotypes were characterized by exceptional cold tolerance of neonatal chicks; their snowy-white down at day-old presumably caused by a recessive mutant gene, sw, for snow-white down [14,15]; and elevated resistance to a few neoplastic diseases, such as Marek’s disease, leukemia, and carcinomas [11]. The current RG population was more recently derived from the RS as a result of a one-time telic crossing with White Leghorns in 2006 not selected further for cold tolerance and subjected to random mating in 2000–2013 [12,16,17,18].
Presently, the small RG population of cold-tolerant RW chickens (25 males and 234 females) is kept in the Genetic Collection of Rare and Endangered Chicken Breeds of the RRIFAGB. In the last two decades, the RW breeding program has been aimed at improved egg production and egg weight and increased yield of vaccine raw material, i.e., extraembryonic (allanto-amniotic) fluid and titers of vaccine viruses in it while maintaining cold tolerance of neonatal chickens [15,19,20]. There is also one more RW population maintained at the All-Russian Poultry Research and Technological Institute (ARPRTI) collection farm, Sergiev Posad.
Because of importance of the yield of extraembryonic fluid (YEF) to produce vaccines, in-depth molecular genetic studies were undertaken to pinpoint markers for YEF in the RW population at RRIFAGB. Its examination for the effects of indels in the PRL (prolactin) and DRD2 (dopamine receptor D2) genes on YEF showed significant associations of the PRL insertion variant with greater YEF and egg weight (EW) [16]. In a genome-wide association study (GWAS) using the Illumina Chicken 60K SNP iSelect BeadChip, several suggestive SNP loci and candidate genes were detected for YEF as well as EW, egg number, age at first egg, body weight, and day-old chick down color [15]. The RW demographic history was explored at the genomic level using the same Illumina BeadChip. Within the current RG population, a heterogeneity of SNP genotypes was revealed, suggesting its subdivision into four subgroups: two relatively homogenous, one heterogenous, and one phylogenetically closer to the historical RS subpopulation. The latter, in turn, was distinguished by more numerous monomorphic markers and longer linkage disequilibrium (LD) regions as well a greater number of runs of homozygosity (ROHs), their greater mean length, and a higher ROH-based inbreeding coefficient. The RG population had the smallest LD values and the largest effective population size [12,17,18]. In an additional GWAS using the Illumina Chicken 60K SNP iSelect BeadChip, nine SNPs were reported in the RG population that demonstrated significant associations with egg production traits and coincided with ROHs [21]. A sampling of RW chickens was also tested for copy number variation (duplication) at the AvBD7 (avian beta-defensin 7) gene relevant to the innate immune system, with a ratio of three duplication carriers to four nonduplication individuals [22].
An old meat-type breed of White Cornish (WC) chickens was created by crossing the English Game breed with Aseel or Malay fowls. It was recognized as a breed in England in 1886 under the name of Indian Game. Unlike RW chickens, the WC breed has been specifically selected for growth performance, feed efficiency, and carcass traits [23]. Such intensive selection for the limited number of traits in meat-type (broiler) chickens has led to the occurrence of increased frequencies of undesirable traits such as low fertility, reduced fitness, increased disease susceptibility, skeletal weakness, etc. [23,24,25,26]. In Russia, a few WC crossbred strains are maintained in the RRIFAGB gene pool collection and have been explored in the genome-wide scanning studies using the Illumina BeadChip [17,18]. They demonstrated a reduced genetic diversity because of small population size, and lower ROH metrics due to crossbreeding effects [17]. A small population of purebred WC chickens is also kept in the ARPRTI gene pool collection.
Comparison of genomic footprints in two chicken breeds with different selection history, i.e., native egg-type RW and meat-type WC, may be helpful in elucidating genomic regions that have been targeted by recent and more ancient selection. Detecting regions of the genome that have undergone artificial selection will expand our understanding of the domestication history and selective breeding in chickens. Identification of genes within such genomic regions will aid in development of effective breeding programs based on marker-assisted and genomic selection.
Different methodologies have been utilized for defining regions of the genome that exhibit evidence of having been under selection. One of the most used methods is based on estimating the fixation index (FST), which quantifies the differences in allele frequencies between populations [27,28]. The fixation index is a single SNP test, which is routinely used for identifying highly differentiated alleles [29]. Genetic differentiation between populations is expected to be low in neutral regions of the genome or in regions under balancing selection, whereas high genetic differentiation indicates regions that have undergone divergent selection among populations. Another metric used for determining genomic regions that have been subjected to selection is the hapFLK analysis. This is a haplotype-based statistic for detecting positive selection using multiple population data [30,31]. For the calculation of hapFLK, both population structure and haplotype information are taken into consideration. The identification of ROH islands was proposed as one more useful indicator of selection signals in livestock populations [32,33]. ROH islands are genomic regions with high homozygosity around a selected locus that might harbor targets of positive selection [34]. Combining different statistics for defining selection signatures can improve the reliability of identified genomic regions [35,36].
Several genome-wide studies have been undertaken to detect the genomic footprints of artificial selection in large numbers of chicken breeds (e.g., [37,38,39,40,41]). Despite the initial chicken genome research efforts undertaken so far at the RRIFAGB [17,18], a comparative in-depth search for selection sweeps in chicken breeds of Russian origin at the genome-wide level have not been carried out and would be much anticipated.
In the present study, we aimed to examine and compare the trajectories of artificial selection in the genomes of two chicken breeds with different selection history and breeding objectives, i.e., the native egg-type RW and meat-type WC breeds. We performed the structural annotation of discovered genomic regions, suggesting multiple key candidate genes associated with egg/meat production and other economically important traits. Our results elucidate the breeding history of RW and WC chickens and provide a useful information for further conservation of their genomic diversity and sustainable breeding.
2. Materials and Methods
2.1. Experimental Birds and Ethics Statement
Young chickens of the RW and CW breeds (Figure 1) were purchased and housed in the Gene Pool Collection of Farm and Wild Animals and Birds of the L.K. Ernst Federal Research Center for Animal Husbandry (LKEFRCAH) with the aim of establishing an F2 resource population to be used for subsequent GWAS experiments. RW chickens of the cold-tolerant line were provided by the RRIFAGB. Nonpedigreed crossbred CW chickens were provided by the Breeding Genetic Center “Smena”, a subsidiary of the Federal Science Center for Poultry Science, of the Russian Academy of Sciences. Individuals of both breeds were grown to the age of 6 months and used to produce F1 generation. No chickens used in the present studies were subject to further pure breeding. Feather samples were collected by trained lab personnel following the LKEFRCAH ethical guidelines for minimizing any possible bird discomfort or distress.

Figure 1.
Two studied chicken breeds: Russian White (a) and White Cornish (b).
2.2. Sampling and DNA Extraction
Feather samples were obtained from 54 individuals including 31 samples of the RW chickens and 23 samples of the WC breed. DNA extraction was performed using Nexttec columns (Nexttec Biotechnologie GmbH, Leverkusen, Germany) according to manufacturer’s instructions. Concentration of dsDNA solutions was determined using a Qubit 3.0 fluorometer (Thermo Fisher Scientific, Wilmington, DE, USA). To check the purity of extracted DNA, OD260/280 ratio was tested using NanoDrop-2000 (Thermo Fisher Scientific, Wilmington, DE, USA).
2.3. SNP Genotyping and Quality Control
Individual sample genotyping was carried out using Chicken 50K_CobbCons chip (Illumina, San Diego, CA, USA). Input files were created using R software [42]. To determine valid genotypes for each SNP, we set a cut-off for the GenCall (GC) and GenTrain (GT) scores of 0.5 [43]. Using PLINK 1.9 software [44], SNP quality control was performed. All chicken genotypes passed the filtering for genotyping efficiency (--mind 0.2). Only SNPs located on autosomes from GGA1 to GGA28 were used for analysis, and SNPs genotyped in less than 90% of the samples (--geno 0.1) were excluded from the analysis. The final data set used for analyzing signatures of selection included 44,728 autosomal SNPs. Additional filters for LD values were used for performing calculation of genetic diversity, principal component analysis (PCA), Neighbor-Net tree construction, and admixture clustering that resulted in 25,768 SNPs. One SNP from each pair of neighbored SNPs where LD (r2) value exceeded 0.5 within 50 SNP windows was removed using--indep-pairwise 50 5 0.5 flag, where 50 is size of the sliding window, 5 is the number of SNPs shifted in each step, and 0.5 is the r2 threshold. Positions of SNPs were assigned according to the GGA reference genome assembly GRCg6a [45].
2.4. Genetic Diversity
PLINK 1.9 [44] was used to evaluated within-population genetic diversity. We calculated the observed heterozygosity (HO), unbiased expected heterozygosity (UHE) [46], rarefied allelic richness (AR) [47], and inbreeding coefficient (UFIS) based on the unbiased expected heterozygosity by using R package diveRsity [48].
2.5. PCA, Neighbor-Net, and Admixture
Procedures of PCA, Neighbor-Net clustering, and model-based clustering (Admixture) were exploited to select the individuals for the analysis of selection sweeps. PCA was performed using PLINK v1.9 software. An R package ggplot2 was used to visualize the results [49]. Pairwise identical-by-state (IBS) distances calculated in SplitsTree4 [50] were used to construct a Neighbor-Net tree. Admixture v1.3 software [51] was employed for genetic admixture analysis and an R package pophelper [52] for plotting the results. A standard admixture cross-validation procedure [53] was used to calculate the number of ancestral populations (K). The assumed number of K corresponded to the lowest value of cross-validation (CV) error as compared to other K values.
2.6. Selection Signature Analysis
Three different statistics were used for detecting the signatures of selection in the genome of chicken: calculation of FST values for each SNP when comparing pairs of breeds, hapFLK analysis, and estimation of the ROH islands, which were overlapped among different animals within each breed.
2.6.1. FST Analysis
Pairwise FST-based genetic differentiation [53] was estimated between all SNPs using PLINK 1.9. We applied a low threshold for minor allele frequency (MAF) of less than 5% (--maf 0.05), because the filtering of SNPs based on MAF may affect the probability of identifying alleles related to selection [54]. The dataset used for FST analysis included 44,728 autosomal SNPs. The top 0.1% FST values were used to represent a selection signature, as was previously proposed by Kijas et al. [55] and Zhao et al. [56].
2.6.2. Runs of Homozygosity
A window-free method for consecutive SNP-based detection, i.e., consecutive runs method [57] implemented in an R package detectRUNS [58], was used for the estimation of ROHs. We allowed one SNP with missing genotype and up to one possible heterozygous genotype in one run to avoid the underestimation of the number of ROHs that were longer than 8 Mb [59]. Because of strong linkage disequilibrium (LD), typically extending up to about 100 kb [60], and for excluding short and very common ROHs, we set the minimum length for an ROH at 500 kb. For minimizing false-positive results, we calculated the minimum number of SNPs (l) as was initially evaluated by Lencz et al. [61] and later modified by Purfield et al. [62]:
where ns is number of genotyped SNPs per individual, ni is number of genotyped individuals, α is percentage of false-positive ROHs (set to 0.05 in our study), and is mean heterozygosity across all SNPs. In our case, the minimum number of SNPs was 23.
We estimated the number and length of ROHs for each individual and then averaged these per individual within each breed. Additionally, we computed the genomic inbreeding coefficient based on ROH (FROH) as the ratio of the sum of the length of all ROHs per animal to the total autosomal SNP coverage (0.94 Gb).
Number of ROHs in the genomes of two studied breeds was examined using the following ROH length classes: 0.5–1, 1–2, 2–4, 4–8, 8–16, and >16 Mb. To define the proportion of genome covered by different ROH segments, we calculated the sum of ROHs for the following different minimum lengths: >0.5, >1, >2, >4, >8, and >16 Mb.
Putative ROH islands were defined as overlapping homozygous regions that shared by more than 50% of analyzed individuals within each breed, as this was suggested in other studies [63,64]. We applied the threshold of 0.3 Mb for the minimal overlapping length size, because it was previously shown that shorter segments of 0.3–1 Mb are predominant in genome of white layers [41].
2.6.3. HapFLK Analysis
To detect the signatures of selection through haplotype differentiation among the studied breeds, we also employed hapFLK 1.4 program [30,31]. The number of haplotype clusters per chromosome was determined in fastPHASE by using cross-validation-based estimation and was set at 35 [65]. For detailed analyses, we selected the hapFLK regions containing at least one SNP with p-value threshold of 0.01 (−log10(p) > 2).
2.7. Search for Genes and QTLs Localized within Identified Genomic Regions
For candidate gene mining in the genomic regions under putative selection, we used the genomic localization of the regions as detected by three different statistics, i.e., FST, ROH, and hapFLK methods. We prioritized those regions that were overlapped and revealed at least by two different techniques. Borders of these regions as localized in the GRCg6a reference assembly chromosomes were used as a query list for retrieving chicken genes and their human orthologs using the web-based Ensembl Genes release 103 database and Ensembl BioMart data mining tool [66]. Results retrieved from the Ensembl BioMart browser for each genomic region of selection signatures were manually sieved and compared to relevant published investigations to identify main candidate genes and other genes of interest.
Additionally, a broader gene mining was carried out that also included the regions found by one method to expand the candidate list with more previously discovered and important genes. Where needed for comparisons with other studies, we used the UCSC liftOver tool [67] to convert genome coordinates between earlier chicken genome builds and the reference assembly GRCg6a. If an older build could not be directly compared to GRCg6a, a sequential lift of coordinates between assemblies was applied.
For specifying the presence of quantitative trait loci (QTLs) and associated genes overlapped with the identified genomic regions, we also analyzed a publicly available chicken database, QTLdb [68].
2.8. Gene Ontology Mining
To perform functional annotation and gene ontology (GO) term enrichment analysis within the determined selective sweep regions, we exploited the Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.8 [69,70]. Using our in-house GO mining pipeline consisted of the BioMart and DAVID tools, the gene and background lists were generated following the procedure described in detail elsewhere [71]. Briefly, chicken–human orthologs of the maximum orthology confidence served as entries for the gene and background lists, applying the 70% gene identity threshold to the gene list and reducing the background list to chromosomes containing the selection footprints. Significant annotation clusters were selected using an enrichment score of more than 1.3 and a p-value < 0.05.
3. Results
3.1. Genetic Diversity
The RW chickens were characterized by significantly lower level of genetic diversity assessed by the level of unbiased expected heterozygosity (UHE = 0.339 vs. 0.383, p < 0.001) and allelic richness (AR = 1.937 vs. 1.982, p < 0.001) as compared to the WC breed. We observed significant deviation in the number of heterozygotes from the Hardy–Weinberg equilibrium in both studied populations. The negative value of the inbreeding coefficient UFIS indicates a slight excess of heterozygotes in the RW population (UFIS = –0.016), while the WC breed showed a slighter deficiency of heterozygotes (UFIS = 0.009) than it would be expected under Hardy–Weinberg equilibrium (Table 1).

Table 1.
Summary of genetic/genomic diversity statistics 1 calculated in the studied Russian White (RW) and White Cornish (WC) breeds based on SNP genotypes.
3.2. Breed Relationship and Admixture
As assessed by PCA, the first component, which is responsible for 9.34% of the genetic variability, clearly differentiated the RW chickens from the WC breed (Figure 2a). The neighbor-joining tree constructed based on pairwise IBS distances showed the breed-specific distribution of individuals between the two groups (Figure 2b). The calculations of CV error for the different number of clusters (from 1 to 5) showed that the probable number of clusters (K) was equal to 2 (Figure S1). The admixture clustering at K = 2 clearly distinguished the RW and WC populations at K = 2 (Figure 2c), indicating a very low level of admixture among the studied populations.
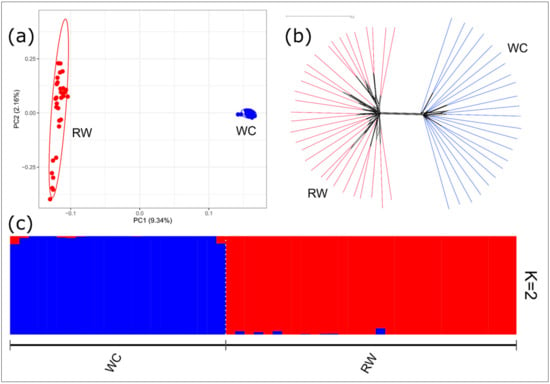
Figure 2.
Genetic relationships between the Russian White (RW) and White Cornish (WC) chicken populations: (a) a principal component analysis (PCA) plot showing the distribution of RW and WC individuals in the dimensions of two coordinates, i.e., the first (PC1; X-axis) and second (PC2; Y-axis) principal components, with percentage of total genetic variability, which can be explained by each of the two components, being indicated within the parentheses; (b) a Neighbor-Net tree constructed based on the FST genetic distances among the studied populations; and (c) an admixture plot representing cluster structure of the studied populations if the number of clusters K = 2.
3.3. ROH Distribution in the Genomes of Studied Chicken Breeds
We revealed a lower mean number of ROH segments in the RW breed genome as compared to the WC breed (109.10 vs. 119.09). In contrast, RW chickens had a greater coverage of genome by ROHs (183.41 vs. 161.97 Mb), which resulted in a higher value of inbreeding coefficient calculated based on ROHs (FROH = 0.195 vs. 0.172) (Table 2).
Table 2.
Summary of the runs of homozygosity (ROH) descriptive statistics 1 calculated in the studied Russian White (RW) and White Cornish (WC) breeds based on SNP genotypes.
Short ROH segments (0,5–1 Mb) were the most distributed throughout the genome and accounted for 49.26% and 54.73% of all ROHs identified in the WC and RW breeds, respectively. The proportion of ROH segments of the greater length (4–8, 8–16, and >16 Mb), typically caused by inbreeding to a more recent ancestor, was higher in RW as compared to WC (5.56 vs. 3.50% for 4–8 Mb; 1.51 vs. 0.44 for 8–16 Mb; and 0.33 vs. 0.07 for ROH > 16 Mb). The genome coverage by the longer ROH segments was greater in RW than that in WC (18.9 vs. 9.69%; 0.66 vs. 0.13%; and 18.49 vs. 10.87%, respectively) (Figure 3, Table S1).
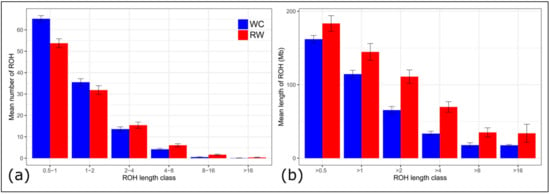
Figure 3.
Descriptive statistics of the runs of homozygosity (ROH) by ROH length class in the breeds of Russian White (RW) and White Cornish (WC) chickens: (a) mean number of ROHs (Y-axis) by ROH length class (X-axis; 0.5–1, 1–2, 2–4, 4–8, 8–16, and >16 Mb); (b) overall mean length of ROHs (Y-axis) by ROH length class (X-axis; >0.5 Mb, >1 Mb, >2 Mb, >4 Mb, >8 Mb, and >16 Mb).
3.4. Analysis of the Signatures of Selection
To detect the signatures of selection in the genome of the studied chicken populations, we calculated FST values for each SNP, performed hapFLK analysis, and estimated ROH islands, which were overlapped among different individuals within each population.
Average pairwise FST value between the studied populations equaled 0.152. We identified 45 SNPs with FST beyond the cut-off value (top 0.1%, FST > 0.92) that were distributed among twelve autosomes (GGA1, GGA2, GGA3, GGA4, GGA5, GGA6, GGA8, GGA9, GGA15, GGA18, GGA26, and GGA28). The greatest number of SNPs was found on GGA5 (16 SNPs) and GGA8 (10 SNPs). Fourteen SNPs localized on GGA5 and all of SNPs on GGA8 were presented by blocks of neighboring SNPs (Figure 4, Table S2).
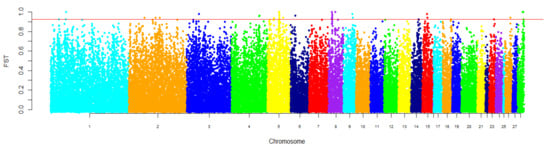
Figure 4.
Genomic distribution of FST values estimated between the Russian White and White Cornish populations. Values for the X-axis are chicken autosomes (breadth of autosomes corresponds to their length) and those for the Y-axis are FST values. SNPs were plotted relative to their positions within each autosome. The threshold, which was estimated as the top 0.1% for FST values, is indicated by a horizontal line.
We found the overlapping ROH islands observed in more than 50% of samples within each population (Table S3, Figure S2). We detected 23 ROH islands in the WC breed, which covered 25.987 Mb of genome and were distributed among nine chromosomes (GGA1, GGA2, GGA3, GGA4, GGA5, GGA7, GGA8, GGA18, and GGA28). Average length of ROH islands was 1.130 Mb, with their size ranging from 0.405 to 3.240 Mb. Eighteen ROH islands covering 14.530 Mb of the genome and distributed among seven chromosomes (GGA1, GGA2, GGA3, GGA4, GGA5, GGA8, and GGA9) were found in the RW population. Length of ROH islands varied between 0.301 and 2.472 Mb and averaged 0.807 Mb. Most of the identified ROH islands were population-specific, except for two ROH islands on GGA1 and GGA2, which partly overlapped in the two studied populations. The ROH islands on GGA1 covered a region between 143,268,071 and 144,065,046 in the genome of the WC breed and between 143,047,114 and 143,929,272 in the genome of the RW breed. The positions of ROH islands on GGA2 were 78,935,731 to 79,963,158 for the WC breed and 79,022,396 to 79,487,897 for the RW chickens.
The hapFLK analysis resulted in identification of 15 putative regions affected by selection, which were distributed among 10 autosomes and covered 20.095 Mb of the chicken genome. Length of the putative regions under selection pressure ranged between 0.022 and 3.764 Mb and averaged 1.340. Twelve regions were breed-specific, including four regions on GGA1, GGA7, GGA13, and GGA14 in the WC breed (coverage of 2.412 Mb) and eight regions on GGA1, GGA3, GGA4, GGA5, GGA8, GGA9, and GGA13 (coverage of 10.572 Mb) in RW chickens. Three genomic regions identified by hapFLK analysis on GGA2 (positions from 69,808,276 to 72,600,534 and from 76,552,199 to 80,316,420) and GGA3 (positions from 29,203,776 to 29,759,199) were common for both populations (Figure 5 and Figure S3, Table S4). Three genomic regions with the significance level p < 0.001, which were detected on GGA1 (positions from 41,095,175 to 44,484,517) and GGA8 (positions from 28,777,070 to 30,174,896) in RW chickens, as well as one common region on GGA2 (positions from 76,552,199 to 80,316,420) were defined as strongest hapFLK regions.
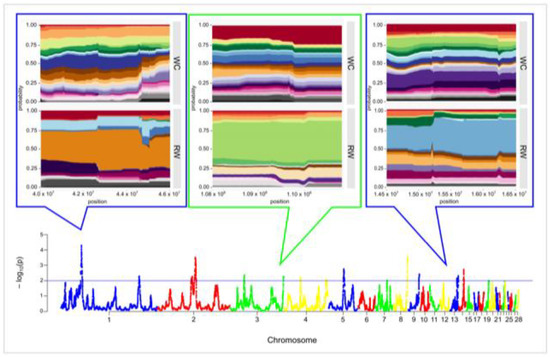
Figure 5.
Signatures of selection in the genomes of the studied Russian White (RW) and White Cornish (WC) chicken populations based on the hapFLK statistics. Values for the X-axis are chicken autosomes, and those for the Y-axis are values of statistical significance (−log10 p-values). The blue line indicates threshold of significance at p < 0.01 (i.e., −log10(p) > 2). Magnified plots of the few most representative chromosome areas containing the hapFLK regions are presented above the plot. Values for the X-axis are the genomic positions on corresponding autosome. Each color band corresponds to one haplotype cluster, and the height of a band shows the cluster frequency. Magnified plots for all 15 putative regions identified by hapFLK analysis are presented in Figure S3.
Comparing genomic localization of the regions under putative selection detected by three different statistics (FST, ROHs, and hapFLK) revealed the presence of 11 overlapped regions, which were identified at least by two different methods (Table 3). The identification of these regions by at least two detection methods implies that such regions are likely to bear true signatures of selection.
Table 3.
Overlapped genomic regions under putative selection identified at least by two different statistics in the Russian White (RW) and White Cornish (WC) breeds.
Thirty-one genomic regions identified in our study by either one or more metrics were overlapped with regions, which were previously detected in other investigations in different chicken breeds (e.g., [15,38,39,40,41,72,73,74,75]). These selection sweeps were localized on GGA1 (three regions), GGA2 (seven regions), GG3 (three regions), GGA4 (five regions), GGA5 (three regions), GGA7 (two regions), GGA13 (two regions), and one region on each of GGA8, GGA9, GGA15, GGA18, and GGA28 (Table 4 and Table S5).
Table 4.
Overlapped genomic regions identified in the present and previous studies.
3.5. Identification of Key Candidate Genes Affected by Selection
As a result of sieving and examining the genomic regions of selective sweeps determined by at least one method, we structurally annotated these regions and revealed the presence of 881 candidate genes in the two studied chicken populations (Table 5 and Table S6). Those involved 548 annotated genes, including 524 protein coding genes and 24 micro RNAs, with many of them being previously described in other relevant studies (e.g., ALX1, DIO2, GJD2, KITLG, TSHR, etc.). The rest of them were 316 novel Ensembl genes, 10 uncharacterized NCBI loci, and 7 uncharacterized chicken homologs to human genes. In all, there were 516 chicken–human orthologs used later for the GO analysis.
Table 5.
Prioritized annotated genes within the overlapped genomic regions affected by putative selection in the Russian White (RW) and White Cornish (WC) chicken breeds and localized by two or three methods.
Among the discovered 881 candidate genes, 762 genes were found on nine macrochromosomes (GGA1–9) and 119 genes on six microchromosomes (GGA13, GGA14, GGA15, GGA18, GGA26, and GGA28). Total length of presumptive selection footprints identified on macro- and microchromosomes was respectively 46.46 and 3.32 Mb. This candidate gene mining survey resulted in an estimate of ~16.4 genes on an average every 1 Mb of macrochromosomal DNA and ~35.9 genes per 1 Mb on microchromosomes, or a ~2.2-fold difference.
In Table 5, a shorter list is presented for 188 prioritized candidate genes that were annotated in either chicken or human genomes and retrieved from the 11 overlapped regions of putative selection as identified at least by two techniques. These included the following 50 previously known candidate genes (Table 5, shown in bold; see also their details in Table S6): ALX1, ANKRD13C, BTG1, FGF3, FGF4, KITLG, LEPR, MGAT4C, NAT10, RPE65, SHANK2, and TYW3 (identified in the RW breed); AP1M1, AVEN, CALR3, ELL, FMN1, GREM1, INSR, KLF2, LOC420160, MIR6666, MIR6693, MYDGF, MYO9B, PLIN3, PTPRS, PCDH7, RAB8A, RABGAP1L, RYR3, SMIM7, SPRED1, TICAM1, TMEM38A, and USE1 (in the WC breed); and ACTC1, BTBD9, CCT5, CMBL, CTNND2, DAP, FASTKD3, GLP1R, GJD2, MEIS2, MTRR, ROPN1L, SEMA5A, and SRD5A1 (in both the RW and WC breeds).
Many other key candidate genes, including those suggested in relevant studies (e.g., ANKH, ANXA10, BMPR1B, CASP6, CD36, CHST11, COL6A1, COL6A2, CRY1, DHX36, DIO2, FGA, FGB, FGG, GTF2A1, IGF1, IGSF10, MMP16, MYF5, MYF6, NUAK1, OVST, PLRG1, PMCH, SFRP2, SH3RF2, SLC2A14, SPOCK3, TSHR, TMEM263, WWP1, etc.) were determined in other genomic regions with selective sweeps in the RW and/or WC chickens using either method (Tables S5 and S6).
3.6. QTLs Overlapped with the Identified Genomic Regions
Using Chicken QTLdb [68], we completed a search that showed the presence of 198 known QTLs overlapped with 45 identified genomic regions localized on GGA1 (six regions), GGA2 (five regions), GGA3 (five regions), GGA4 (seven regions), GGA5 (three regions), GGA6 (one region), GGA7 (five regions), GGA8 (two regions), GGA9 (three regions), GGA13 (one region), GGA14 (one region), GGA15 (one region), GGA18 (two regions), GGA26 (one region), and GGA28 (two regions) (Table 6 and Table S7).
Table 6.
Selective sweeps identified in genomes of the Russian White (RW) and White Cornish (WC) chicken breeds, which are overlapped with known QTLs and associated genes.
3.7. Functional Annotation and GO Term Enrichment
Using the DAVID web tool and a list of 516 chicken–human orthologous genes found in the genomic regions with selection signatures (Table S6), we performed the analysis of functional annotation and enrichment of GO terms. A number of key candidate genes underlying the selective sweeps were determined as summarized in Table 7.
Table 7.
Functional annotation and enrichment of gene ontology (GO) terms among the identified genes within the sweep regions as ascertained by DAVID.
4. Discussion
4.1. Genetic Diversity and Evolutionary Relationships
In the current study, we have taken advantage of examining and comparing two breeds that are typical representatives of two major, different, and, in a certain sense, opposite evolutionary lineages occurred in the process of chicken domestication and breeding [9]. That is, one was selected for egg-type traits (RW) and the other for meat-type traits (WC). We found that 15.2% of variability was caused by genetic differences between these two studied breeds (FST = 0.152), and the remaining 84.8% was due to allelic variation within the breeds. We observed a significantly lower level of genetic diversity in RW chickens as compared to WC (UHE = 0.339 vs. 0.383) (Table 1). A possible reason for this can be the genetic drift occurred in the population of RW chickens, which has a very small size (~260 heads) and has been subjected to a long-term breeding as a closed population. At the same time, a higher level of genetic diversity in WC chickens may also reflect a crossbred origin of the individuals used in the present study. A higher value of FROH inbreeding coefficient, which was observed in RW chickens (0.195 vs. 0.172) (Table 2), may reflect the origin of the studied RW population from a small number of founders [11]. A slight excess of heterozygotes observed in the RW population may be a consequence of a lower selection pressure and selection for a variety of breeding traits [15,18] as compared to WC [23]. Genetic drift can be considered another possible reason that led to a significant deviation in the number of heterozygotes among RW chickens as compared to that expected under Hardy–Weinberg equilibrium.
The results of the PCA plotting, Neighbor-Net analysis, and admixture clustering (Figure 2a–c) clearly distinguished the RW and WC breeds, confirming their different genetic origin.
As the egg-type breed, the RW chickens, exposed also in the past to crossing with a typical egg layer breed of White Leghorns, were selected for fecundity, egg number, and other egg performance traits. In contrast, the WC breed was derived from crosses between meat and game breeds and selected for growth, muscle development, and other meat production traits. Both breeds manifesting different phenotypic traits were previously included by Moiseyeva et al. [9] in a detailed comparative phylogenetic survey of various chicken breeds using two sets of morphological discrete characters, body measurements, biochemical markers, and the activity of serum esterase-1 (currently, CES1L1, carboxylesterase 1 like 1). In the generated dendrograms for evolutionary relationships in chickens, RW clustered with the White Leghorn and other egg-type breeds, whereas WC formed common clusters with game and meat-type breeds. Lately, using the Illumina Chicken 60K SNP iSelect BeadChip, Dementieva et al. [17] localized RW and WC on the opposite branches of an FST-based Neighbor Joining tree. Our data based on Chicken 50K_CobbCons chip-assisted SNP genotypes strongly supported these previous phylogenetic relationship assessments for the RW and WC breeds that exemplify typical egg- and meat-type chickens.
4.2. Genomic Trajectories of Selection
To the best of our knowledge, this is the first study of putative signatures of selection in the genomes of the native egg-type RW breed (Figure 1a). We performed it based on the analysis of genome-wide SNP genotypes using 44,728 autosomal SNPs. In the time of the Soviet Union, RW was the major layer breed used for egg production [10]. Currently, only a small number of cold-tolerant and disease-resistant RW chickens are kept in the gene pool collection [19,20]. Identifying genomic regions affected by natural and artificial selection in breeds with different genetic backgrounds may give an insight into the history of domestication and selection for economically important traits [76,77,78,79]. Therefore, we chose for comparison the meat-type WC breed, which is characterized by high growth rate and outstanding meat productivity [23]. We believe that the breed-specific genomic regions found as being under selection pressure are most likely related to recent artificial selection and harbor genes and their variants associated with breeding traits. On the contrary, the regions, which overlap in the two breeds that manifest contrasting phenotypic traits, may reflect more ancient evolution events prior to domestication or breed specialization.
Three different statistics (selecting top 0.1% SNPs by FST value, hapFLK analysis, and detection of ROH islands shared by more than 50% of individuals) were applied to detect the genomic regions and genes that are affected by selection in RW and WC chickens (Table 3, Table 4 and Table 5, Tables S5 and S6). Using multiple methods based on different approaches may complement each other and therefore have higher informative power [35,80]. Among 45 SNPs that were selected by FST value (Table S2), 33 were localized within the regions identified by hapFLK or ROH analysis (Table S5). Among 15 genomic regions identified by hapFLK analysis, five were overlapped with ROH islands (Table 3). In total, we identified 11 true genomic regions under selection pressure (so were identified by at least two different methods), including four RW-specific selection sweeps on GGA1, GGA5, GGA8, and GGA9; four WC-specific signatures of selection on GGA1, GGA5, GGA8, and GGA28; and three regions on GGA2 (two sweeps) and GGA3, which were common for both breeds (Table 3). Comparing selection signatures identified in the present study with the results of previous investigations performed in the RW, WC, and other chicken breeds that support our findings [15,21,38,39,40,41,72,73,74,75], we showed the presence of 31 overlapped genomic regions distributed among twelve chromosomes (Table 4 and Table S5).
4.3. Key Candidate Genes and Overlapping QTLs within Sweep Regions
The present study provided a rich estimation of coding genes that underlie certain regions subject to selection in the chicken genome. Of especial interest are those that formed a set of 188 prioritized candidate genes as detected by more than two approaches to hunting for selection signatures in the genomes of the two studied breeds with contrasting phenotypes (Table 5). Of particular note are the genes ascertained in other genome-wide or more focused investigations targeting selection footprints, ROHs, QTLs, and candidate genes associated with important phenotypes and functions (shown in Table 5 in bold). Among them, we emphasized ten genes specific to the egg-type RW breed sweep regions. For the meat-type WC breed regions, there were 24 specific genes. In both breeds, we observed the following 14 candidates: ACTC1, BTBD9, CCT5, CMBL, CTNND2, DAP, FASTKD3, GLP1R, GJD2, MEIS2, MTRR, ROPN1L, SEMA5A, and SRD5A1. We outline these genes below in a more detail-oriented manner, along with characterizing the appropriate QTLs known within the same regions.
4.3.1. GGA1 Region Candidate Genes and QTLs
The ALX1 gene (41,898,277 … 41,919,541 bp), a member of the aristaless-like homeobox (ALX) family, is associated with craniofacial and limb development [81]. In chickens, the gene is involved in beak morphology and was shown to have been under selection prior to domestication, being colocalized with a selective sweep on GGA1 [40], i.e., similarly to our finding. Within the same genomic region, there are three other genes, BTG1 (44,429,339 … 44,433,309 bp), KITLG (43,015,486 … 43,066,975 bp), and MGAT4C (42,251,047 … 42,358,204 bp). BTG anti-proliferation factor 1 (BTG1) is a candidate gene expressed during early chick development [82], being related to muscle structure growth and development at early stages [83]. The KITLG (ligand of tyrosine–kinase receptor) gene was cytogenetically mapped to GGA1 [84] and more recently ascertained as a gene related to pigmentation traits, having been under selection before chicken domestication and overlapping a selective sweep [40]. MGAT4C (MGAT4 family member C) was recognized as a differentially expressed gene associated with nonsynonymous SNPs and putative selective signaling, suggesting its relation to chicken adaptation to high-altitude conditions [85].
Six selective sweeps (two RW-specific and four WC-specific) identified on GGA1 were overlapped with 28 known QTLs (Table S7). The region of 42.2 … 44.5 Mb under selection in RW chickens was associated with multiple QTLs for egg yolk weight (QTL:24938, QTL:24939, and QTL:24940; within 42,044,310 … 42,991,116 bp) that might be a result of recent selection for increased egg weight. Selective sweeps found in WC chickens mainly covered QTLs for growth, carcass traits, and feed efficiency including body weight at 9 days (QTL:96626, 45,501,491 … 54,773,135 bp), feed conversion ratio (QTL:139668, 54,874,224 … 54,874,264 bp; and QTL:139747, 76,365,781 … 76,365,821 bp), abdominal fat weight (QTL:19362553, 914,126 … 55,103,802 bp; and QTL:193624, 52,797,908 … 53,910,398 bp), and carcass fat content (QTL:193637, 52,797,908 … 53,910,398 bp) (Table 6 and Table S7).
4.3.2. GGA2 Region Candidate Genes and QTLs
Previously, Qanbari et al. [38] detected signals of selective sweeps in this region embracing candidate genes in laying hens that might be under selection pressure. In our study, we identified a number of these genes including CCT5 (chaperonin-containing TCP1 subunit 5; 78,405,510 … 78,413,636 bp), CMBL (carboxymethylenebutenolidase homolog; 78,327,060 … 78,338,570 bp), DAP (death-associated protein; 78,125,211 … 78,169,242 bp), FASTKD3 (FAST kinase domains 3; 79,210,863 … 79,217,888 bp), and ROPN1L (rhophilin associated tail protein 1 like; 78,263,682 … 78,272,601 bp). Additionally, we established that this region contains some other important genes such as CTNND2 (77,868,375 … 77,907,969 bp), MTRR (79,184,684 … 79,211,008 bp), SEMA5A (78,655,148 … 78,914,889 bp), and SRD5A1. In another study [86], the CTNND2 (catenin delta 2; 79,772,665 … 79,786,504 bp) gene was shown to have an extremely strong sweeping signal, with highest zFst score in chromosome 2, though with no relationship to muscle development, growth, or other economic traits. This gene was suggested to be responsible for the evolutionary changes of domestic chickens associated with vision deterioration [86]. MTRR (5-methyltetrahydrofolate-homocysteine methyltransferase reductase) is a SNP-containing candidate gene for dermatological diseases/conditions, being also associated with amino acid changes [87]. The gene was also found to be expressed in liver of growing broilers after in ovo injection of folic acid [88]. SEMA5A (semaphorin 5A) is a strong candidate gene with selective sweep that is responsible for response to selection on antibody response to sheep red blood cells [73]. In the SRD5A1 (steroid-5-alpha-reductase, alpha polypeptide 1) gene, a differentially methylated region was found as the most likely biomarker of inbreeding depression of reproduction in Langshan chickens [89,90].
In total, we identified five selective sweeps on GGA2 overlapped with nine known QTLs. Among them, two were specific for RW, one for WC, and two were found in both studied breeds (Table 6 and Table S7). Interestingly, the genomic region within 123.0 … 123.7 Mb identified in RW chickens was overlapped with a previously detected QTL for body temperature (QTL:30853, 121,250,696 … 123,202,074 bp), which could reflect the long-term selection of RW chickens for cold tolerance at the earlier stage of breed development [11,12,13]. Moreover, this region contains a causal SNP associated with day-old chick down color in RW chickens [15].
4.3.3. GGA3 Region Candidate Genes and QTLs
Of a particular interest within this region are two candidates for the development and weight of the comb, BTBD9 (BTB domain containing 9; 13,189,289 … 13,199,549 bp) and GLP1R (glucagon like peptide 1 receptor; 29,410,438 … 29,492,850 bp) [91]. For the latter gene, a signal of selective sweeps was also determined by Qanbari et al. [40].
Among five selective sweeps overlapped with 16 known QTLs, three were specific for the RW breed, one for WC, and two were common for both studied breeds (Table 6 and Table S7). Identification of additional regions in WC chickens (12.8 … 13.5 and 29.2 … 29.8 Mb) associated with QTLs for feed conversion ratio (QTL:139401, 13,278,977 … 13,279,017 bp; and QTL:139333, 29,681,999 … 29,682,039 bp, respectively) suggested that genes associated with feed efficiency could be involved in the artificial selection.
4.3.4. GGA4 Region Candidate Genes and QTLs
The region contains the PCDH7 (protocadherin 7; 71,575,850 … 71,827,560 bp) gene, a notable positional candidate associated with internal organ traits in chickens and located within a QTL for intestine length and gizzard weight [92] (Table S7).
We also found seven selective sweeps in the genomes of RW and WC chickens (five RW-specific and two WC-specific), which covered 31 previously described QTLs (Table 6 and Table S7). A selective sweep within 38.0 … 38.6 Mb identified in the genome of RW chickens was overlapped with multiple QTLs for feed intake (QTL:195036 to QTL:195049, QTL:195085, QTL:195087, and QTL:195096). This observation could reflect a selective advantage of RW chickens with high eating capacity that would be able to accumulate more energy necessary to survive at low-temperature environment and maintain a high egg productivity.
4.3.5. GGA5 Region I Candidate Genes and QTLs
A genomic region on GGA5 within 17.8 … 18.8 Mb, which was found to be under selection pressure in RW chickens (Table S5), contains two closely located genes, FGF3 and FGF4 (fibroblast growth factors 3 and 4; 17,872,481 … 17,877,619 and 17,843,044 … 17,845,980 bp, respectively), suggested as candidates for the feathered-leg trait [93]. NAT10 (N-acetyltransferase 10; 18,749,501 … 18,771,124 bp), an additional known candidate gene in laying hens, displays a signal of selective sweeps [38] and contributes to feed conversion ratio [94]. One more gene, SHANK2 (SH3 and multiple ankyrin repeat domains 2; 18,179,810 … 18,393,764 bp), was localized within a QTL associated with metabolizable efficiency traits in broilers [75].
4.3.6. GGA5 Region II Candidate Genes and QTLs
This region covers 29.6 … 32.9 Mb and comprises a series of vital genes we identified in both the RW and WC chickens. In particular, ACTC1 (actin alpha cardiac muscle 1; 32,480,478 … 32,485,436 bp) expressed in heart and skeletal muscle was identified as a candidate gene in laying hens that possesses a signal of selective sweeps [38] and is differentially expressed in breast muscle of growing chickens [95]. The GJD2 (gap junction protein delta 2; 32,502,596 … 32,505,180 bp) gene also has a signal of sweeps, being expressed in brain and retina and potentially involved in adaptation process during chicken domestication [41,96]. An overlapping with a signal of selective sweeps was also found at RYR3 (ryanodine receptor 3; 30,373,917 … 30,541,375 bp), a supposedly candidate gene for pigment synthesis [97] that is expressed in broiler breast meat in response to heat stress and methionine dipeptide-deficient diet [98].
Interestingly, the region we described here overlaps with ROH islands that were shared between commercial lines and red junglefowls [41] and contain, among other genes, AVEN (apoptosis and caspase activation inhibitor; 30,287,850 … 30,373,018 bp), FMN1 (formin 1; 30,598,320 … 30,751,690 bp), GREM1 (gremlin 1, DAN family BMP antagonist; 30,776,671 … 30,777,369 bp), MEIS2 (Meis homeobox 2; 31,437,974 … 31,606,534 bp), and SPRED1 (sprouty-related EVH1 domain containing 1; 30,948,808 … 31,002,826 bp). Both AVEN expressed in adult brain, heart, intestine, kidney, lung, stomach, and spleen, as well as in whole embryos [99], and GREM1 negatively regulate apoptosis [41]. GREM1 and FMN1 play an important role in mouse and chick limb development [100], with the former being located within a chicken QTL related to limb development [41]. MEIS2 is related to angiogenesis at tibial lesions in broiler chickens [101] and overlapped with a QTL for body weight [41] (Table S7).
4.3.7. GGA8 Region I Candidate Genes and QTLs
Among the genes we localized within this region (7.1 … 7.8 Mb) in the WC genome, RABGAP1L (RAB GTPase activating protein 1 like) was lately suggested as a chicken candidate gene for plasma very low-density lipoprotein concentration in a respective GWAS carried out by Zhang et al. [102]. This blood characteristic is thought to be useful for selecting lean meat-type lines [102].
The region found as affected by selection in WC chickens also overlaps with several QTLs associated with feed conversion ratio (QTL:139410; 7,689,799 … 7,689,839 bp), breast muscle pH (QTL:157180; 7,219,671 … 7,569,140 bp), shank circumference (QTL:213550; 7,521,372 … 7,569,140 bp), VLDL cholesterol level (QTL:170802; 7,550,094 … 7,550,134 bp), and antibody titer to IBV (QTL: 24340; 7,500,998 … 7,501,038 bp) (Table S7).
4.3.8. GGA8 Region II Candidate genes
This region within 28.6 … 30.1 Mb that we determined in the RW genome includes few genes of interest, e.g., LEPR (leptin receptor; 28,687,054 … 28,717,255 bp), a candidate gene in laying hens harboring a signal of selective sweeps [38]. As a candidate gene suggestive of production-oriented selection [40], it is also associated with growth and feed efficiency in meat-type chickens [103]. Another selection sweep in this candidate region was defined at the TYW3 (tRNA-yW synthesizing protein 3 homolog; 29,988,144 … 29,991,956 bp) gene [104]. ANKRD13C (ankyrin repeat domain 13C; 29,389,240 … 29,401,259 bp) gene was suggested to be a candidate gene for feed conversion ratio [94]. The RPE65 (retinoid isomerohydrolase; 29,124,295 … 29,130,623 bp) gene is related to retinol metabolism, its expression being downregulated in rose-comb chickens [105].
4.3.9. GGA28 Region Candidate Genes and QTLs
We identified within this region (4.0 … 5.0 Mb) several other recently suggested candidate genes for plasma very low-density lipoprotein concentration as determined by Zhang et al. [102], including AP1M1 (adaptor-related protein complex 1 mu 1 subunit; 4,510,264 … 4,518,819 bp), CALR3 (calreticulin 3; 4,437,755 … 4,441,529 bp), ELL (elongation factor for RNA polymerase II; 3,997,087 … 4,031,848 bp), KLF2 (Kruppel-like factor 2; 4,484,819 … 4,487,194 bp), MIR6666 (microRNA 6666; 4,808,669 … 4,808,772 bp), MIR6693 (microRNA 6693; 4,387,579 … 4,387,688 bp), MYO9B (myosin IXB; 4,263,083 … 4,303,548 bp), PTPRS (protein tyrosine phosphatase, receptor type S; 4,727,381 … 4,835,713 bp), RAB8A (RAB8A, member RAS oncogene family; 4,524,693 … 4,537,499 bp), SMIM7 (small integral membrane protein 7; 4,391,143 … 4,393,274 bp), TICAM1 (toll-like receptor adaptor molecule 1; 4,961,810 … 4,965,486 bp), TMEM38A (transmembrane protein 38A; 4,384,819 … 4,391,056 bp), and USE1 (unconventional SNARE in the ER 1; 4,258,017 … 4,262,921 bp). AP1M1 is involved in endosome to melanosome transport [106] and USE1 [38] is overlapped with signals of selective sweeps. ELL is also a positional candidate gene for egg number in broiler breeders [107] and a candidate gene associated with egg quality (yolk weight) [108]. The KLF2 gene is related to angiogenesis at tibial lesions in broiler chickens [101]. Our findings extend knowledge about genes under selection within this region on GGA28 in meat-type chickens.
In addition, this region embraced few other important genes as demonstrated in our and other investigations. INSR (insulin receptor; 4,218,437 … 4,253,117 bp) is known as a candidate gene in laying hens that contains a signal of selective sweeps [38]. LOC420160 (cathepsin L1-like; 5,030,693 … 5,034,171 bp) is a chicken ortholog to human CTSV (cathepsin V) and CTSL (cathepsin L), cysteine cathepsins being attributed to extracellular matrix degradation and tissue remodeling [109]. Initially, the chicken CTSL (now CTSV) gene that has an increased expression during oviduct regression [110] was mapped to the Z chromosome [111], while LOC420160 is considered as its paralog on GGA28. MYDGF (myeloid-derived growth factor; 5,034,853 … 5,039,077 bp) is a candidate gene related to growth and development at early stages [83]. The PLIN3 (perilipin 3; 4,952,051 … 4,961,161 bp) gene is associated with immunity and enhances growth performance in broilers [112].
The region selected in WC chickens overlapped with several previously described QTLs associated with fat content (QTL:193631 for abdominal fat content; QTL:193655 and QTL:193647 for carcass fat content; within 3,753,016 … 4,712,620 bp) and blood parameters including blood pH (QTL:71128; 3,856,132 … 4,097,788 bp), CO2 partial pressure (QTL:71133 and QTL:71137; 3,835,952 … 4,167,579 bp), mean blood cell volume (QTL:71178; 3,944,019 … 4,197,143 bp), and blood hemoglobin level (QTL:71183; 3,944,019 … 4,197,143 bp) (Table S7).
4.3.10. Other Important Candidate Genes
Our genome-wide screening resulted in establishing multiple candidate genes underlying the sweep regions and potentially affected by selection for egg and meat production traits in the genomes of the egg-type RW and meat-type WC chickens as identified by either technique (Tables S5 and S6). Many of them were supported by other previous studies and deserve a further attention. For instance, Qanbari et al. [38,40] reported signals of selective sweeps for the following candidate genes in laying hens (with respective chromosome of their location indicated in the parentheses): ANKH (ANKH inorganic pyrophosphate transport regulator; GGA1), IGF1 (insulin-like growth factor 1; GGA1), BMPR1B (bone morphogenetic protein receptor type 1B; GGA4), TSHR (thyroid stimulating hormone receptor; GGA5), and IGSF10 (immunoglobulin superfamily member 10; GGA9). Among these, IGF1 is also an important growth factor [40] associated with abdominal fat weight/deposition, body weight, and other traits in chickens [113,114], and BMPR1B is a candidate gene for low methylation related to hypoxic adaptation in Tibetan chickens [115]. The BMPR1B gene was also found within a feed intake-associated QTL [68] (Table 6).
On GGA1, we defined a few more candidates under selection in the egg-type breed. In particular, signals of selective sweeps were observed at the CD36 (CD36 molecule) gene [116] and at the closely located MYF5 and MYF6 (myogenic factors 5 and 6) genes [106]. CD36 is a candidate gene for pendulous comb [116] and related to adipogenesis and lipogenesis in pectoralis muscle tissue [117]. In the meat-type breed, signals of selective sweeps overlapped with OVST (ovostatin) [118], PMCH (promelanin concentrating hormone) [37,119], and SLC2A14 (solute carrier family 2 member 14) [120]. The OVST gene is involved in oviduct development and eggshell formation [118]. PMCH is a candidate gene involved in chicken growth control and overexpressed in low growth rate broiler cockerels [119] that also contributes to appetite and food intake [37,118]. SLC2A14 is supposedly a candidate gene for pigment synthesis [120].
Among other candidates, we also discovered on GGA2 that there is the WWP1 (WW domain containing E3 ubiquitin protein ligase 1) gene, a selection candidate for muscularity in broilers detected as signal of parallel divergence [96]. Quite close to this gene, Kudinov et al. [15] found a SNP rs15151359 that was one of eight markers suggestively associated in RW chickens with the snow-white down color at day-old. This SNP is flanked by the MMP16 (matrix metallopeptidase 16) gene [15], although the latter overlaps with a novel gene ENSGALG00000053033 (Table S6) that is homologous to RNA-directed DNA polymerase from mobile element jockey-like. We identified MMP16 and ENSGALG00000053033 in the RW-specific selective sweep region. Because we have no other relevant information about these two genes in chickens, further speculation about their contribution to phenotypic traits in the RW breed would require an additional investigation.
Within a GGA4-specific ROH, we identified ANXA10 (annexin A10), a candidate gene for abdominal fat in a copy number variation (CNV) region overlapping with a QTL and a selective sweep [121]. Among other genes in RW-specific regions under putative selection on GGA4, there was a trio of the linked fibrinogen alpha, beta, and gamma chain genes (FGA, FGB, FGG) that are involved in cell differentiation and had signals of selective sweeps [106]. FGA is also related to response to Marek’s disease [122], while FGB is a candidate gene overexpressed in low growth rate broiler cockerels [119]. Two more closely located genes, PLRG1 (pleiotropic regulator 1) and SFRP2 (secreted frizzled related protein 2), also overlap with signals of selective sweeps and contribute to cell differentiation. SFRP2 is involved in inbreeding depression of reproduction [90] and embryogenesis (development of the neural system, eyes, muscles, and limbs) [123], and was described as a candidate gene for adaptation to solar radiation in North- and East-African chickens [106]. SPOCK3 (SPARC/osteonectin, cwcv, and kazal-like domains proteoglycan 3) is a candidate gene for abdominal fat within a CNV region overlapped with a QTL and a selective sweep [121]. An ROH island on GGA4 found in the WC genome harbored the CASP6 (caspase 6) gene that is involved in regulation of apoptosis and was reported to be upregulated in broiler chickens in response to Salmonella and phytobiotic intake [124].
Most intriguingly, we identified a ROH on GGA5 in the egg-type RW breed that involves the TSHR gene related to reproductive machinery and most prominent for being coined as a domestication gene in chickens [37,40,118,125]. Moreover, we also confirmed the known candidate genes located within the same LD block as TSHR, such as CEP128 (centrosomal protein 128), DIO2 (deiodinase, iodothyronine type II), GTF2A1 (general transcription factor IIA subunit 1), SEL1L (SEL1L adaptor subunit of ERAD E3 ubiquitin ligase), and STON2 (stonin 2) [126]. DIO2 is a candidate gene, overexpressed in low growth rate broiler cockerels [120], involved in regulation of hormone levels and possibly affected by the domesticated mutation at TSHR [126]. GTF2A1 is a candidate gene for egg production traits [127] colocalized with a signal of selective sweeps [104]. The SEL1L and STON2 genes are candidates associated with egg number in laying hens [127], and STON2 is also a candidate gene related to yolk weight [108].
Next, we detected a ROH on GGA7 in the meat-type WC breed that contains two notable genes, COL6A1 and COL6A2 (collagen type VI alpha 1 chain and collagen type VI alpha 2 chain). These are candidate genes involved in skeletal system development and overexpressed in low growth rate broiler cockerels [120]. They also contribute to meat quality and bear strong selection signals in Chinese indigenous chicken genomes [97]. Through the DNA methylation mechanism, the COL6A1 gene modification affects intramuscular fat deposition in chicken [128] and relates to meat quality of breast muscle [129]. The COL6A2 gene was also listed among candidates for feed conversion ratio [94].
Two more genes are noteworthy to discuss here: DHX36 (DEAH-box helicase 36) and SH3RF2 (SH3 domain containing ring finger 2). The DHX36 gene is located on GGA9 within breakpoint of evolutionary conservation between human and chicken chromosomes [130] and serves as an RNA sensor in innate immunity for recognizing viral RNA [131]. Within a WC-specific genomic region on GGA13, we also determined SH3RF2, a strong candidate gene in broilers that has a signal of selective sweeps and gene deletion associated with increased growth, while lying within a QTL region for body weight [37].
4.4. GO Term Annotation Clustering of Candidate Genes
As a result of GO analysis, we found four significant annotation clusters enriched with GO terms that underlay key candidate genes of interest derived from the regions of selection footprints in the RW and WC breeds. Remarkably, these GO term clusters involved certain important genes, most of which we have outlined above, e.g., WWP1, IGF1, INSR, SEMA5A, RPE65, and COL6A2. A couple of noteworthy candidates can also be mentioned here, including PIK3CB (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta) and TGFBI (transforming growth factor beta-induced). The PIK3CB gene is associated with white/red earlobe color formation in chicken [132]. The TGFBI gene is related to angiogenesis at tibial lesions in broiler chickens [102] and was reported to be a candidate gene for Müllerian duct development and its disorders [133].
4.5. Gene Richness
In terms of comparing gene density on chicken macro- and microchromosomes, we found that microchromosomes contained 2.2 times more genes in the genomic regions harboring selection signatures. This observation is highly concordant with the previous general estimates of two- to three-fold gene richness on microchromosomes as compared to that on macrochromosomes (e.g., [134]), demonstrating a clear-cut negative correlation between gene density and chromosome length in the chicken genome [8].
5. Conclusions
Using three complementary statistical methods (FST at pairwise breed comparison, hapFLK analysis, and identification of ROH islands), we performed the search for genomic footprints in two contrasting chicken breeds, the native egg-type RW and meat-type WC. Eleven true genomic regions under selection pressure were identified by at least two different methods as distributed on seven G. gallus autosomes. Four of such selective sweep regions were breed-specific for each of RW and WC chickens, while three remaining regions were common for both breeds. Several identified genomic regions were overlapped with known QTLs. In RW chickens, these regions included known QTLs for body temperature, egg production rate, egg yolk weight, and feed intake that are in accordance with the breed origin and the history of its artificial selection for cold tolerance and egg laying. Selective sweeps identified in the genome of WC chickens were mainly overlapped with QTLs responsible for growth, meat and carcass traits, and feed conversion ratio that can reflect the long-term selection of this breed for increased growth rate, meat productivity, and feed efficiency. We determined a set of 188 prioritized candidate genes localized within selected genomic regions and reviewed their functional relevance in both breeds. One of RW-specific sweep regions contained the TSHR gene known for being associated with domestication and reproduction in chickens. Examination of gene ontology in the sweep regions added more information for their functional annotation in two breeds. We also suggested a reasonable estimate of greater candidate gene richness within sweep regions on microchromosomes vs. macrochromosomes. These findings extend our knowledge about genomic diversity and genes under selection in the genomes of two chicken breeds with different selection histories and contrasting phenotypic traits. The research results will be useful for conservation, sustainable breeding, and efficient selection of the Russian White chicken breed.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/biology10090876/s1. Table S1: Number and overall length of ROHs by ROH length class. Table S2: Top 0.1% SNPs by FST values. Table S3: ROH islands identified in genome of the White Cornish and (WC) and Russian White (RW) chicken populations. Table S4: Regions in genome of the White Cornish (WC) and Russian White (RW) chicken breeds as identified by the hapFLK analysis. Table S5: Summary of selective sweeps and candidate SNPs observed in the present study as compared with other related investigations. Table S6: Summary of chicken genes and their human orthologs within genomic regions of selective sweeps as retrieved from BioMart (Ensembl Genes release 103). Table S7: Genomic regions identified in the RW and WC chicken breeds, which are overlapped with known associations and QTLs. Figure S1: A plot showing cross-validation (CV) error (Y-axis) for different K-values (X-axis). Figure S2: Distribution of ROHs within chromosomes. Figure S3: Plots of the chromosome areas containing the hapFLK regions.
Author Contributions
Conceptualization, N.A.Z. and G.B.; methodology, A.S.A., A.V.D., M.N.R., and N.A.Z.; software, A.S.A., A.V.D., and M.N.R.; validation, A.V.D., M.N.R., and N.A.Z.; formal analysis, A.N.V., N.A.V., and E.S.F.; investigation, A.N.R. and E.A.G.; resources, A.V.D., I.V.G., and N.A.Z.; data curation, A.V.D.; writing—original draft preparation, A.S.A., M.N.R., and N.A.Z.; writing—review and editing, A.S.A., A.V.D., M.N.R., O.I.S., D.K.G., G.B., and N.A.Z.; visualization, A.S.A.; supervision, N.A.Z.; project administration, N.A.Z.; funding acquisition, A.V.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 21-66-00007 (genotyping of RW chickens, data processing, and analysis). Genotypes of WC chickens were produced with financial support of the Ministry of Science and Higher Education of the Russian Federation within theme 0445-2019-0024. The work of M.N.R. was funded by the Ministry of Science and Higher Education of the Russian Federation, grant agreement number 14.W03.31.0013.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and the LKEFRCAH ethical guidelines. Protocol No. 3/1 was approved by the LKEFRCAH Commission on the Ethics of Animal Experiments on 4 December 2019.
Informed Consent Statement
Not applicable.
Data Availability Statement
The genotyping data presented in this study can be shared with the third parties upon approval with the GWMAS Consortium.
Acknowledgments
We thank the USDA Chicken GWMAS Consortium, Cobb Vantress, and Hendrix Genetics for access to the developed 50K_CobbCons chicken array produced by Illumina Inc. for the GWMAS Consortium.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Weigend, S.; Romanov, M.N. Molekulare Charakterisierung genetischer Vielfalt beim Geflügel—Molecular characterization of genetic diversity in chicken. In Jahresbericht 1998; Bundesforschungsanstalt Für Landwirtschaft (FAL): Braunschweig, Germany, 1999; p. 66. [Google Scholar]
- Romanov, M.N.; Weigend, S. Using RAPD markers for assessment of genetic diversity in chickens. Arch. Geflügelkunde 2001, 65, 145–148. [Google Scholar]
- Weigend, S.; Romanov, M.N. Genetische Diversitätsanalysen bei Hühnern mit Hilfe molekularer Marker—Assessment of genetic diversity in chickens using molecular markers. In Jahresbericht 2001; Bundesforschungsanstalt Für Landwirtschaft (FAL): Braunschweig, Germany, 2002; p. 67. [Google Scholar]
- Weigend, S.; Romanov, M.N.; Rath, D. Methodologies to identify, evaluate and conserve poultry genetic resources. In Proceedings of the XXII World’s Poultry Congress & Exhibition: Participant List & Full Text CD + Book of Abstracts, Istanbul, Turkey, 8–13 June 2004; WPSA—Turkish Branch: Istanbul, Turkey, 2004; p. 84. [Google Scholar]
- Deniskova, T.E.; Dotsev, A.V.; Selionova, M.I.; Kunz, E.; Medugorac, I.; Reyer, H.; Wimmers, K.; Barbato, M.; Traspov, A.A.; Brem, G.; et al. Population structure and genetic diversity of twenty-five Russian sheep breeds based on whole-genome genotyping. Genet. Sel. Evol. 2018, 50, 1–16. [Google Scholar] [CrossRef]
- Zinovieva, N.A.; Dotsev, A.V.; Sermyagin, A.A.; Deniskova, T.E.; Abdelmanova, A.S.; Kharzinova, V.R.; Sölkner, J.; Reyer, H.; Wimmers, K.; Brem, G. Selection signatures in two oldest Russian native cattle breeds revealed using high-density single nucleotide polymorphism analysis. PLoS ONE 2020, 15, e0242200. [Google Scholar] [CrossRef]
- Zinovieva, N.A.; Sheiko, I.P.; Dotsev, A.V.; Sheiko, R.I.; Mikhailova, M.E.; Sermyagin, A.A.; Abdelmanova, A.S.; Kharzinova, V.R.; Reyer, H.; Wimmers, K.; et al. Genome-wide SNP analysis clearly distinguished the Belarusian Red cattle from other European cattle breeds. Anim. Genet. 2021. [Google Scholar] [CrossRef]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- Moiseyeva, I.G.; Romanov, M.N.; Nikiforov, A.A.; Sevastyanova, A.A.; Semyenova, S.K. Evolutionary relationships of Red Jungle Fowl and chicken breeds. Genet. Sel. Evol. 2003, 35, 403–423. [Google Scholar] [CrossRef]
- Paronyan, I.A.; Yurchenko, O.P. Domestic fowl. In Animal Genetic Resources of the USSR; Dmitriev, N.G., Ernst, L.K., Eds.; Food and Agriculture Organization of the United Nations: Rome, Italy, 1989; FAO Animal Production and Health Paper 65, Chapter 13; pp. 437–468. [Google Scholar]
- Sokolova, A.N. Genetic and Selection Methods of Creation of a Chicken Population with an Increased Resistance to Neoplasms: Author’s Abstract. Dissertation of the Doctor of Agricultural Sciences; RRIFAGB: Pushkin, St. Petersburg, Russia, 1999; 56p. [Google Scholar]
- Dementeva, N.V.; Romanov, M.N.; Kudinov, A.A.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Terletsky, V.P.; Fedorova, E.S.; Nikitkina, E.V.; Plemyashov, K.V. Studying the structure of a gene pool population of the Russian White chicken breed by genome-wide SNP scan. Agric. Biol. 2017, 52, 1166–1174. [Google Scholar] [CrossRef]
- Sokolova, A.N. Selection as a function of thermoregulation productivity. In Proceedings of the 13th World’s Poultry Science Congress, Kiev, Ukraine, 15–21 August 1966; Kolos: Moscow, Russia, 1966; pp. 151–155. [Google Scholar]
- Hutt, F.B. Snow-white down in the chick. J. Hered. 1951, 42, 117–120. [Google Scholar] [CrossRef]
- Kudinov, A.A.; Dementieva, N.V.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Fedorova, E.S.; Larkina, T.A.; Mishina, A.I.; Plemyashov, K.V.; Griffin, D.K.; Romanov, M.N. Genome-wide association studies targeting the yield of extraembryonic fluid and production traits in Russian White chickens. BMC Genom. 2019, 20, 270. [Google Scholar] [CrossRef] [PubMed]
- Dementieva, N.V.; Fedorova, E.S.; Krutikova, A.A.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Pleshanov, N.V.; Smaragdov, M.G.; Kudinov, A.A.; Terletsky, V.P.; Romanov, M.N. Genetic variability of indels in the prolactin and dopamine receptor D2 genes and their association with the yield of allanto-amniotic fluid in Russian White laying hens. Tarım Bilim. Derg.—J. Agric. Sci. 2020, 26, 373–379. [Google Scholar] [CrossRef]
- Dementieva, N.V.; Kudinov, A.A.; Larkina, T.A.; Mitrofanova, O.V.; Dysin, A.P.; Terletsky, V.P.; Tyshchenko, V.I.; Griffin, D.K.; Romanov, M.N. Genetic variability in local and imported germplasm chicken populations as revealed by analyzing runs of homozygosity. Animals 2020, 10, 1887. [Google Scholar] [CrossRef]
- Dementieva, N.V.; Mitrofanova, O.V.; Dysin, A.P.; Kudinov, A.A.; Stanishevskaya, O.I.; Larkina, T.A.; Plemyashov, K.V.; Griffin, D.K.; Romanov, M.N.; Smaragdov, M.G. Assessing the effects of rare alleles and linkage disequilibrium on estimates of genetic diversity in the chicken populations. Animal 2021, 15, 100171. [Google Scholar] [CrossRef]
- Stanishevskaya, O.I.; Fedorova, E.S. Comparative evaluation of the peculiarities of stress reactivity of the Russian white breed chicken with sw+ mutation and Amrox in hypothermia conditions during embryonal and early postnatal periods of ontogenesis. Agric. Biol. [Sel’skokhozyaistvennaya Biol.] 2019, 54, 1135–1143. [Google Scholar] [CrossRef]
- Stanishevskaya, O.I.; Fedorova, E.S. Dosed exposure to low temperature as a breeding background in the selection of gene pool breeds of chickens for viral vaccines production. Open Agric. J. 2020, 14, 345–351. [Google Scholar] [CrossRef]
- Mitrofanova, O.V.; Dementieva, N.V.; Fedorova, E.S.; Pozovnikova, M.V.; Tyshchenko, V.I.; Shcherbakov, Y.S.; Plemyashov, K.V. Assessment of variability of egg production traits based on analysis of SNP markers and search for traces of selection in the genome of Russian white chickens. Ekol. Genet. 2020, 18, 423–432. [Google Scholar] [CrossRef]
- Lee, M.O.; Romanov, M.N.; Plemyashov, K.V.; Dementieva, N.V.; Mitrofanova, O.V.; Barkova, O.Y.; Womack, J.E. Haplotype structure and copy number polymorphism of the beta-defensin 7 genes in diverse chicken breeds. Anim. Genet. 2017, 48, 490–492. [Google Scholar] [CrossRef]
- Crawford, R.D. (Ed.) Poultry Breeding and Genetics; Elsevier: New York, NY, USA, 1990. [Google Scholar]
- Julian, R.J. Rapid growth problems: Ascites and skeletal deformities in broilers. Poult. Sci. 1998, 77, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, E.; Bruggeman, V.; Barbato, G.F.; Buyse, J. Growth and reproduction problems associated with selection for increased broiler meat production. In Poultry Genetics, Breeding and Biotechnology; Muir, W.M., Aggrey, S.E., Eds.; CABI Publishing: Wallingford, UK, 2003; pp. 13–28. [Google Scholar] [CrossRef]
- Song, B.; Tang, D.; Yan, S.; Fan, H.; Li, G.; Shahid, M.S.; Mahmood, T.; Guo, Y. Effects of age on immune function in broiler chickens. J. Anim. Sci. Biotechnol. 2021, 12, 42. [Google Scholar] [CrossRef]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef]
- Fariello, M.I.; Boitard, S.; Naya, H.; SanCristobal, M.; Servin, B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 2013, 193, 929–941. [Google Scholar] [CrossRef]
- Bonhomme, M.; Chevalet, C.; Servin, B.; Boitard, S.; Abdallah, J.; Blott, S.; Sancristobal, M. Detecting selection in population trees: The Lewontin and Krakauer test extended. Genetics 2010, 186, 241–262. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2016, 48, 255–271. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Cadzow, M.; Boocock, J.; Nguyen, H.T.; Wilcox, P.; Merriman, T.R.; Black, M.A. A bioinformatics workflow for detecting signatures of selection in genomic data. Front. Genet. 2014, 5, 293. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ding, X.; Qanbari, S.; Weigend, S.; Zhang, Q.; Simianer, H. Properties of different selection signature statistics and a new strategy for combining them. Heredity 2015, 115, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.-J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.S.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef]
- Qanbari, S.; Strom, T.M.; Haberer, G.; Weigend, S.; Gheyas, A.A.; Turner, F.; Burt, D.W.; Preisinger, R.; Gianola, D.; Simianer, H. A high resolution genome-wide scan for significant selective sweeps: An application to pooled sequence data in laying chickens. PLoS ONE 2012, 7, e49525. [Google Scholar] [CrossRef]
- Elferink, M.G.; Megens, H.-J.; Vereijken, A.; Hu, X.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Signatures of selection in the genomes of commercial and non-commercial chicken breeds. PLoS ONE 2012, 7, e32720. [Google Scholar] [CrossRef] [PubMed]
- Qanbari, S.; Rubin, C.J.; Maqbool, K.; Weigend, S.; Weigend, A.; Geibel, J.; Kerje, S.; Wurmser, C.; Peterson, A.T.; Brisbin, I.L.; et al. Genetics of adaptation in modern chicken. PLoS Genet. 2019, 15, e1007989. [Google Scholar] [CrossRef]
- Talebi, R.; Szmatoła, T.; Mészáros, G.; Qanbari, S. Runs of homozygosity in modern chicken revealed by sequence data. G3 2020, 10, 4615–4623. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2018. Available online: https://www.R-project.org/ (accessed on 8 May 2021).
- Fan, J.-B.; Oliphant, A.; Shen, R.; Kermani, B.G.; Garcia, F.; Gunderson, K.L.; Hansen, M.; Steemers, F.; Butler, S.L.; Deloukas, P.; et al. Highly parallel SNP genotyping. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Genome Reference Consortium Chicken Build 6a. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000002315.6 (accessed on 1 May 2021).
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodohl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Francis, R.M. POPHELPER: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Iso-Touru, T.; Tapio, M.; Vilkki, J.; Kiseleva, T.; Ammosov, I.; Ivanova, Z.; Popov, R.; Ozerov, M.; Kantanen, J. Genetic diversity and genomic signatures of selection among cattle breeds from Siberia, eastern and northern Europe. Anim. Genet. 2016, 47, 647–657. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; Cristobal, M.S.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. International Sheep Genomics Consortium Members. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsam, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2014, 46, 110–121. [Google Scholar] [CrossRef]
- Biscarini, F.; Paolo Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R Package Version 0.9.5. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 8 May 2021).
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef]
- Wall, J.D.; Pritchard, J.K. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 2003, 4, 587–597. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.D.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Grilz-Seger, G.; Neuditschko, M.; Ricard, A.; Velie, B.; Lindgren, G.; Mesarič, M.; Cotman, M.; Horna, M.; Dobretsberger, M.; Brem, G.; et al. Genome-wide homozygosity patterns and evidence for selection in a set of European and Near Eastern horse breeds. Genes 2019, 10, 491. [Google Scholar] [CrossRef]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef]
- The Chicken Quantitative Trait Locus (QTL) Database (Chicken QTLdb). Available online: https://www.animalgenome.org/cgi-bin/QTLdb/GG/index (accessed on 17 May 2021).
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- O’Connor, R.E.; Romanov, M.N.; Kiazim, L.G.; Barrett, P.M.; Farré, M.; Damas, J.; Ferguson-Smith, M.; Valenzuela, N.; Larkin, D.M.; Griffin, D.K. Reconstruction of the diapsid ancestral genome permits chromosome evolution tracing in avian and non-avian dinosaurs. Nat. Commun. 2018, 9, 1883. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Lee, W.R.; Abasht, B. Detection of genomic signatures of recent selection in commercial broiler chickens. BMC Genet. 2016, 17, 122. [Google Scholar] [CrossRef]
- Lillie, M.; Sheng, Z.Y.; Honaker, C.F.; Dorshorst, B.J.; Ashwell, C.M.; Siegel, P.B.; Carlborg, Ö. Genome-wide standing variation facilitates long-term response to bidirectional selection for antibody response in chickens. BMC Genom. 2017, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, L.; Su, Z.; Zhu, M.; Li, W.; Wu, K.; Deng, X. Genome-wide scan and analysis of positive selective signatures in Dwarf Brown-egg Layers and Silky Fowl chickens. Poult. Sci. 2017, 96, 4158–4171. [Google Scholar] [CrossRef]
- Li, W.; Liu, R.; Zheng, M.; Feng, F.; Liu, D.; Guo, Y.; Zhao, G.; Wen, J. New insights into the associations among feed efficiency, metabolizable efficiency traits and related QTL regions in broiler chickens. J. Anim. Sci. Biotechnol. 2020, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- de Simoni Gouveia, J.J.; da Silva, M.V.; Paiva, S.R.; de Oliveira, S.M. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014, 37, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.; Lee, W.; Jeon, S.; Yoon, J.; Dessie, T.; Hanotte, O.; Mwai, O.A.; Kemp, S.; Cho, S.; Oh, S.J.; et al. Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm. Genome 2017, 28, 528–541. [Google Scholar] [CrossRef]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 2019, 20 (Suppl. 3), 294. [Google Scholar] [CrossRef]
- Deniskova, T.E.; Petrov, S.N.; Sermyagin, A.A.; Dosev, A.V.; Fornara, M.S.; Reyer, H.; Wimmers, K.; Bagirov, V.A.; Brem, G.; Zinovieva, N.A. A search for genomic variants associated with body weight in sheep based on high-density SNP genotypes analysis. Agric. Biol. [Sel’skokhozyaistvennaya Biol.] 2021, 56, 279–291. [Google Scholar] [CrossRef]
- François, O.; Martins, H.; Caye, K.; Schoville, S.D. Controlling false discoveries in genome scans for selection. Mol. Ecol. 2016, 25, 454–469. [Google Scholar] [CrossRef]
- Beverdam, A.; Meijlink, F. Expression patterns of group-I aristaless-related genes during craniofacial and limb development. Mech. Dev. 2001, 107, 163–167. [Google Scholar] [CrossRef]
- Kamaid, A.; Giráldez, F. Btg1 and Btg2 gene expression during early chick development. Dev. Dyn. 2008, 237, 2158–2169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wu, P.; Zhou, K.; He, M.; Zhang, X.; Qiu, C.; Li, T.; Zhang, T.; Xie, K.; Dai, G.; et al. Study on the transcriptome for breast muscle of chickens and the function of key gene RAC2 on fibroblasts proliferation. BMC Genom. 2021, 22, 157. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, A.A.; Sazanova, A.L.; Tzareva, V.A.; Kozyreva, A.A.; Smirnov, A.F.; Romanov, M.N.; Price, J.A.; Dodgson, J.B. Refined localization of the chicken KITLG, MGP and TYR genes on GGA1 by FISH mapping using BACs. Anim. Genet. 2004, 35, 148–150. [Google Scholar] [CrossRef]
- Zhang, Q.; Gou, W.; Wang, X.; Zhang, Y.; Ma, J.; Zhang, H.; Zhang, Y.; Zhang, H. Genome resequencing identifies unique adaptations of Tibetan chickens to hypoxia and high-dose ultraviolet radiation in high-altitude environments. Genome Biol. Evol. 2016, 8, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Li, D.; Wang, Y.; Zhu, Q. Whole-genome resequencing analysis of Pengxian Yellow Chicken to identify genome-wide SNPs and signatures of selection. 3 Biotech 2019, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.M.; Erf, G.F.; Rowland, K.C.; Kong, B.W. Genome resequencing and bioinformatic analysis of SNP containing candidate genes in the autoimmune vitiligo Smyth line chicken model. BMC Genom. 2014, 15, 707. [Google Scholar] [CrossRef]
- Li, S.; Zhi, L.; Liu, Y.; Shen, J.; Liu, L.; Yao, J.; Yang, X. Effect of in ovo feeding of folic acid on the folate metabolism, immune function and epigenetic modification of immune effector molecules of broiler. Br. J. Nutr. 2016, 115, 411–421. [Google Scholar] [CrossRef]
- Han, W.; Xue, Q.; Li, G.; Yin, J.; Zhang, H.; Zhu, Y.; Xing, W.; Cao, Y.; Su, Y.; Wang, K.; et al. Genome-wide analysis of the role of DNA methylation in inbreeding depression of reproduction in Langshan chicken. Genomics 2020, 112, 2677–2687. [Google Scholar] [CrossRef]
- Xue, Q.; Li, G.; Cao, Y.; Yin, J.; Zhu, Y.; Zhang, H.; Zhou, C.; Shen, H.; Dou, X.; Su, Y.; et al. Identification of genes involved in inbreeding depression of reproduction in Langshan chickens. Anim. Biosci. 2021, 34, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Liu, Y.; Zhang, M.; Shan, Y.; Ji, G.; Ju, X.; Zou, J.; Shu, J. Identifying signatures of selection related to comb development. J. Poult. Sci. 2021, 58, 5–11. [Google Scholar] [CrossRef]
- Moreira, G.C.M.; Salvian, M.; Boschiero, C.; Cesar, A.S.M.; Reecy, J.M.; Godoy, T.F.; Ledur, M.C.; Garrick, D.; Mourão, G.B.; Coutinho, L.L. Genome-wide association scan for QTL and their positional candidate genes associated with internal organ traits in chickens. BMC Genom. 2019, 20, 669. [Google Scholar] [CrossRef]
- Yang, S.; Shi, Z.; Ou, X.; Liu, G. Whole-genome resequencing reveals genetic indels of feathered-leg traits in domestic chickens. J. Genet. 2019, 98, 47. [Google Scholar] [CrossRef]
- Shah, T.M.; Patel, N.V.; Patel, A.B.; Upadhyay, M.R.; Mohapatra, A.; Singh, K.M.; Deshpande, S.D.; Joshi, C.G. A genome-wide approach to screen for genetic variants in broilers (Gallus gallus) with divergent feed conversion ratio. Mol. Genet. Genom. 2016, 291, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhang, X.; Zhang, G.; Chen, F.; He, M.; Zhang, T.; Wang, J.; Xie, K.; Dai, G. Transcriptome for the breast muscle of Jinghai yellow chicken at early growth stages. PeerJ 2020, 8, e8950. [Google Scholar] [CrossRef] [PubMed]
- Qanbari, S.; Seidel, M.; Strom, T.-M.; Mayer, K.F.; Preisinger, R.; Simianer, H. Parallel selection revealed by population sequencing in chicken. Genome Biol. Evol. 2015, 7, 3299–3306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, X.; Otecko, N.O.; Peng, M.; Weng, Z.; Li, W.; Chen, J.; Zhong, M.; Zhong, F.; Jin, S.; Geng, Z.; et al. Genome-wide genetic structure and selection signatures for color in 10 traditional Chinese yellow-feathered chicken breeds. BMC Genom. 2020, 21, 316. [Google Scholar] [CrossRef] [PubMed]
- de Freitas Dionizio, A.; de Souza Khatlab, A.; Alcalde, C.R.; Gasparino, E.; Feihrmann, A.C. Supplementation with free methionine or methionine dipeptide improves meat quality in broilers exposed to heat stress. J. Food Sci. Technol. 2021, 58, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Vezyri, E.; Mikrou, A.; Athanassiadou, A.; Zarkadis, I.K. Molecular cloning and expression of Aven gene in chicken. Protein J. 2011, 30, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, B.I.; Voet, T.; De Smet, L.; Vermeesch, J.R.; Devriendt, K.; Fryns, J.-P.; Debeer, P. Genomic rearrangements of the GREM1-FMN1 locus cause oligosyndactyly, radio-ulnar synostosis, hearing loss, renal defects syndrome and Cenani-Lenz-like non-syndromic oligosyndactyly. J. Med. Genet. 2010, 47, 569–574. [Google Scholar] [CrossRef]
- Jahejo, A.R.; Zhang, D.; Niu, S.; Mangi, R.A.; Khan, A.; Qadir, M.F.; Khan, A.; Chen, H.-C.; Tian, W.-X. Transcriptome-based screening of intracellular pathways and angiogenesis related genes at different stages of thiram induced tibial lesions in broiler chickens. BMC Genom. 2020, 21, 50. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, L.; Li, Y.; Xu, Z.; Zhang, X.; Yu, J.; Cao, Z.; Luan, P. Genome-wide association study for plasma very low-density lipoprotein concentration in chicken. J. Anim. Breed. Genet. 2019, 136, 351–361. [Google Scholar] [CrossRef]
- El Moujahid, E.M.; Chen, S.; Jin, S.; Lu, Y.; Zhang, D.; Ji, C.; Yang, N. Association of leptin receptor gene polymorphisms with growth and feed efficiency in meat-type chickens. Poult. Sci. 2014, 93, 1910–1915. [Google Scholar] [CrossRef]
- Lawal, R.A.; Al-Atiyat, R.M.; Aljumaah, R.S.; Silva, P.; Mwacharo, J.M.; Hanotte, O. Whole-genome resequencing of red junglefowl and indigenous village chicken reveal new insights on the genome dynamics of the species. Front. Genet. 2018, 9, 264. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Feng, C.; Zhao, Y.; Hu, X.; Li, N. Transcriptome analysis of comb and testis from Rose-comb Silky chicken (R1/R1) and Beijing Fatty wild type chicken (r/r). Poult. Sci. 2017, 96, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Elbeltagy, A.R.; Bertolini, F.; Fleming, D.S.; Van Goor, A.; Ashwell, C.M.; Schmidt, C.J.; Kugonza, D.R.; Lamont, S.J.; Rothschild, M.F. Natural selection footprints among African chicken breeds and village ecotypes. Front. Genet. 2019, 10, 376. [Google Scholar] [CrossRef]
- Tarsani, E.; Kranis, A.; Maniatis, G.; Avendano, S.; Hager-Theodorides, A.L.; Kominakis, A. Deciphering the mode of action and position of genetic variants impacting on egg number in broiler breeders. BMC Genom. 2020, 21, 512. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lu, J.; Yi, G.; Yuan, J.; Duan, Z.; Qu, L.; Xu, G.; Wang, K.; Yang, N. Promising loci and genes for yolk and ovary weight in chickens revealed by a genome-wide association study. PLoS ONE 2015, 10, e0137145. [Google Scholar] [CrossRef][Green Version]
- Obermajer, N.; Jevnikar, Z.; Doljak, B.; Kos, J. Role of cysteine cathepsins in matrix degradation and cell signalling. Connect. Tissue Res. 2008, 49, 193–196. [Google Scholar] [CrossRef]
- Bae, S.-M.; Lim, W.; Jeong, W.; Kim, J.; Bazer, F.W.; Song, G. Expression and regulation of avian cathepsin L in the oviduct during molting. Gen. Comp. Endocrinol. 2014, 204, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, A.A.; Sazanova, A.L.; Stekolnikova, V.A.; Kozyreva, A.A.; Smirnov, A.F.; Romanov, M.N.; Dodgson, J.B. Chromosomal localization of CTSL: Expanding of the region of evolutionary conservation between GGAZ and HSA9. Anim. Genet. 2004, 35, 260. [Google Scholar] [CrossRef]
- Zou, A.; Nadeau, K.; Wang, P.W.; Lee, J.Y.; Guttman, D.S.; Sharif, S.; Korver, D.R.; Brumell, J.H.; Parkinson, J. Accumulation of genetic variants associated with immunity in the selective breeding of broilers. BMC Genet. 2020, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Guo, Y.Q.; Xie, K.J.; Li, Y.D.; Li, Z.W.; Wang, N.; Xiao, F.; Guo, H.S.; Li, H.; Wang, S.Z. A functional variant in the promoter region of IGF1 gene is associated with chicken abdominal fat deposition. Domest. Anim. Endocrinol. 2021, 75, 106584. [Google Scholar] [CrossRef]
- Kulibaba, R.A.; Tereshchenko, A.V. Transforming growth factor β1, pituitary-specific transcriptional factor 1 and insulin-like growth factor I gene polymorphisms in the population of the Poltava clay chicken breed: Association with productive traits. Agric. Sci. Pract. 2015, 2, 67–72. [Google Scholar] [CrossRef]
- Zhang, Y.; Gou, W.; Ma, J.; Zhang, H.; Zhang, Y.; Zhang, H. Genome methylation and regulatory functions for hypoxic adaptation in Tibetan chicken embryos. PeerJ 2017, 5, e3891. [Google Scholar] [CrossRef]
- Guo, X.; Wang, J.; Ma, C.; Wang, Z.; Chen, H.; Su, H.; Wan, Y.; Jiang, R. Genome-wide re-sequencing and transcriptome analysis reveal candidate genes associated with the pendulous comb phenotype in domestic chickens. Anim. Sci. J. 2020, 91, e13308. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, X.; Cui, H.; Liu, R.; Zhao, G.; Wen, J. Transcriptional insights into key genes and pathways controlling muscle lipid metabolism in broiler chickens. BMC Genom. 2019, 20, 863. [Google Scholar] [CrossRef]
- Ma, Y.; Gu, L.; Yang, L.; Sun, C.; Xie, S.; Fang, C.; Gong, Y.; Li, S. Identifying artificial selection signals in the chicken genome. PLoS ONE 2018, 13, e0196215. [Google Scholar] [CrossRef] [PubMed]
- Piórkowska, K.; Żukowski, K.; Połtowicz, K.; Nowak, J.; Ropka-Molik, K.; Derebecka, N.; Wesoły, J.; Wojtysiak, D. Identification of candidate genes and regulatory factors related to growth rate through hypothalamus transcriptome analyses in broiler chickens. BMC Genom. 2020, 21, 509. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Pu, Y.; Song, C.; Sheng, Z.; Hu, X. A quantitative trait locus on chromosome 2 was identified that accounts for a substantial proportion of phenotypic variance of the yellow plumage color in chicken. Poult. Sci. 2020, 99, 2902–2910. [Google Scholar] [CrossRef]
- Zhang, H.; Du, Z.-Q.; Dong, J.-Q.; Wang, H.-X.; Shi, H.-Y.; Wang, N.; Wang, S.-Z.; Li, H. Detection of genome-wide copy number variations in two chicken lines divergently selected for abdominal fat content. BMC Genom. 2014, 15, 517. [Google Scholar] [CrossRef]
- Dong, K.; Chang, S.; Xie, Q.; Black-Pyrkosz, A.; Zhang, H. Comparative transcriptomics of genetically divergent lines of chickens in response to Marek’s disease virus challenge at cytolytic phase. PLoS ONE 2017, 12, e0178923. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-T.; Lin, Y.-T.; Kuo, T.-F. Investigation of mRNA expression for secreted frizzled-related protein 2 (sFRP2) in chick embryos. J. Reprod. Dev. 2007, 53, 801–810. [Google Scholar] [CrossRef]
- Laptev, G.Y.; Filippova, V.A.; Kochish, I.I.; Yildirim, E.A.; Ilina, L.A.; Dubrovin, A.V.; Brazhnik, E.A.; Novikova, N.I.; Novikova, O.B.; Dmitrieva, M.E.; et al. Examination of the expression of immunity genes and bacterial profiles in the caecum of growing chickens infected with Salmonella Enteritidis and fed a phytobiotic. Animals 2019, 9, 615. [Google Scholar] [CrossRef] [PubMed]
- Grottesi, A.; Gabbianelli, F.; Valentini, A.; Chillemi, G. Structural and dynamic analysis of G558R mutation in chicken TSHR gene shows altered signal transduction and corroborates its role as a domestication gene. Anim. Genet. 2020, 51, 51–57. [Google Scholar] [CrossRef]
- Karlsson, A.-C.; Fallahshahroudi, A.; Johnsen, H.; Hagenblad, J.; Wright, D.; Andersson, L.; Jensen, P. A domestication related mutation in the thyroid stimulating hormone receptor gene (TSHR) modulates photoperiodic response and reproduction in chickens. Gen. Comp. Endocrinol. 2016, 228, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Sun, C.; Dou, T.; Yi, G.; Qu, L.; Qu, L.; Wang, K.; Yang, N. Identification of promising mutants associated with egg production traits revealed by genome-wide association study. PLoS ONE 2015, 10, e0140615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, D.; Zhai, Y.; Wang, Z.; Ma, X.; Zhang, D.; Li, G.; Han, R.; Jiang, R.; Li, Z.; et al. The landscape of DNA methylation associated with the transcriptomic network of intramuscular adipocytes generates insight into intramuscular fat deposition in chicken. Front. Cell Dev. Biol. 2020, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, F.-B.; Li, F.; Jiang, K.-R.; Li, D.-H.; Han, R.-L.; Li, Z.-J.; Jiang, R.-R.; Liu, X.-J.; Kang, X.-T.; et al. Genome-wide DNA methylation profiles reveal novel candidate genes associated with meat quality at different age stages in hens. Sci. Rep. 2017, 7, 45564. [Google Scholar] [CrossRef]
- Sazanov, A.A.; Sazanova, A.L.; Stekol’nikova, V.A.; Kozyreva, A.A.; Romanov, M.N.; Malewski, T.; Smirnov, A.F. Chromosomal localization of seven HSA3q13→q23 NotI linking clones on chicken microchromosomes: Orthology of GGA14 and GGA15 to a gene-rich region of HSA3. Cytogenet. Genome Res. 2005, 111, 128–133. [Google Scholar] [CrossRef]
- Sato, H.; Oshiumi, H.; Takaki, H.; Hikono, H.; Seya, T. Evolution of the DEAD box helicase family in chicken: Chickens have no DHX9 ortholog. Microbiol. Immunol. 2015, 59, 633–640. [Google Scholar] [CrossRef]
- Luo, W.; Xu, J.; Li, Z.; Xu, H.; Lin, S.; Wang, J.; Ouyang, H.; Nie, Q.; Zhang, X. Genome-wide association study and transcriptome analysis provide new insights into the white/red earlobe color formation in chicken. Cell. Physiol. Biochem. 2018, 46, 1768–1778. [Google Scholar] [CrossRef]
- Roly, Z.Y.; Godini, R.; Estermann, M.A.; Major, A.T.; Pocock, R.; Smith, C.A. Transcriptional landscape of the embryonic chicken Müllerian duct. BMC Genom. 2020, 21, 688. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Bruley, C.K.; Paton, I.R.; Dunn, I.; Jones, C.T.; Windsor, D.; Morrice, D.R.; Law, A.S.; Masabanda, J.; Sazanov, A.; et al. Differences in gene density on chicken macrochromosomes and microchromosomes. Anim. Genet. 2000, 31, 96–103. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).