
Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials & Methods
2.1. Experimental Animals and Sample Collections
2.2. RNA Extraction
2.3. Library Preparation and Illumina HiSeq 4000 Sequencing
2.4. Read Mapping and Transcriptome Assembly
2.5. Identification of circRNAs
2.6. Differential Expression Analysis and Functional Enrichment
2.7. Time-Series Analysis
2.8. Analysis of circRNAs Regulatory Network
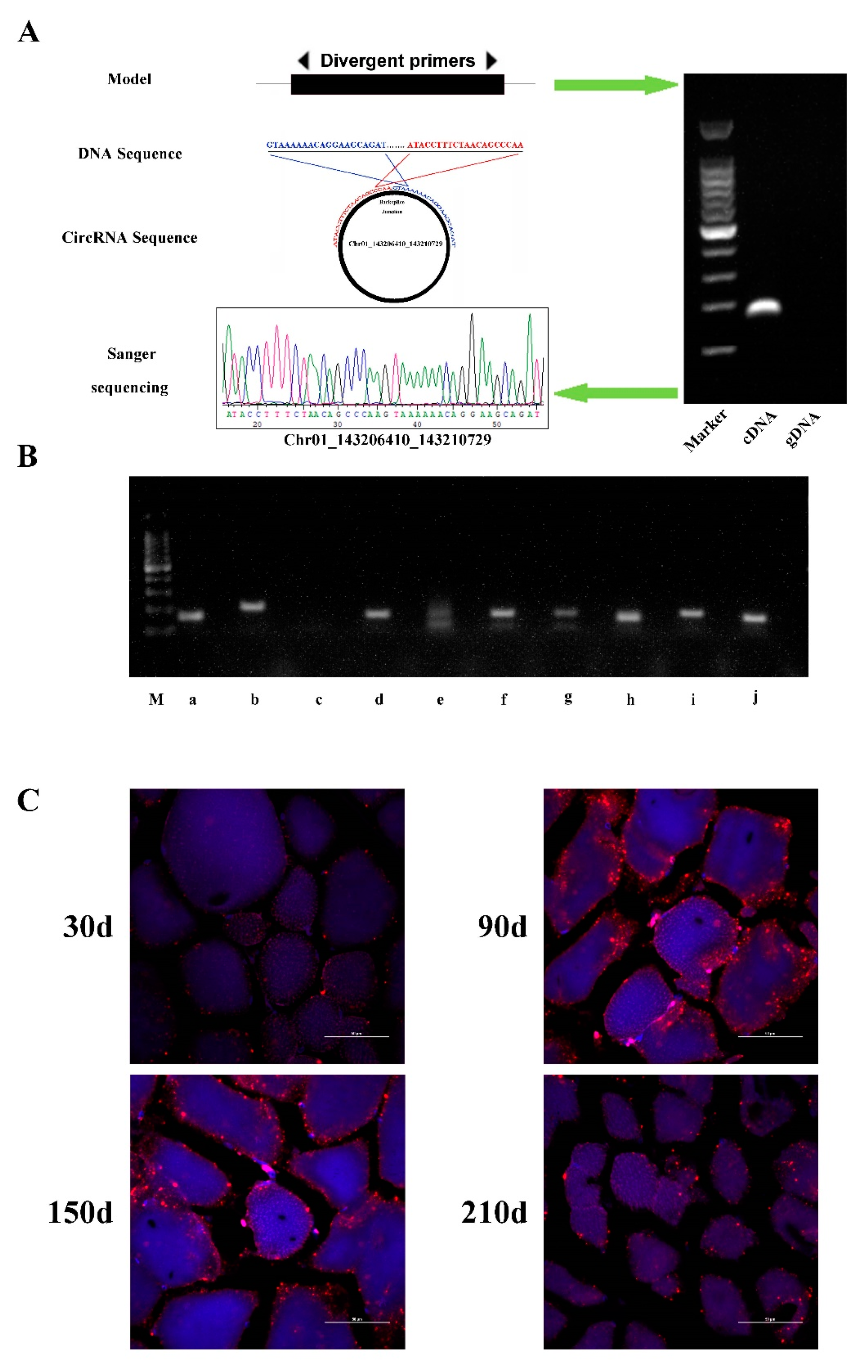
2.9. Validation of Expression by PCR
2.10. Fluorescence In Situ Hybridization (FISH)
2.11. Statistical Analysis
3. Results
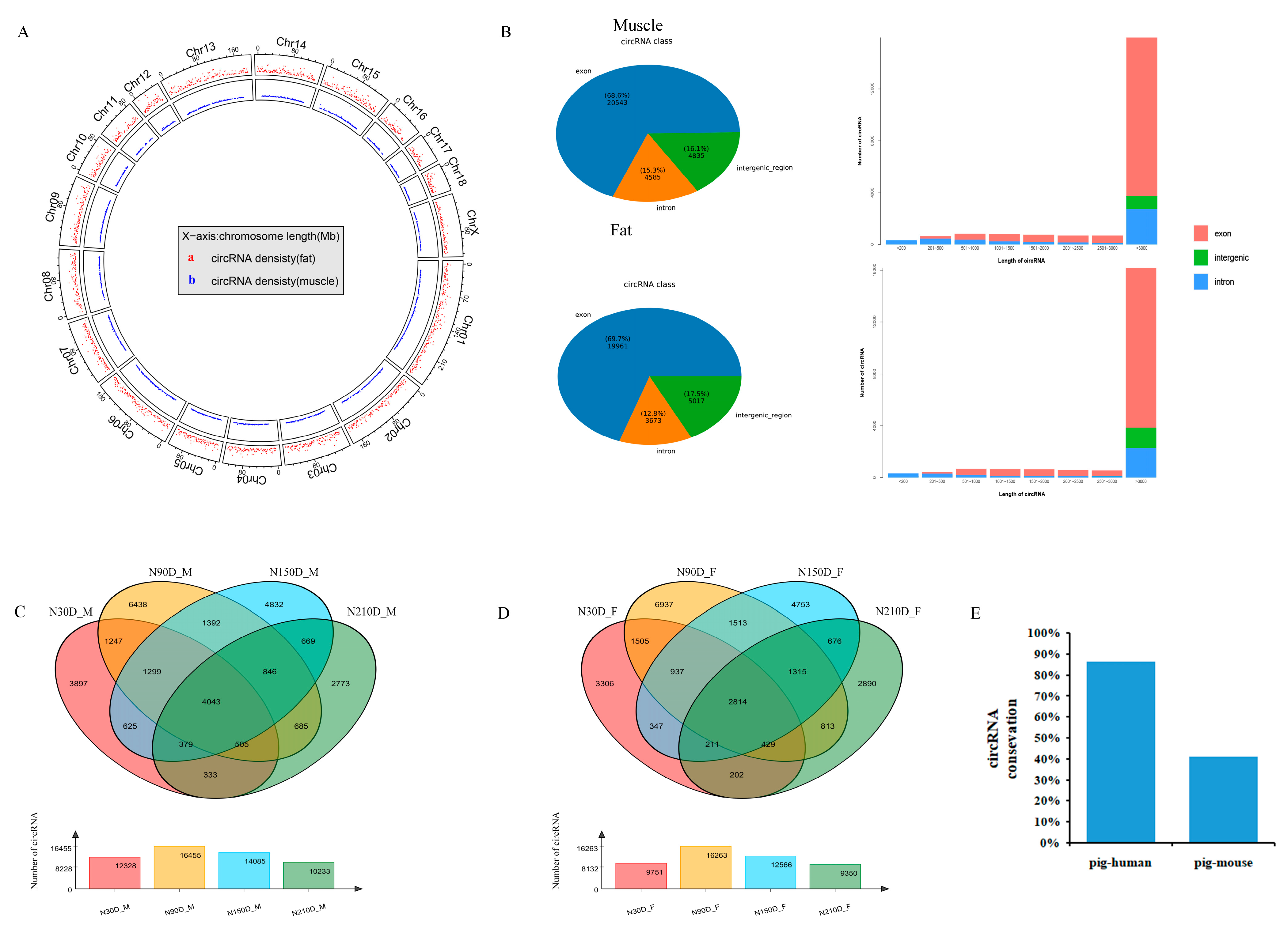
3.1. Identifications and Characteristics of circRNAs
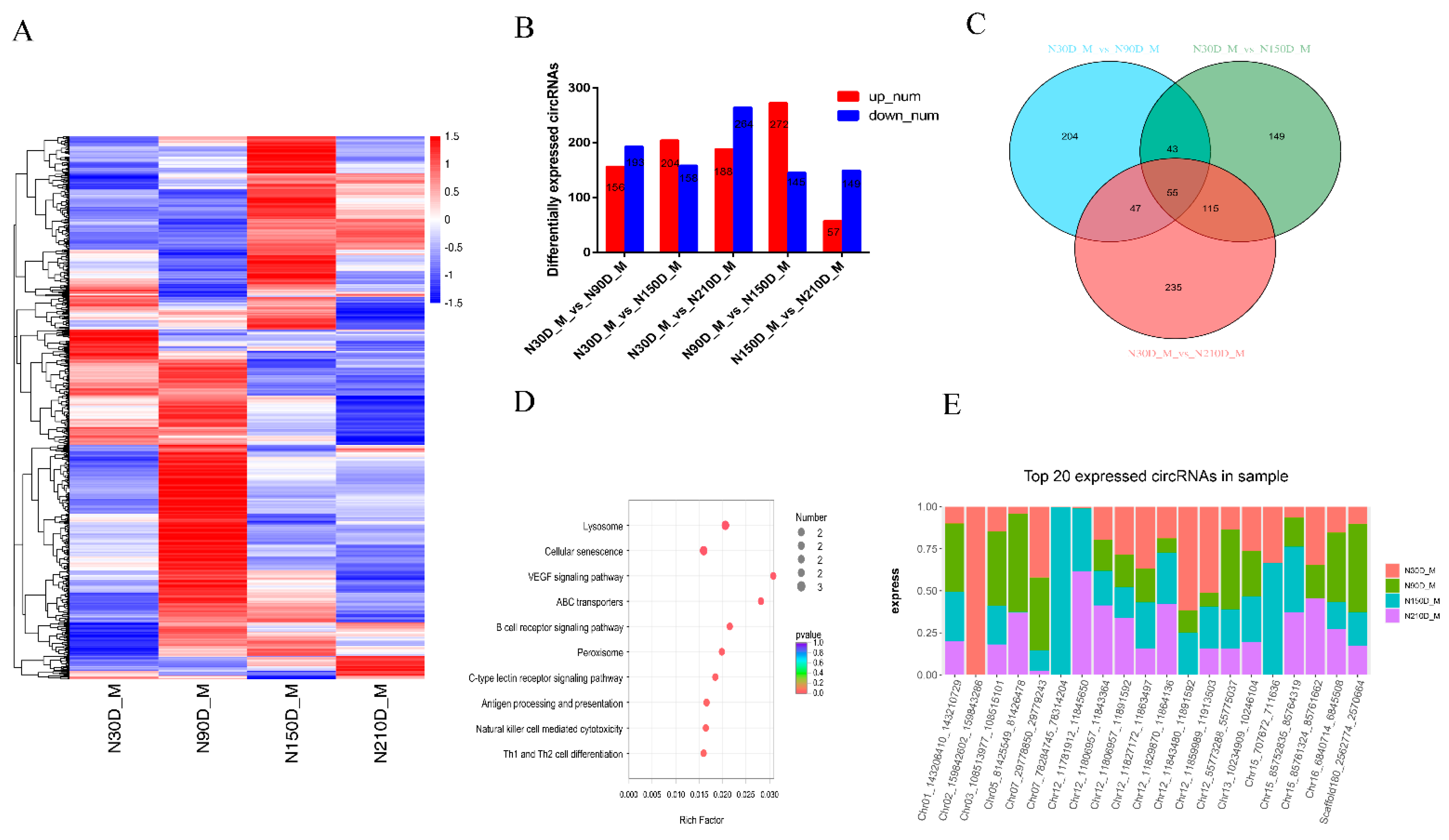
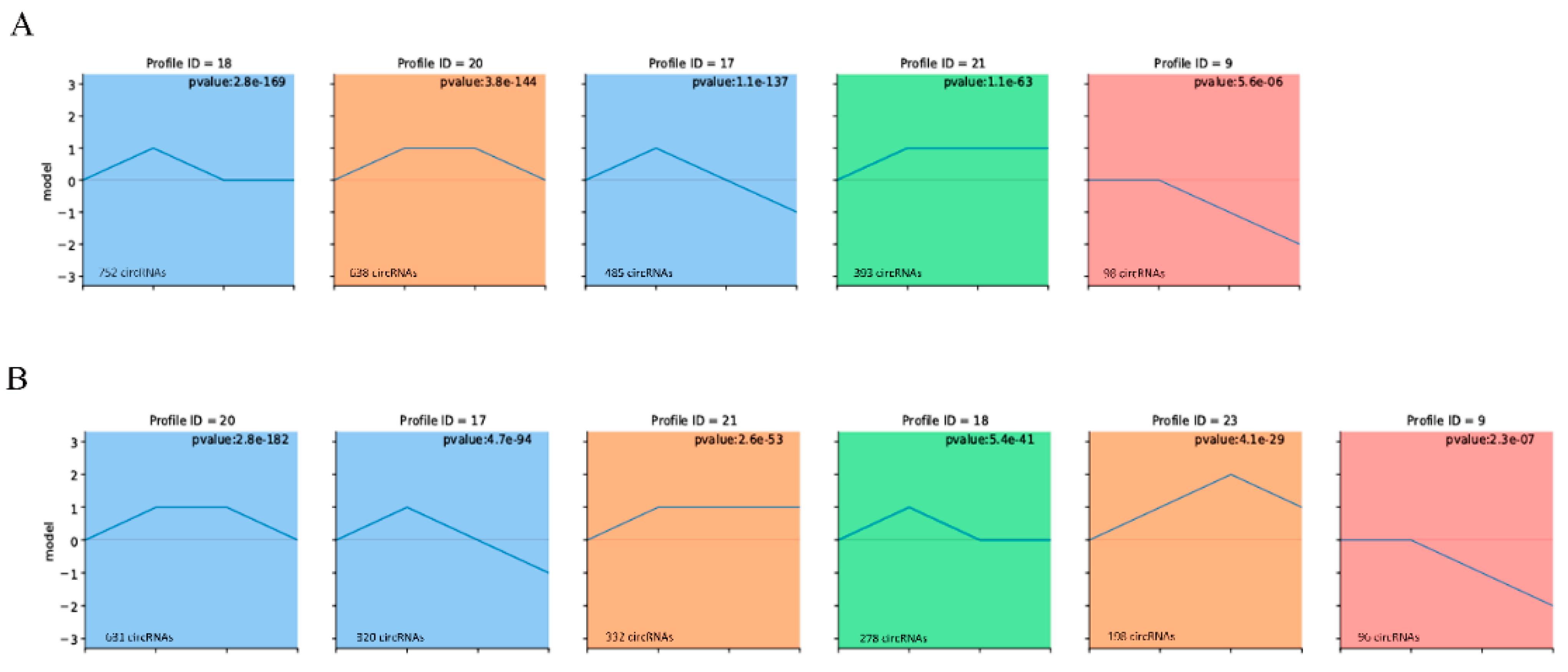
3.2. Expression Patterns of circRNAs during Muscle Growth
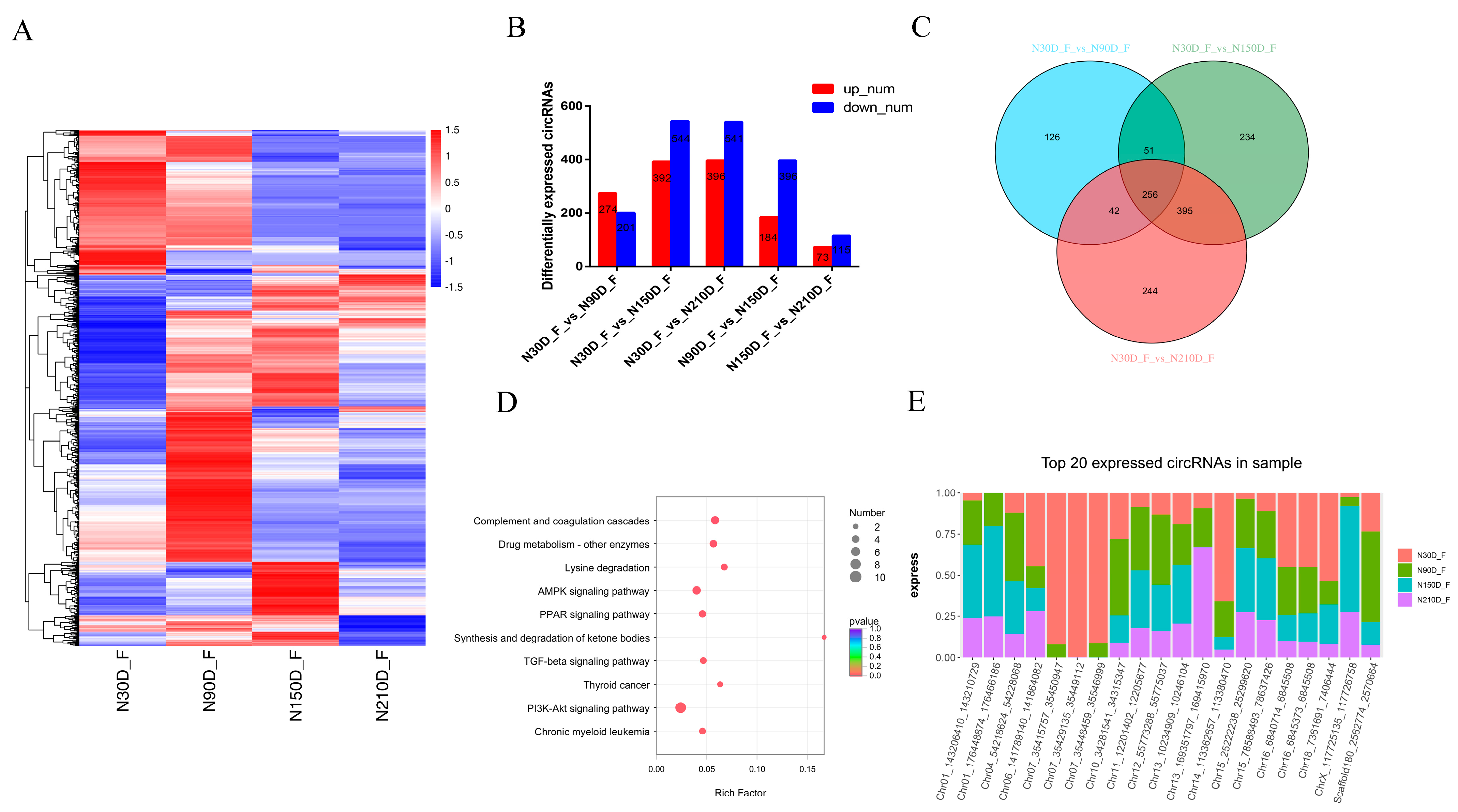
3.3. Expression Patterns of circRNAs in Adipose Tissue
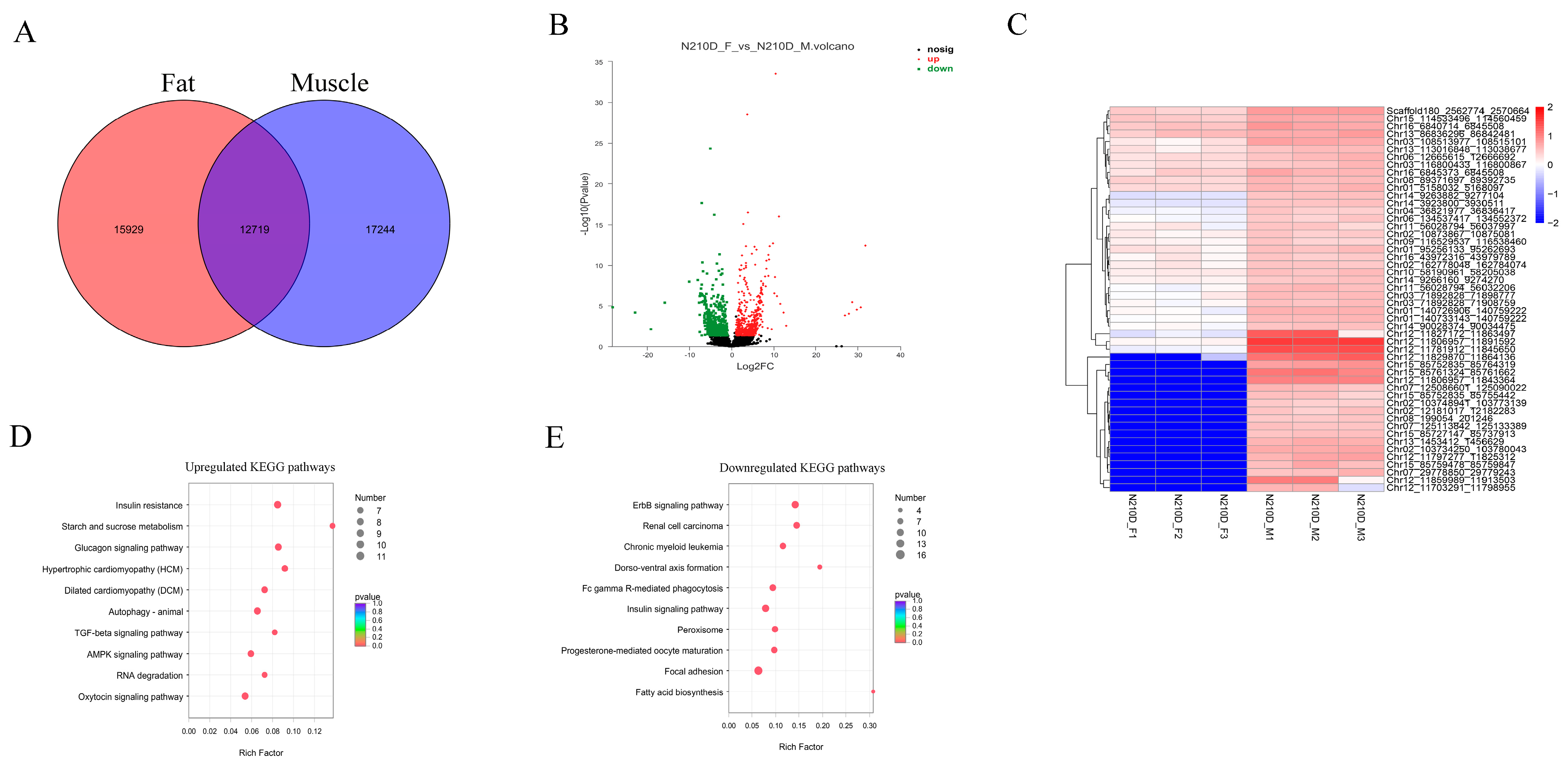
3.4. Differentially Expressed circRNAs between Muscle and Adipose Tissue
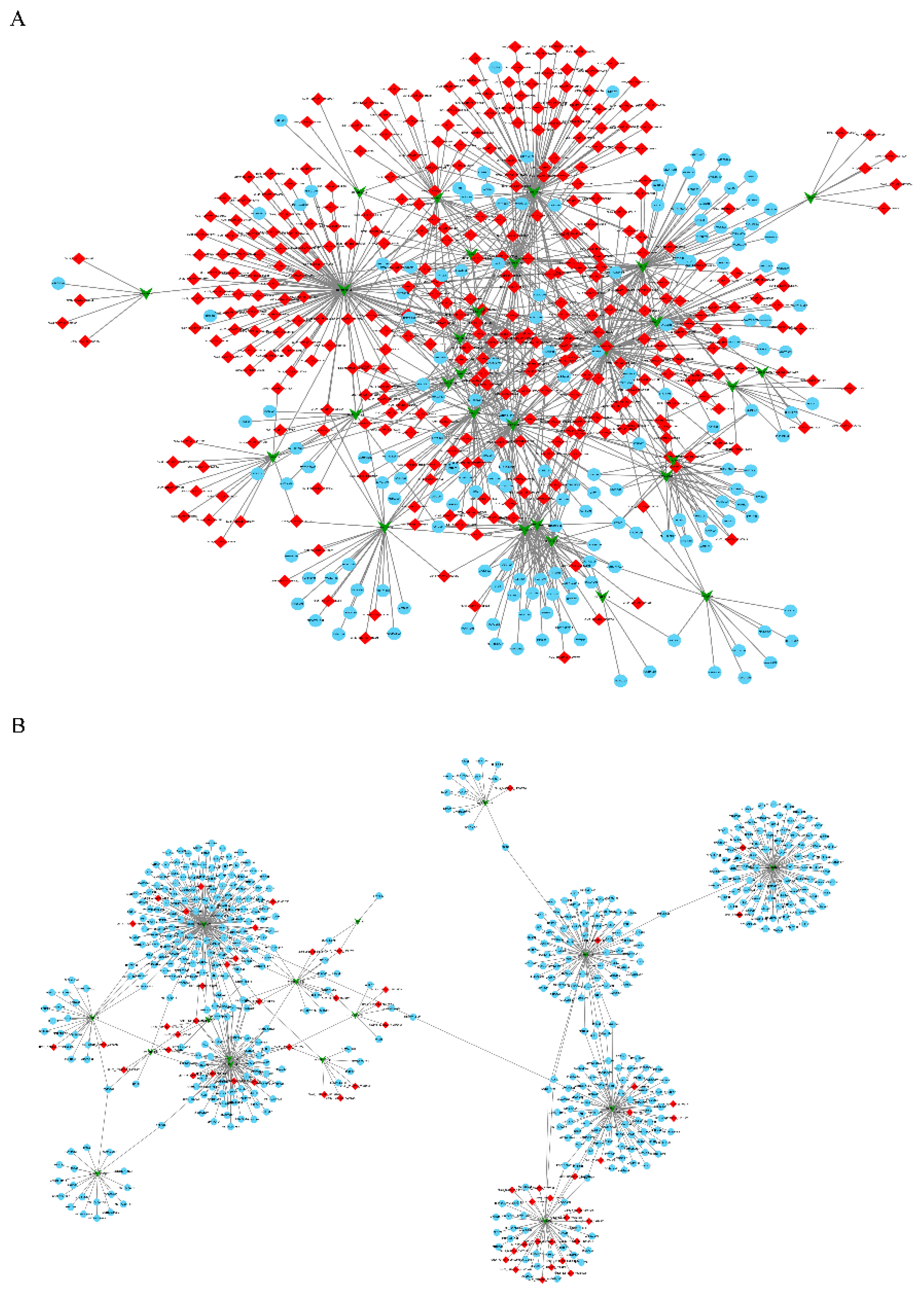
3.5. Construction of the circRNA-miRNA-mRNA Co-Expression Networks through Time-Series Analysis
3.6. Verification of Identified circRNAs
4. Discussion
4.1. Ningxiang Pig Circular RNAs
4.2. Circular RNAs in Muscle and Fat
4.3. Network of circRNA, miRNA and mRNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sun, J.; Xie, M.; Huang, Z.; Li, H.; Chen, T.; Sun, R.; Wang, J.; Xi, Q.Y.; Wu, T.; Zhang, Y. Integrated analysis of non-coding RNA and mRNA expression profiles of 2 pig breeds differing in muscle traits. J. Anim. Sci. 2017, 95, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive analysis of differentially expressed mRNA, lncRNA and circRNA and their ceRNA networks in the longissimus dorsi muscle of two different pig breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef]
- Han, P.; Li, P.; Zhou, W.; Fan, L.; Wang, B.; Liu, H.; Gao, C.; Du, T.; Pu, G.; Wu, C.; et al. Effects of various levels of dietary fiber on carcass traits, meat quality and myosin heavy chain I, IIa, IIx and IIb expression in muscles in Erhualian and large white pigs. Meat Sci. 2020, 169, 108160. [Google Scholar] [CrossRef]
- Jiang, Q.M.; Li, C.Y.; Yu, Y.N.; Xing, Y.T.; Xiao, D.F.; Zhang, B. Comparison of fatty acid profile of three adipose tissues in Ningxiang pigs. Anim. Nutr. 2018, 4, 256–259. [Google Scholar] [CrossRef]
- He, Q.H.; Ren, P.P.; Kong, X.F.; Wu, Y.N.; Wu, G.Y.; Li, P.; Hao, F.H.; Tang, H.R.; Blachier, F.; Yin, Y.L. Comparison of serum metabolite compositions between obese and lean growing pigs using an NMR-based metabonomic approach. J. Nutr. Biochem. 2012, 23, 133–139. [Google Scholar] [CrossRef]
- Schmitz, G.; Ecker, J. The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid Res. 2008, 47, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, P.; Davidson, N.C.; Schmidt, E.B.; Calder, P.C. Cardiovascular effects of marine omega-3 fatty acids. Lancet 2010, 376, 540–550. [Google Scholar] [CrossRef]
- Gerfault, V.; Louveau, I.; Mourot, J.; Le Dividich, J. Lipogenic enzyme activities in subcutaneous adipose tissue and skeletal muscle from neonatal pigs consuming maternal or formula milk. Reprod. Nutr. Dev. 2000, 40, 103–112. [Google Scholar] [CrossRef]
- Boone, C.; Mourot, J.; Gregoire, F.; Remacle, C. The adipose conversion process: Regulation by extracellular and intracellular factors. Reprod. Nutr. Dev. 2000, 40, 325–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kershaw, E.E.; Hamm, J.K.; Verhagen, L.A.; Peroni, O.; Katic, M.; Flier, J.S. Adipose triglyceride lipase: Function, regulation by insulin, and comparison with adiponutrin. Diabetes 2006, 55, 148–157. [Google Scholar] [CrossRef]
- Amills, M.; Vidal, O.; Varona, L.; Tomas, A.; Gil, M.; Sanchez, A.; Noguera, J.L. Polymorphism of the pig 2,4-dienoyl CoA reductase 1 gene (DECR1) and its association with carcass and meat quality traits. J. Anim. Sci. 2005, 83, 493–498. [Google Scholar] [CrossRef]
- Vidal, O.; Varona, L.; Oliver, M.A.; Noguera, J.L.; Sànchez, A.; Amills, M. Malic enzyme 1 genotype is associated with backfat thickness and meat quality traits in pigs. Anim. Genet. 2006, 37, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Huang, W.L.; Zhang, X.X.; Xie, L.L.; Miao, X.Y. Identification and characterization of CircRNAs of two pig breeds as a new biomarker in metabolism-related diseases. Cell. Physiol. Biochem. 2018, 47, 2458–2470. [Google Scholar] [CrossRef]
- Kokta, T.A.; Dodson, M.V.; Gertler, A.; Hill, R.A. Intercellular signaling between adipose tissue and muscle tissue. Domest. Anim. Endocrinol. 2004, 27, 303–331. [Google Scholar] [CrossRef]
- Liu, C.D.; Shen, L.Y.; Du, J.J.; Wu, X.Q.; Luo, J.; Pu, Q.; Tan, Z.D.; Cheng, X.; Du, J.G.; Yang, Q.; et al. The effect of lipid metabolism-related genes on intramuscular fat content and fatty acid composition in multiple muscles. Anim. Prod. Sci. 2018, 58, 2003–2010. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Vijay, D.; Federowicz, S.; Pico, A.R.; Ferrin, T.E. CyAnimator: Simple animations of cytoscape networks. F1000Research 2015, 4, 482. [Google Scholar] [CrossRef]
- Liang, G.; Yang, Y.; Niu, G.; Tang, Z.; Li, K. Genome-wide profiling of Sus scrofa circular RNAs across nine organs and three developmental stages. DNA Res. 2017, 24, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef]
- Guo, J.U.; Agarwal, V.; Guo, H.L.; Bartel, D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef]
- Dang, Y.; Yan, L.; Hu, B.; Fan, X.; Ren, Y.; Li, R.; Lian, Y.; Yan, J.; Li, Q.; Zhang, Y.; et al. Tracing the expression of circular RNAs in human pre-implantation embryos. Genome Biol. 2016, 17, 130. [Google Scholar] [CrossRef]
- Cameron, N.D.; Enser, M.B. Fatty acid composition of lipid in Longissimus dorsi muscle of Duroc and British Landrace pigs and its relationship with eating quality. Meat Sci. 1991, 29, 295–307. [Google Scholar] [CrossRef]
- Delgado, G.L.; Gomez, C.S.; Rubio, L.M.; Capella, V.S.; Mendez, M.D.; Labastida, R.C. Fatty acid and triglyceride profiles of intramuscular and subcutaneous fat from fresh and dry-cured hams from Hairless Mexican Pigs. Meat Sci. 2002, 61, 61–65. [Google Scholar] [CrossRef]
- Martins, A.P.; Lopes, P.A.; Madeira, M.S.; Martins, S.V.; Santos, N.C.; Moura, T.F.; Prates, J.A.; Soveral, G. Differences in lipid deposition and adipose membrane biophysical properties from lean and obese pigs under dietary protein restriction. Biochem. Biophys. Res. Commun. 2012, 423, 170–175. [Google Scholar] [CrossRef]
- Kouba, M.; Enser, M.; Whittington, F.M.; Nute, G.R.; Wood, J.D. Effect of a high-linolenic acid diet on lipogenic enzyme activities, fatty acid composition, and meat quality in the growing pig. J. Anim. Sci. 2003, 81, 1967–1979. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, J.; Neumann-Haefelin, C.; Belz, U.; Kalisch, J.; Juretschke, H.P.; Stein, M.; Kleinschmidt, E.; Kramer, W.; Herling, A.W. Intramyocellular lipid and insulin resistance: A longitudinal in vivo 1H-spectroscopic study in Zucker diabetic fatty rats. Diabetes 2003, 52, 138–144. [Google Scholar] [CrossRef]
- Kitessa, S.M.; Abeywardena, M.Y. Lipid-induced insulin resistance in skeletal muscle: The chase for the culprit goes from total intramuscular fat to lipid intermediates, and finally to species of lipid intermediates. Nutrients 2016, 8, 466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, J.R.; Sun, C.H.; Andrews, E.; Chao, R.F.; Lin, F.M.; Weng, S.L.; Hsu, S.D.; Huang, C.C.; Cheng, C.; et al. CircNet: A database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res. 2016, 44, D209–D215. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Veno, M.T.; Hansen, T.B.; Veno, S.T.; Clausen, B.H.; Grebing, M.; Finsen, B.; Holm, I.E.; Kjems, J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015, 16, 245. [Google Scholar] [CrossRef]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs Reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Wu, W.; Hou, L.; Chen, H.; Zuo, B.; Xiong, Y.; Yang, J. Skeletal muscle-specific overexpression of PGC-1α induces fiber-type conversion through enhanced mitochondrial respiration and fatty acid oxidation in mice and pigs. Int. J. Biol. Sci. 2017, 13, 1152–1162. [Google Scholar] [CrossRef]
- Yang, H.; Xu, Z.Y.; Lei, M.G.; Li, F.E.; Deng, C.Y.; Xiong, Y.Z.; Zuo, B. Association of 3 polymorphisms in porcine troponin I genes (TNNI1 and TNNI2) with meat quality traits. J. Appl. Genet. 2010, 51, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.J.; Jin, J.P. TNNI1, TNNI2 and TNNI3, evolution, regulation, and protein structure-function relationships. Gene 2016, 576, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Xing, Y.P.; Muramatsu, Y.; Ohta, T.; Kose, H.; Zhou, H.M.; Yamada, T. Association of expression levels in skeletal muscle and a SNP in the MYBPC1 gene with growth-related trait in Japanese Black beef cattle. J. Genet. 2015, 94, 135–137. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, T.J.; Li, J.; Gao, Y.S.; Meng, F.G.; Yan, Y.B.; Zhou, H.M. Slow skeletal muscle myosin-binding protein-C (MyBPC1) mediates recruitment of muscle-type creatine kinase (CK) to myosin. Biochem. J. 2011, 436, 437–445. [Google Scholar] [CrossRef]
- Sasaki, Y.; Nagai, K.; Nagata, Y.; Doronbekov, K.; Nishimura, S.; Yoshioka, S.; Fujita, T.; Shiga, K.; Miyake, T.; Taniguchi, Y.; et al. Exploration of genes showing intramuscular fat deposition-associated expression changes in musculus longissimus muscle. Anim. Genet. 2006, 37, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Cao, X.K.; Dong, D.; Shen, X.M.; Cheng, J.; Jiang, R.; Yang, Z.X.; Peng, S.J.; Huang, Y.Z.; Lan, X.Y.; et al. Circular RNA TTN Acts As a miR-432 sponge to facilitate proliferation and differentiation of myoblasts via the IGF2/PI3K/AKT Signaling pathway. Mol. Ther.-Nucleic Acids 2019, 18, 966–980. [Google Scholar] [CrossRef]
- Freundt, J.K.; Linke, W.A. Titin as a force-generating muscle protein under regulatory control. J. Appl. Physiol. 2019, 126, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Liu, X.; Zhou, L.; Yang, J.; Yang, B.; Ma, H.; Xie, X.; Huang, Y.; Fang, S.; Xiao, S.; et al. Genome-wide association analysis reveals genetic loci and candidate genes for meat quality traits in Chinese Laiwu pigs. Mamm Genome 2015, 26, 181–190. [Google Scholar] [CrossRef]
- Zhou, S.L.; Li, M.Z.; Li, Q.H.; Guan, J.Q.; Li, X.W. Differential expression analysis of porcine MDH1, MDH2 and ME1 genes in adipose tissues. Genet. Mol. Res. 2012, 11, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Debril, M.B.; Dubuquoy, L.; Feige, J.N.; Wahli, W.; Desvergne, B.; Auwerx, J.; Gelman, L. Scaffold attachment factor B1 directly interacts with nuclear receptors in living cells and represses transcriptional activity. J. Mol. Endocrinol. 2005, 35, 503–517. [Google Scholar] [CrossRef]
- Omura, Y.; Nishio, Y.; Takemoto, T.; Ikeuchi, C.; Sekine, O.; Morino, K.; Maeno, Y.; Obata, T.; Ugi, S.; Maegawa, H.; et al. SAFB1, an RBMX-binding protein, is a newly identified regulator of hepatic SREBP-1c gene. BMB Rep. 2009, 42, 232–237. [Google Scholar] [CrossRef]
- Garee, J.P.; Oesterreich, S. SAFB1’s multiple functions in biological control-lots still to be done! J. Cell Biochem. 2010, 109, 312–319. [Google Scholar] [CrossRef]
- Chan, C.W.; Lee, Y.B.; Uney, J.; Flynn, A.; Tobias, J.H.; Norman, M. A novel member of the SAF (scaffold attachment factor)-box protein family inhibits gene expression and induces apoptosis. Biochem. J. 2007, 407, 355–362. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Peng, S.; Song, C.; Li, H.; Cao, X.; Ma, Y.; Wang, X.; Huang, Y.; Lan, X.; Lei, C.; Chaogetu, B.; et al. Circular RNA SNX29 sponges miR-744 to regulate proliferation and differentiation of myoblasts by activating the Wnt5a/Ca(2+) signaling pathway. Mol. Nucleic Acids 2019, 16, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Liu, H.T.; Li, Y.; Mao, R.; Yang, H.W.; Zhang, Y.C.; Zhang, Y.; Guo, P.S.; Zhan, D.F.; Zhang, T.T. Circular RNA SAMD4A controls adipogenesis in obesity through the miR-138-5p/EZH2 axis. Theranostics 2020, 10, 4705–4719. [Google Scholar] [CrossRef]
- Guo, X.Y.; Sun, F.; Chen, J.N.; Wang, Y.Q.; Pan, Q.; Fan, J.G. circRNA_0046366 inhibits hepatocellular steatosis by normalization of PPAR signaling. World J. Gastroenterol. 2018, 24, 323–337. [Google Scholar] [CrossRef]
- Walker, P.D.; Jarosz, P.A.; Bouhamdan, M.; MacKenzie, R.G. Effects of gender on locomotor sensitivity to amphetamine, body weight, and fat mass in regulator of G protein signaling 9 (RGS9) knockout mice. Physiol. Behav. 2015, 138, 305–312. [Google Scholar] [CrossRef]
- Waugh, J.L.; Celver, J.; Sharma, M.; Dufresne, R.L.; Terzi, D.; Risch, S.C.; Fairbrother, W.G.; Neve, R.L.; Kane, J.P.; Malloy, M.J.; et al. Association between regulator of G protein signaling 9-2 and body weight. PLoS ONE 2011, 6, e27984. [Google Scholar] [CrossRef]
- Smirnova, E.; Goldberg, E.B.; Makarova, K.S.; Lin, L.; Brown, W.J.; Jackson, C.L. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 2006, 7, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, D.G.; Ueda, N.; Yamamoto, S. The fatty acid amide hydrolase (FAAH). Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Deignan, J.L.; Qi, H.; Zhu, J.; Qian, S.; Zhong, J.; Torosyan, G.; Majid, S.; Falkard, B.; Kleinhanz, R.R.; et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat. Genet. 2009, 41, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Oller do Nascimento, C.M.; Ribeiro, E.B.; Oyama, L.M. Metabolism and secretory function of white adipose tissue: Effect of dietary fat. Acad. Bras. Cienc. 2009, 81, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Matsui, T.; Iwabuchi, K.; Date, T. PKU-beta/TLK1 regulates myosin II activities, and is required for accurate equaled chromosome segregation. Mutat. Res. 2008, 657, 63–67. [Google Scholar] [CrossRef]
- Vaicik, M.K.; Thyboll Kortesmaa, J.; Movérare-Skrtic, S.; Kortesmaa, J.; Soininen, R.; Bergström, G.; Ohlsson, C.; Chong, L.Y.; Rozell, B.; Emont, M.; et al. Laminin α4 deficient mice exhibit decreased capacity for adipose tissue expansion and weight gain. PLoS ONE 2014, 9, e109854. [Google Scholar] [CrossRef] [PubMed]
- Vaicik, M.K.; Blagajcevic, A.; Ye, H.; Morse, M.C.; Yang, F.; Goddi, A.; Brey, E.M.; Cohen, R.N. The absence of laminin α4 in male mice results in enhanced energy expenditure and increased beige subcutaneous adipose tissue. Endocrinology 2018, 159, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Adapala, V.J.; Adedokun, S.A.; Considine, R.V.; Ajuwon, K.M. Acute inflammation plays a limited role in the regulation of adipose tissue COL1A1 protein abundance. J. Nutr. Biochem. 2012, 23, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sabeva, N.S.; Bhatnagar, S.; Li, X.A.; Pujol, A.; Graf, G.A. ABCD2 is abundant in adipose tissue and opposes the accumulation of dietary erucic acid (C22, 1) in fat. J. Lipid Res. 2010, 51, 162–168. [Google Scholar] [CrossRef]
- Liu, J.; Liang, S.; Liu, X.; Brown, J.A.; Newman, K.E.; Sunkara, M.; Morris, A.J.; Bhatnagar, S.; Li, X.; Pujol, A.; et al. The absence of ABCD2 sensitizes mice to disruptions in lipid metabolism by dietary erucic acid. J. Lipid Res. 2012, 53, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Buch, A.; Carmeli, E.; Boker, L.K.; Marcus, Y.; Shefer, G.; Kis, O.; Berner, Y.; Stern, N. Muscle function and fat content in relation to sarcopenia, obesity and frailty of old age—An overview. Exp. Gerontol. 2016, 76, 25–32. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | NX30d | NX90d | NX150d | NX210d | ||||
|---|---|---|---|---|---|---|---|---|
| Muscle | Fat | Muscle | Fat | Muscle | Fat | Muscle | Fat | |
| Raw reads number | 65606883 | 57278618 | 50488816 | 53672934 | 53929255 | 45120232 | 56107588 | 58206111 |
| Clean reads number | 64137048 | 56517482 | 48897656 | 52575561 | 52912480 | 44451010 | 55237321 | 57266380 |
| Clean reads rate | 97.76% | 98.67% | 96.85% | 97.96% | 98.11% | 98.52% | 98.45% | 98.39% |
| Clean Q30 bases rate | 95.42% | 95.02% | 95.35% | 95.48% | 95.35% | 95.48% | 95.63% | 95.28% |
| Mapped reads | 120386916 | 106157588 | 91329628 | 97298423 | 99877320 | 81793231 | 104372240 | 105818225 |
| Mapping rate | 93.85% | 93.90% | 93.38% | 92.55% | 94.38% | 92.00% | 94.48% | 92.41% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Yang, J.; He, J.; Gong, Y.; Xiao, Y.; Zeng, Q.; Xu, K.; Duan, Y.; He, J.; Ma, H. Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth. Biology 2021, 10, 841. https://doi.org/10.3390/biology10090841
Li B, Yang J, He J, Gong Y, Xiao Y, Zeng Q, Xu K, Duan Y, He J, Ma H. Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth. Biology. 2021; 10(9):841. https://doi.org/10.3390/biology10090841
Chicago/Turabian StyleLi, Biao, Jinzeng Yang, Jun He, Yan Gong, Yu Xiao, Qinghua Zeng, Kang Xu, Yehui Duan, Jianhua He, and Haiming Ma. 2021. "Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth" Biology 10, no. 9: 841. https://doi.org/10.3390/biology10090841
APA StyleLi, B., Yang, J., He, J., Gong, Y., Xiao, Y., Zeng, Q., Xu, K., Duan, Y., He, J., & Ma, H. (2021). Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth. Biology, 10(9), 841. https://doi.org/10.3390/biology10090841