
In Planta Transcriptome and Proteome Profiles of Spongospora subterranea in Resistant and Susceptible Host Environments Illuminates Regulatory Principles Underlying Host–Pathogen Interaction




Abstract
:Simple Summary
Abstract

1. Introduction
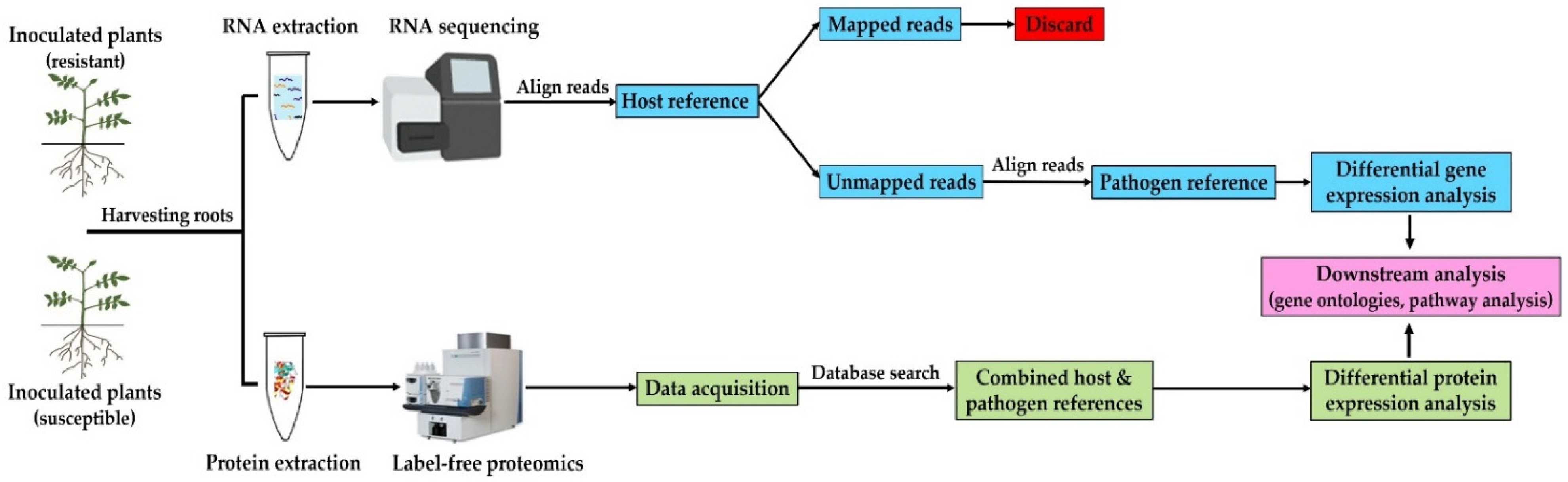
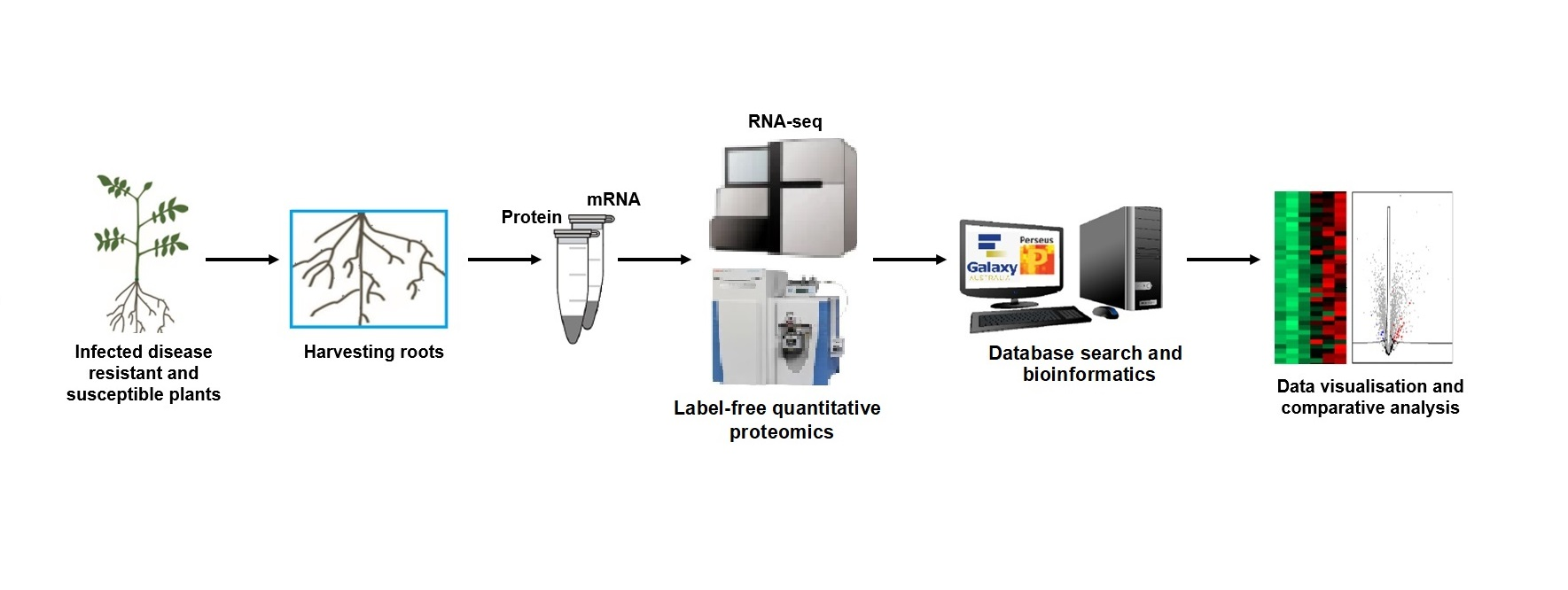
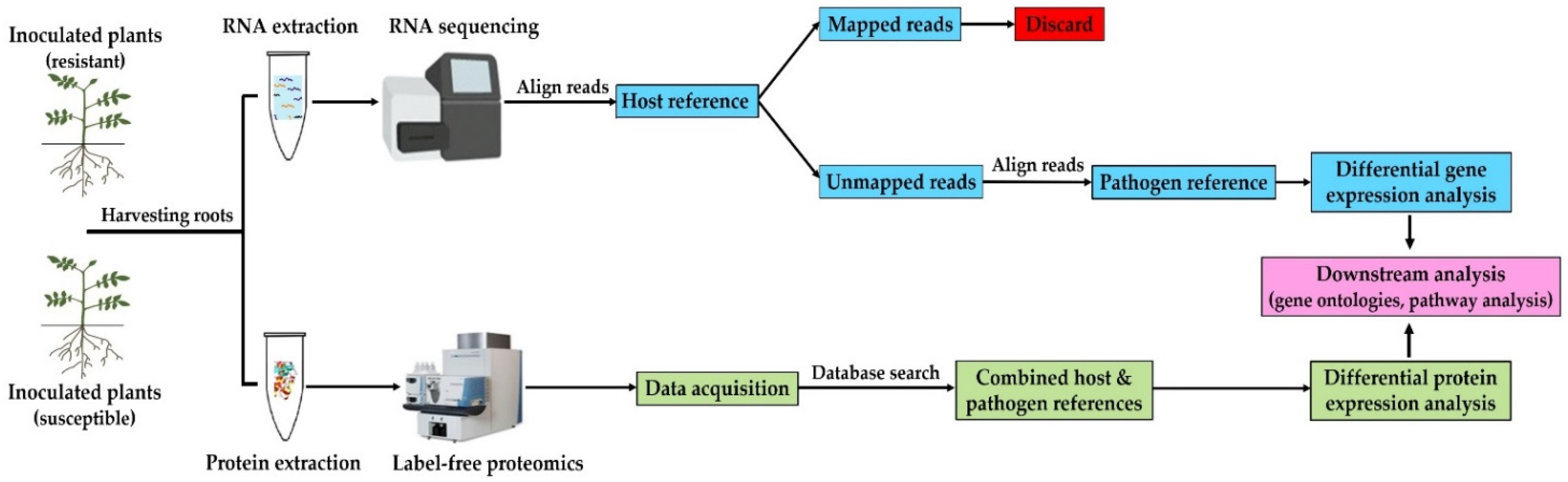
2. Materials and Methods
2.1. Pathogen Source and Potato Cultivars Used in This Study
2.2. Plant Growth and Pathogen Infection
2.3. RNA Extraction and Sequencing
2.4. Transcriptome Assembly and Differential Expression Analysis
2.5. Protein Extraction and Digestion
2.6. Data-Independent Acquisition Mass Spectrometry (DIA-MS)
2.7. Raw Data Processing and Protein Label-Free Quantitation (LFQ)
2.8. Gene Ontology Enrichment Analysis
3. Results and Discussion
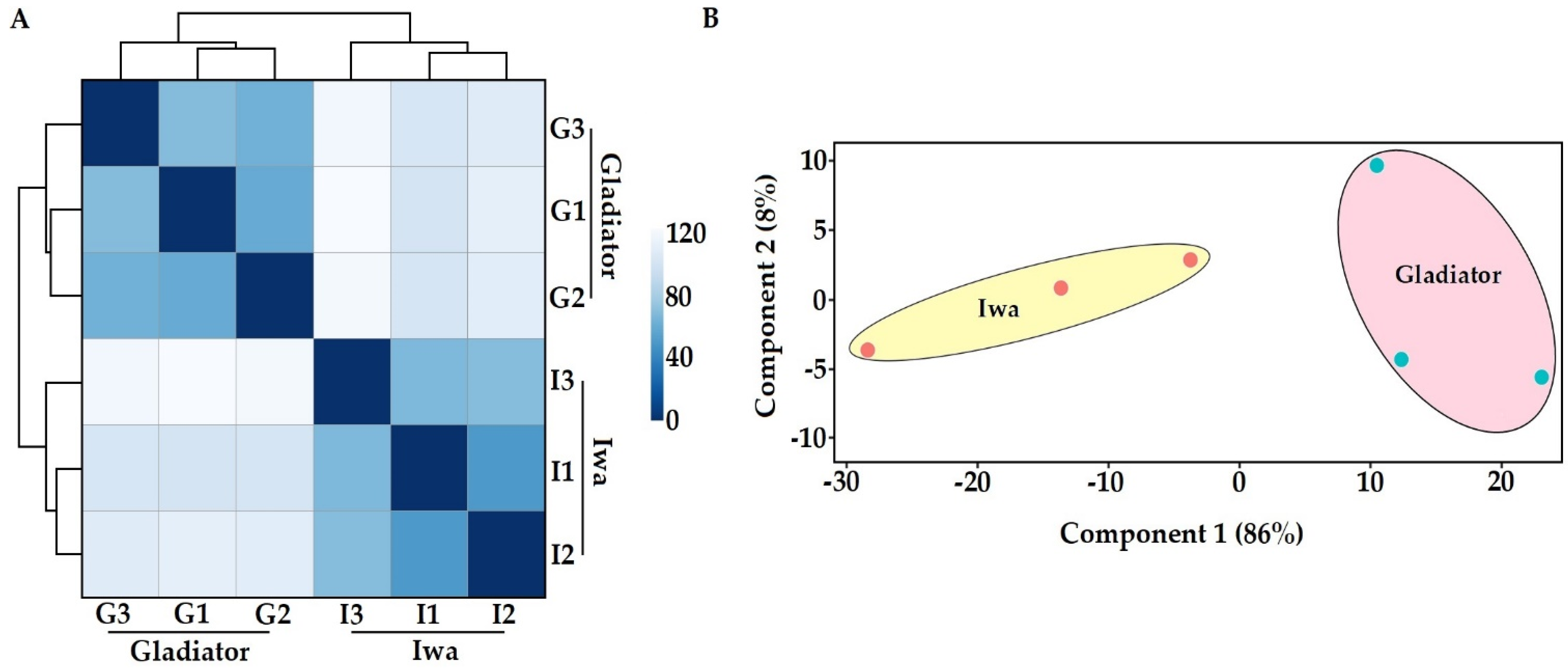
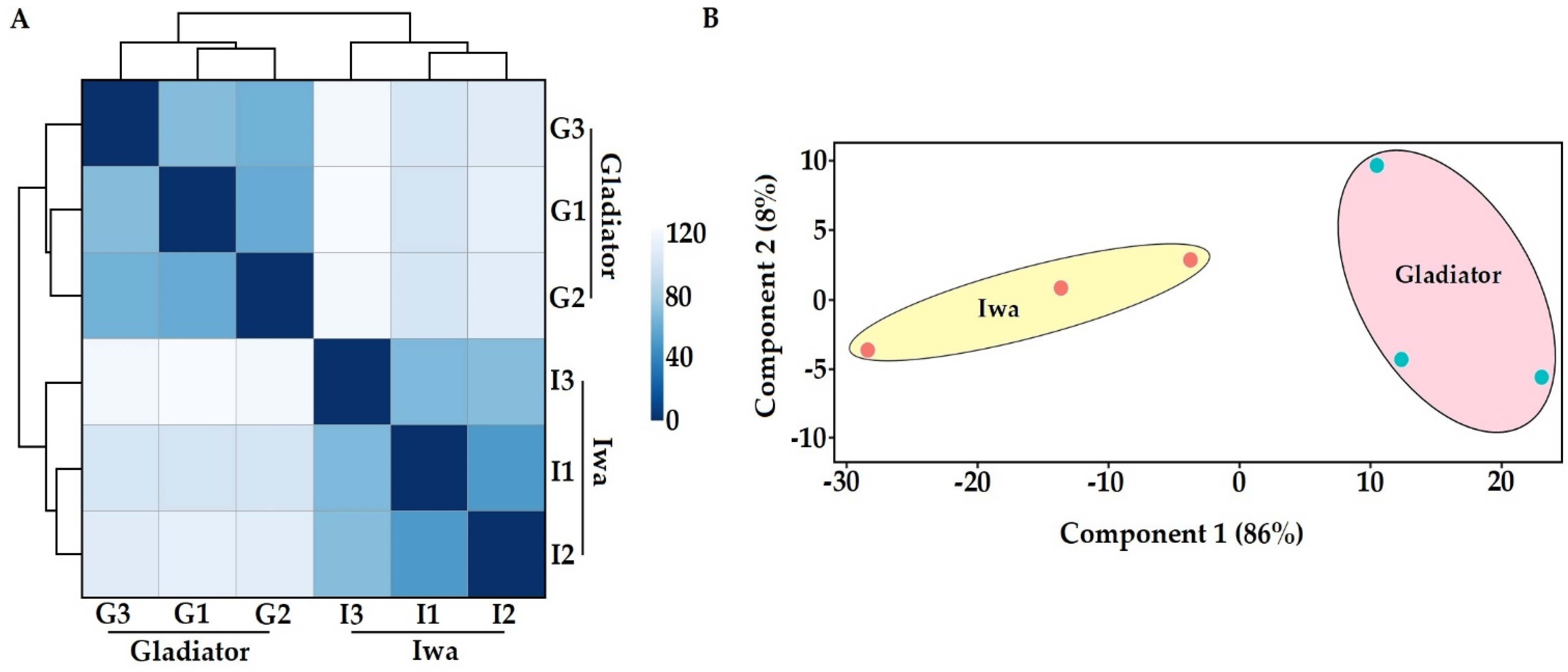
3.1. In Planta Transcriptome and Proteome Profiling of S. subterranea
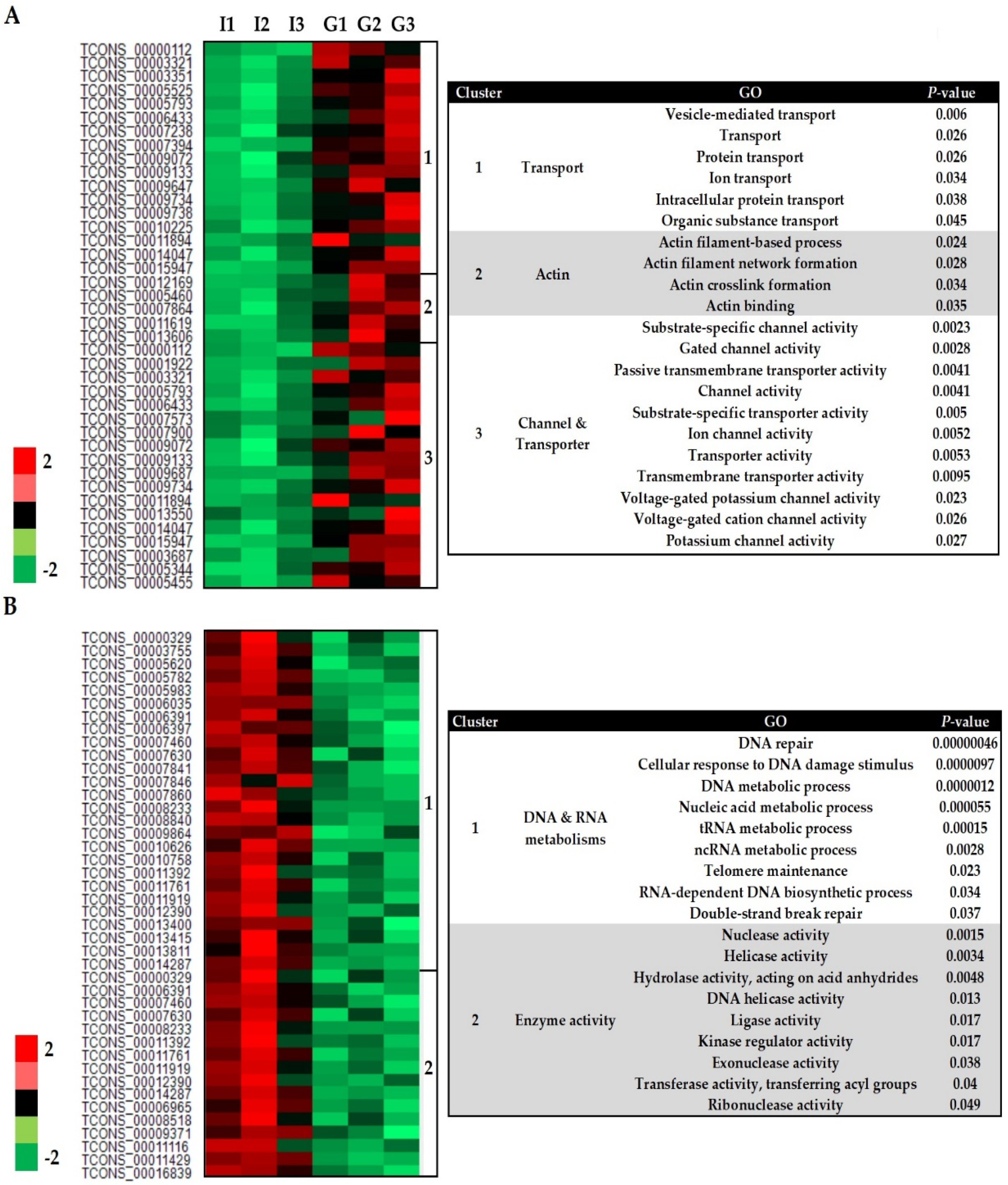
3.2. Transcriptome Profile of S. subterranea inside the Resistant and Susceptible Hosts
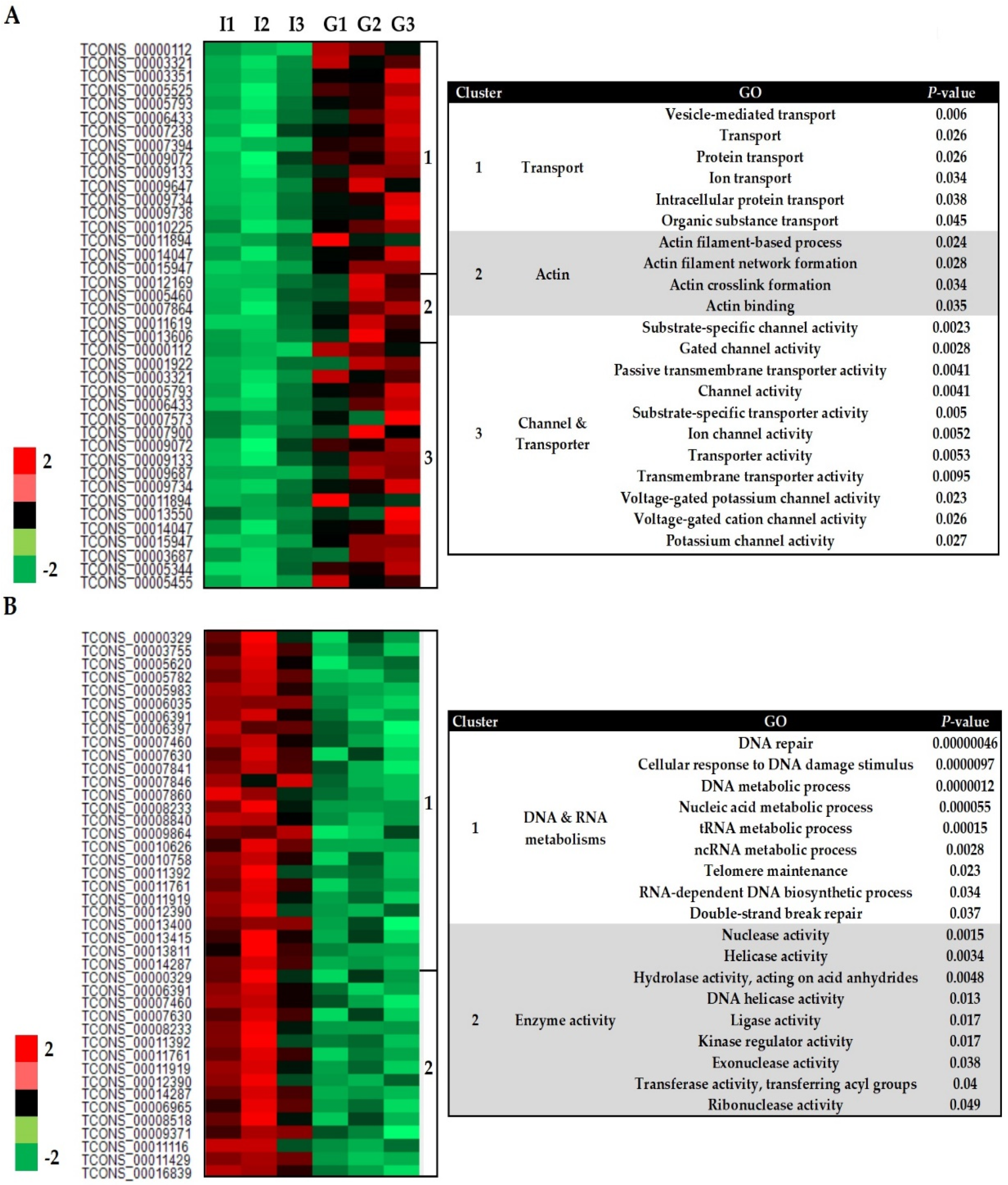
3.3. Functional Annotation of Differently Expressed Genes
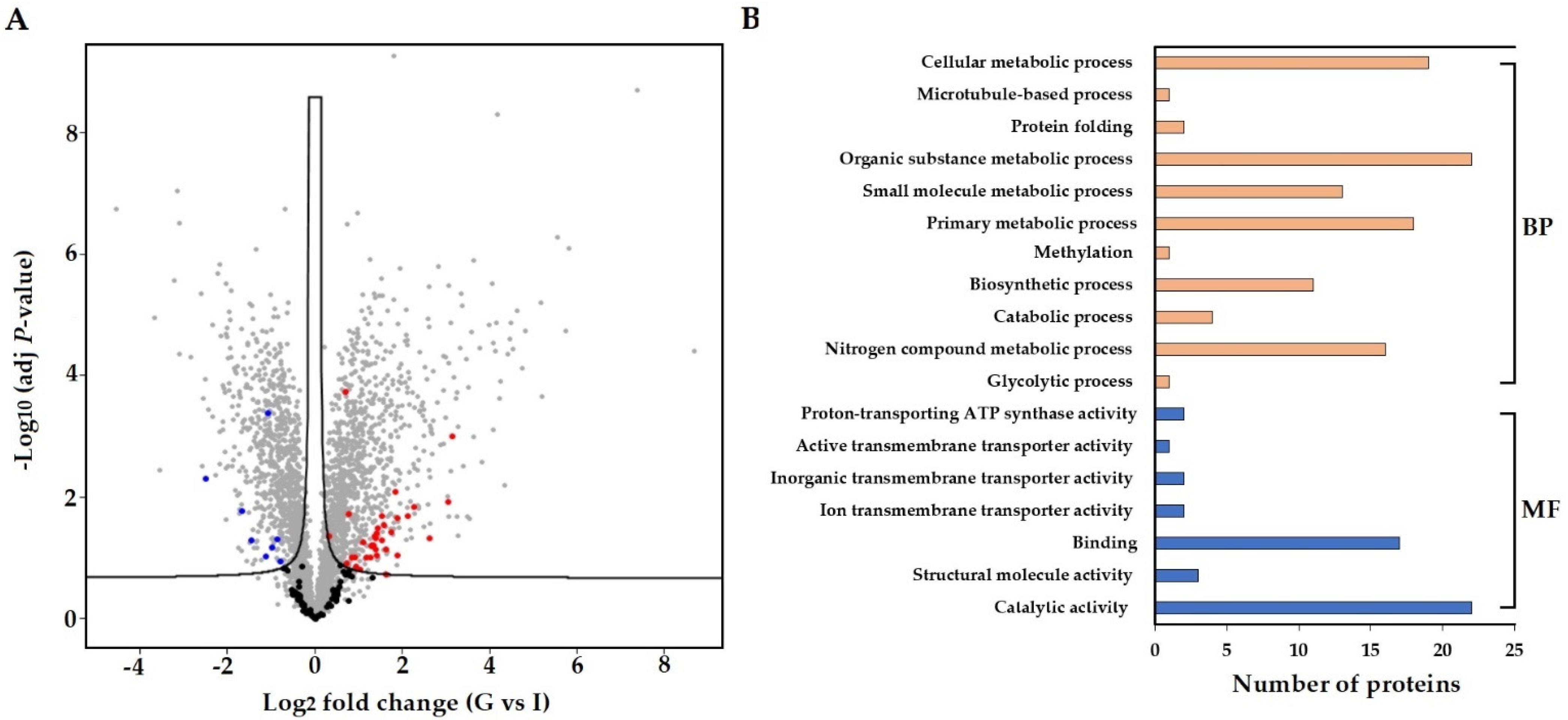
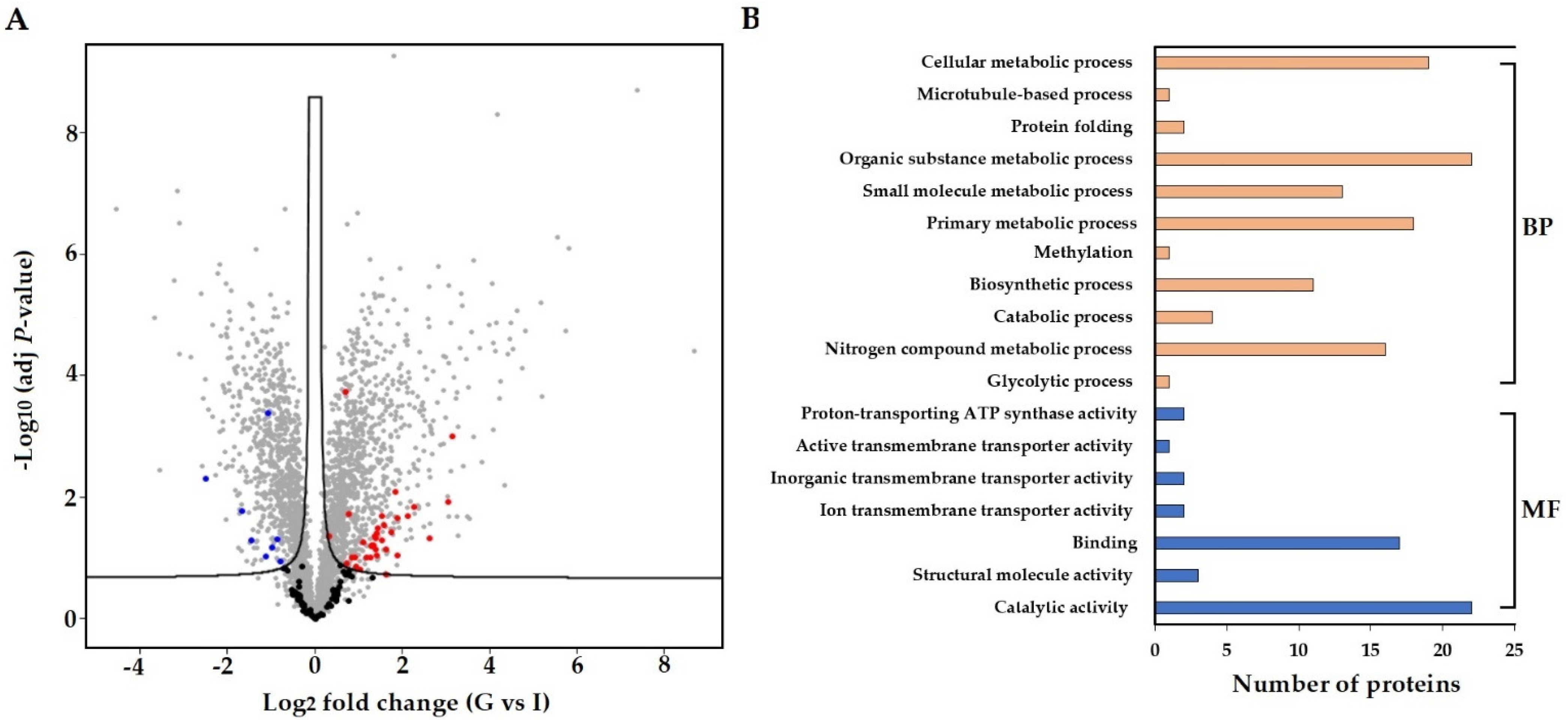
3.4. In Planta Protein Analysis of S. subterranea
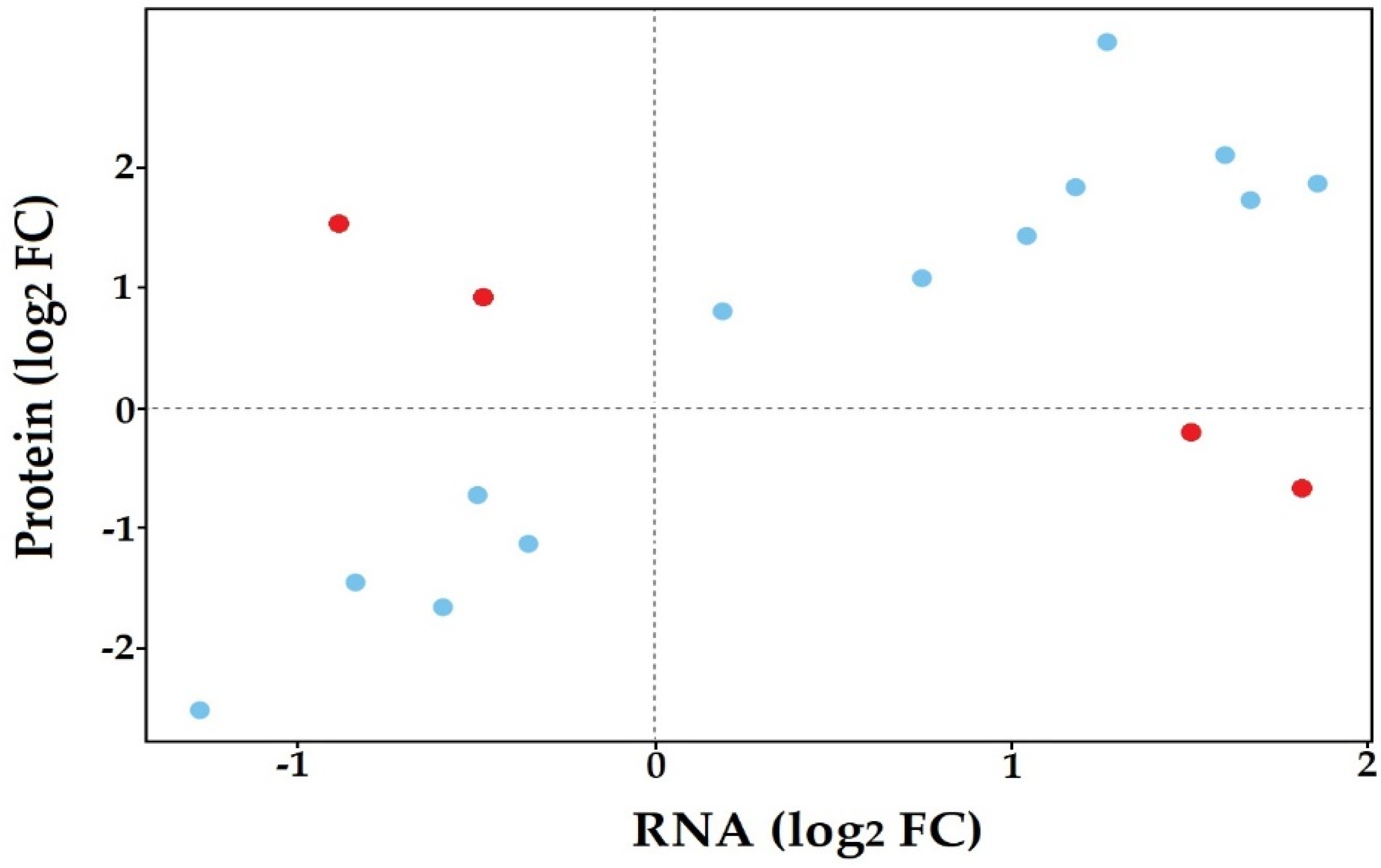
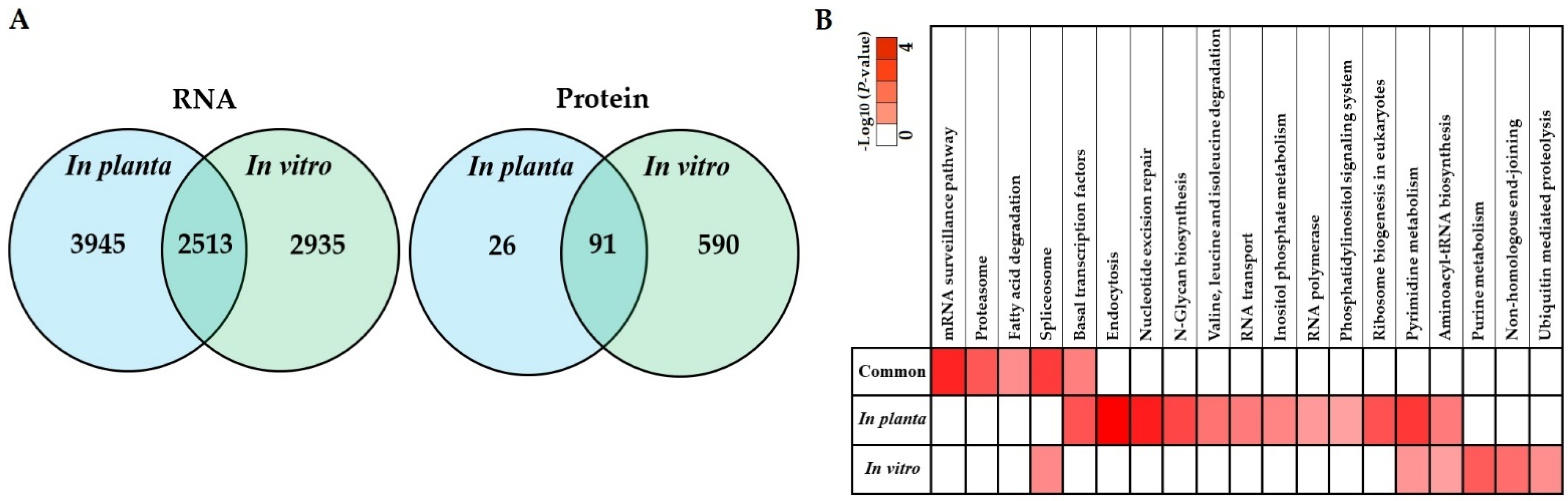
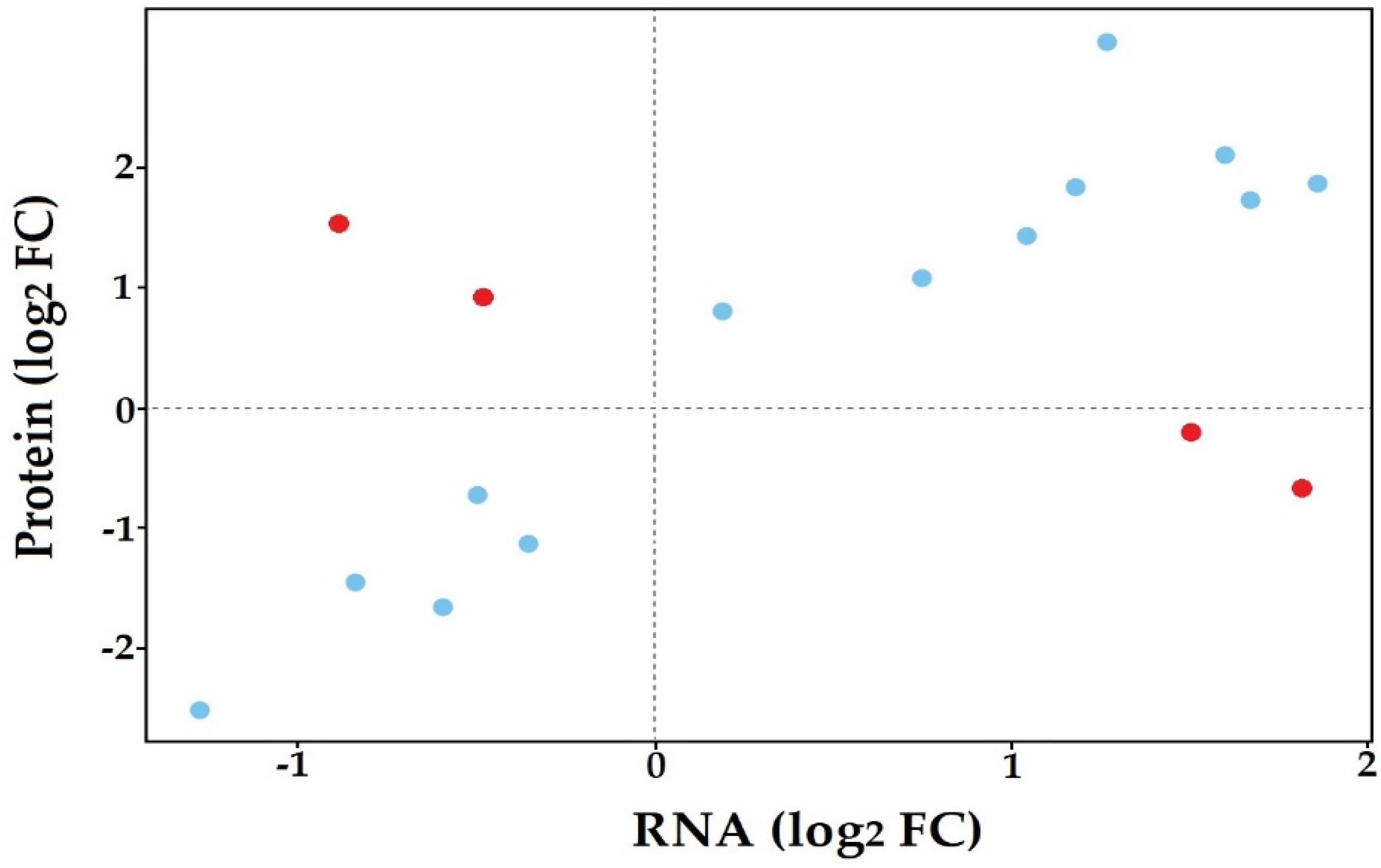
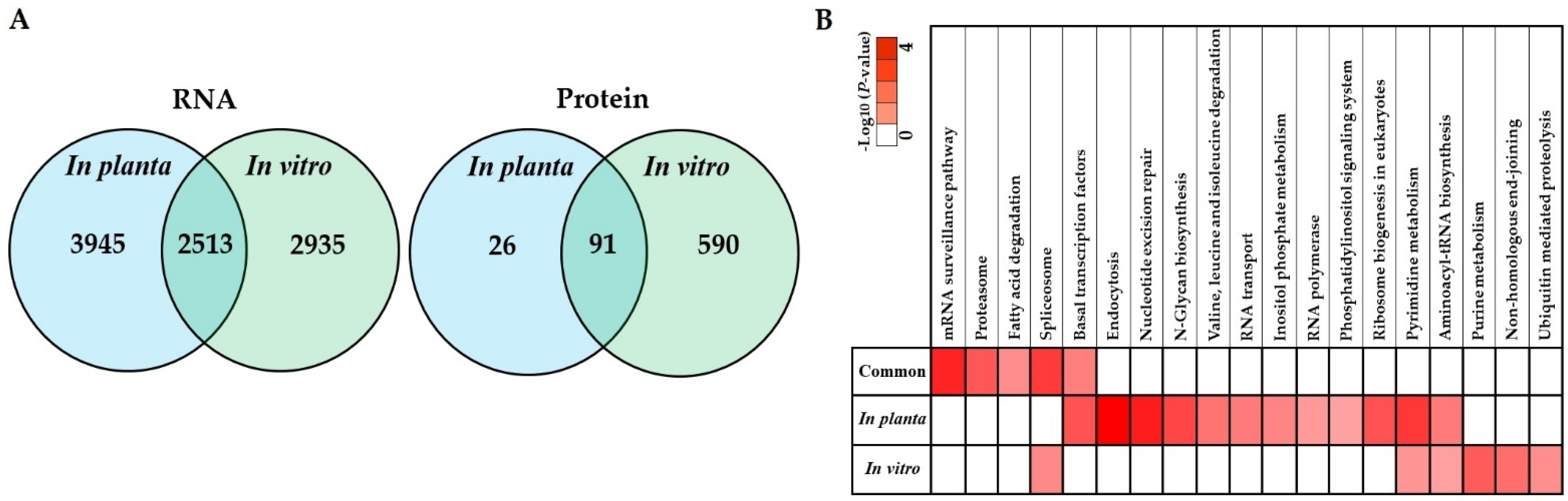
3.5. Co-Expression of In Planta and In Vitro Analysis of S. subterranea Transcriptome and Proteome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amponsah, J.; Tegg, R.S.; Thangavel, T.; Wilson, C.R. Moments of weaknesses–exploiting vulnerabilities between germination and encystment in the Phytomyxea. Biol. Rev. 2021, 96, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Fulladolsa, A.C.; Cordova, A.M.; O’Neill, P.; Gray, S.M.; Charkowski, A.O. Evaluation of effects of chemical soil treatments and potato cultivars on Spongospora subterranea soil inoculum and incidence of powdery scab and potato mop-top virus in potato. Plant Dis. 2020, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Balendres, M.; Tegg, R.; Wilson, C. Key events in pathogenesis of Spongospora diseases in potato: A review. Australas. Plant Pathol. 2016, 45, 229–240. [Google Scholar] [CrossRef]
- Neuhauser, S.; Bulman, S.; Kirchmair, M. Plasmodiophorids: The challenge to understand soil-borne, obligate biotrophs with a multiphasic life cycle. In Molecular Identification of Fungi; Springer: Berlin/Heidelberg, Germany, 2010; pp. 51–78. [Google Scholar]
- Balendres, M.; Clark, T.; Tegg, R.; Wilson, C. Germinate to exterminate: Chemical stimulation of Spongospora subterranea resting spore germination and its potential to diminish soil inoculum. Plant Pathol. 2018, 67, 902–908. [Google Scholar] [CrossRef]
- Falloon, R.; Merz, U.; Butler, R.; Curtin, D.; Lister, R.; Thomas, S. Root infection of potato by Spongospora subterranea: Knowledge review and evidence for decreased plant productivity. Plant Pathol. 2016, 65, 422–434. [Google Scholar] [CrossRef]
- Balendres, M.A.; Tegg, R.S.; Amponsah, J.; Wilson, C.R. Zoosporangial root infection of tomato by Spongospora subterranea in hydroponic and glasshouse culture results in diminished plant growth. J. Phytopathol. 2018, 166, 412–419. [Google Scholar] [CrossRef]
- Gao, L.; Tu, Z.J.; Millett, B.P.; Bradeen, J.M. Insights into organ-specific pathogen defense responses in plants: RNA-seq analysis of potato tuber-Phytophthora infestans interactions. BMC Genom. 2013, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; Oono, Y.; Kanamori, H.; Matsumoto, T.; Itoh, T.; Minami, E. Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS ONE 2012, 7, e49423. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-J.; Sniezko, R.A.; Zamany, A.; Williams, H.; Omendja, K.; Kegley, A.; Savin, D.P. Comparative transcriptomics and RNA-seq-based bulked segregant analysis reveals genomic basis underlying Cronartium ribicola vcr2 Virulence. Front. Microbiol. 2021, 12, 309. [Google Scholar]
- Naidoo, S.; Visser, E.A.; Zwart, L.; Du Toit, Y.; Bhadauria, V.; Shuey, L.S. Dual RNA-seq to elucidate the plant–pathogen duel. Curr. Issues Mol. Biol. 2018, 27, 127–142. [Google Scholar] [CrossRef]
- Yazawa, T.; Kawahigashi, H.; Matsumoto, T.; Mizuno, H. Simultaneous transcriptome analysis of Sorghum and Bipolaris sorghicola by using RNA-seq in combination with de novo transcriptome assembly. PLoS ONE 2013, 8, e62460. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.J.P.L.; de Toledo Thomazella, D.P.; Reis, O.; do Prado, P.F.V.; do Rio, M.C.S.; Fiorin, G.L.; José, J.; Costa, G.G.L.; Negri, V.A.; Mondego, J.M.C. High-resolution transcript profiling of the atypical biotrophic interaction between Theobroma cacao and the fungal pathogen Moniliophthora perniciosa. Plant Cell 2014, 26, 4245–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobori, T.; Velásquez, A.C.; Wu, J.; Kvitko, B.H.; Kremer, J.M.; Wang, Y.; He, S.Y.; Tsuda, K. Transcriptome landscape of a bacterial pathogen under plant immunity. Proc. Natl. Acad. Sci. USA 2018, 115, E3055–E3064. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Chen, M.; Ma, Y.; Du, Z.; Yuan, N.; Li, Y.; Xiao, J.; Zhang, Y. Whole-genome and time-course dual RNA-Seq analyses reveal chronic pathogenicity-related gene dynamics in the ginseng rusty root rot pathogen Ilyonectria robusta. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindschedler, L.V.; Burgis, T.A.; Mills, D.J.; Ho, J.T.; Cramer, R.; Spanu, P.D. In planta proteomics and proteogenomics of the biotrophic barley fungal pathogen Blumeria graminis f. sp. hordei. Mol. Cell. Proteom. 2009, 8, 2368–2381. [Google Scholar] [CrossRef] [Green Version]
- Jashni, M.K.; van der Burgt, A.; Battaglia, E.; Mehrabi, R.; Collemare, J.; de Wit, P.J. Transcriptome and proteome analyses of proteases in biotroph fungal pathogen Cladosporium fulvum. J. Plant Pathol. 2020, 102, 377–386. [Google Scholar] [CrossRef]
- Balotf, S.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Spore germination of the obligate biotroph Spongospora subterranea: Transcriptome analysis reveals germination associated genes. Front. Microbiol. 2021, 12, 1557. [Google Scholar] [CrossRef] [PubMed]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Quantitative proteomics provides an insight into germination-related proteins in the obligate biotrophic plant pathogen Spongospora subterranea. Environ. Microbiol. Rep. 2021, 13, 521–532. [Google Scholar] [CrossRef]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Optimisation of sporosori purification and protein extraction techniques for the biotrophic protozoan plant pathogen Spongospora subterranea. Molecules 2020, 25, 3109. [Google Scholar] [CrossRef] [PubMed]
- Balendres, M.A.; Nichols, D.S.; Tegg, R.S.; Wilson, C.R. Metabolomes of potato root exudates: Compounds that stimulate resting spore germination of the soil-borne pathogen Spongospora subterranea. J. Agric. Food Chem. 2016, 64, 7466–7474. [Google Scholar] [CrossRef]
- Ciaghi, S.; Neuhauser, S.; Schwelm, A. Draft genome resource for the potato powdery scab pathogen Spongospora subterranea. Mol. Plant-Microbe Interact. 2018, 31, 1227–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 1–5. [Google Scholar] [CrossRef]
- Zhou, W.; Eudes, F.; Laroche, A. Identification of differentially regulated proteins in response to a compatible interaction between the pathogen Fusarium graminearum and its host, Triticum aestivum. Proteomics 2006, 6, 4599–4609. [Google Scholar] [CrossRef]
- Gétaz, M.; Puławska, J.; Smits, T.H.; Pothier, J.F. Host–pathogen interactions between Xanthomonas fragariae and its host Fragaria × ananassa investigated with a dual RNA-Seq analysis. Microorganisms 2020, 8, 1253. [Google Scholar] [CrossRef]
- Rudd, J.J.; Kanyuka, K.; Hassani-Pak, K.; Derbyshire, M.; Andongabo, A.; Devonshire, J.; Lysenko, A.; Saqi, M.; Desai, N.M.; Powers, S.J. Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 2015, 167, 1158–1185. [Google Scholar]
- Gao, S.; Zeng, R.; Xu, L.; Song, Z.; Gao, P.; Dai, F. Genome sequence and spore germination-associated transcriptome analysis of Corynespora cassiicola from cucumber. BMC Microbiol. 2020, 20, 1–20. [Google Scholar] [CrossRef]
- Galhano, R.; Illana, A.; Ryder, L.S.; Rodriguez-Romero, J.; Demuez, M.; Badaruddin, M.; Martinez-Rocha, A.L.; Soanes, D.M.; Studholme, D.J.; Talbot, N.J. Tpc1 is an important Zn (II) 2Cys6 transcriptional regulator required for polarized growth and virulence in the rice blast fungus. PLoS Pathog. 2017, 13, e1006516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beis, K. Structural basis for the mechanism of ABC transporters. Biochem. Soc. Trans. 2015, 43, 889–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plante, S.; Labbé, S. Spore germination requires ferrichrome biosynthesis and the siderophore transporter Str1 in Schizosaccharomyces pombe. Genetics 2019, 211, 893–911. [Google Scholar] [CrossRef] [Green Version]
- Doehlemann, G.; Molitor, F.; Hahn, M. Molecular and functional characterization of a fructose specific transporter from the gray mold fungus Botrytis cinerea. Fungal Genet. Biol. 2005, 42, 601–610. [Google Scholar] [CrossRef]
- Chagué, V.; Maor, R.; Sharon, A. CgOpt1, a putative oligopeptide transporter from Colletotrichum gloeosporioides that is involved in responses to auxin and pathogenicity. BMC Microbiol. 2009, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Seong, K.-Y.; Zhao, X.; Xu, J.-R.; Güldener, U.; Kistler, H.C. Conidial germination in the filamentous fungus Fusarium graminearum. Fungal Genet. Biol. 2008, 45, 389–399. [Google Scholar] [CrossRef]
- Sephton-Clark, P.C.; Voelz, K. Spore germination of pathogenic filamentous fungi. Adv. Appl. Microbiol. 2018, 102, 117–157. [Google Scholar]
- Setlow, P. I will survive: Protecting and repairing spore DNA. J. Bacteriol. 1992, 174, 2737–2741. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.A.; Köppe, M.; Grenville-Briggs, L.J.; Avrova, A.O.; Horner, N.R.; McKinnon, A.D.; Whisson, S.C.; Birch, P.R.; Van West, P. A putative DEAD-box RNA-helicase is required for normal zoospore development in the late blight pathogen Phytophthora infestans. Fungal Genet. Biol. 2008, 45, 954–962. [Google Scholar] [CrossRef]
- Valenzuela-García, L.I.; Ayala-García, V.M.; Regalado-García, A.G.; Setlow, P.; Pedraza-Reyes, M. Transcriptional coupling (Mfd) and DNA damage scanning (DisA) coordinate excision repair events for efficient Bacillus subtilis spore outgrowth. MicrobiologyOpen 2018, 7, e00593. [Google Scholar] [CrossRef] [Green Version]
- Miyawaki, K.; Tarkowski, P.; Matsumoto-Kitano, M.; Kato, T.; Sato, S.; Tarkowska, D.; Tabata, S.; Sandberg, G.; Kakimoto, T. Roles of Arabidopsis ATP/ADP isopentenyltransferases and tRNA isopentenyltransferases in cytokinin biosynthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16598–16603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hluska, T.; Hlusková, L.; Emery, R. The Hulks and the Deadpools of the cytokinin universe: A dual strategy for cytokinin production, translocation, and signal transduction. Biomolecules 2021, 11, 209. [Google Scholar] [CrossRef]
- Chanclud, E.; Kisiala, A.; Emery, N.R.J.; Chalvon, V.; Ducasse, A.; Romiti-Michel, C.; Gravot, A.; Kroj, T.; Morel, J.-B. Cytokinin production by the rice blast fungus is a pivotal requirement for full virulence. PLoS Pathog. 2016, 12, e1005457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, M.; Motyka, V.; Weihmann, F.; Malbeck, J.; Deising, H.B.; Wirsel, S.G. Remodeling of cytokinin metabolism at infection sites of Colletotrichum graminicola on maize leaves. Mol. Plant-Microbe Interact. 2012, 25, 1073–1082. [Google Scholar] [CrossRef]
- Hinsch, J.; Galuszka, P.; Tudzynski, P. Functional characterization of the first filamentous fungal tRNA-isopentenyltransferase and its role in the virulence of Claviceps purpurea. New Phytol. 2016, 211, 980–992. [Google Scholar] [CrossRef]
- Keep, N.; Ward, J.; Robertson, G.; Cohen-Gonsaud, M.; Henderson, B. Bacterial resuscitation factors: Revival of viable but non-culturable bacteria. Cell. Mol. Life Sci. 2006, 63, 2555. [Google Scholar] [CrossRef]
- Wrońska, A.K.; Boguś, M.I.; Włóka, E.; Kazek, M.; Kaczmarek, A.; Zalewska, K. Cuticular fatty acids of Galleria mellonella (Lepidoptera) inhibit fungal enzymatic activities of pathogenic Conidiobolus coronatus. PLoS ONE 2018, 13, e0192715. [Google Scholar] [CrossRef] [Green Version]
- Takano, Y.; Kubo, Y.; Shimizu, K.; Mise, K.; Okuno, T.; Furusawa, I. Structural analysis of PKS1, a polyketide synthase gene involved in melanin biosynthesis in Colletotrichum lagenarium. Mol. Gen. Genet. MGG 1995, 249, 162–167. [Google Scholar] [CrossRef]
- Ökmen, B.; Bachmann, D.; De Wit, P.J. A conserved GH17 glycosyl hydrolase from plant pathogenic Dothideomycetes releases a DAMP causing cell death in tomato. Mol. Plant Pathol. 2019, 20, 1710–1721. [Google Scholar] [CrossRef]
- Talukdar, P.K.; Sarker, M.R. The serine proteases CspA and CspC are essential for germination of spores of Clostridium perfringens SM101 through activating SleC and cortex hydrolysis. Food Microbiol. 2020, 86, 103325. [Google Scholar] [CrossRef]
- Tzima, A.; Paplomatas, E.J.; Rauyaree, P.; Kang, S. Roles of the catalytic subunit of cAMP-dependent protein kinase A in virulence and development of the soilborne plant pathogen Verticillium dahliae. Fungal Genet. Biol. 2010, 47, 406–415. [Google Scholar] [CrossRef]
- Nguyen, K.B.; Sreelatha, A.; Durrant, E.S.; Lopez-Garrido, J.; Muszewska, A.; Dudkiewicz, M.; Grynberg, M.; Yee, S.; Pogliano, K.; Tomchick, D.R. Phosphorylation of spore coat proteins by a family of atypical protein kinases. Proc. Natl. Acad. Sci. USA 2016, 113, E3482–E3491. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, M.; Shimoda, C. The cyclic AMP/PKA signal pathway is required for initiation of spore germination in Schizosaccharomyces pombe. Yeast 2001, 18, 207–217. [Google Scholar] [CrossRef]
- Rekadwad, B. Applications of serine/threonine protein kinases (STPK): A bus for dormancy exit. In Quorum Sensing and Its Biotechnological Applications; Springer: Berlin/Heidelberg, Germany, 2018; pp. 271–278. [Google Scholar]
- Pang, Z.; Srivastava, V.; Liu, X.; Bulone, V. Quantitative proteomics links metabolic pathways to specific developmental stages of the plant-pathogenic oomycete Phytophthora capsici. Mol. Plant Pathol. 2017, 18, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.H.; Yarden, O.; Gollop, N.; Chen, S.; Zveibil, A.; Belausov, E.; Freeman, S. Differential protein expression in Colletotrichum acutatum: Changes associated with reactive oxygen species and nitrogen starvation implicated in pathogenicity on strawberry. Mol. Plant Pathol. 2008, 9, 171–190. [Google Scholar] [CrossRef]
- Fernández-Acero, F.J.; Colby, T.; Harzen, A.; Carbú, M.; Wieneke, U.; Cantoral, J.M.; Schmidt, J. 2-DE proteomic approach to the Botrytis cinerea secretome induced with different carbon sources and plant-based elicitors. Proteomics 2010, 10, 2270–2280. [Google Scholar] [CrossRef]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Duplessis, S.; Lorrain, C.; Petre, B.; Figueroa, M.; Dodds, P.N.; Aime, M.C. Host adaptation and virulence in heteroecious rust fungi. Annu. Rev. Phytopathol. 2021, 59. [Google Scholar] [CrossRef]
- Zhou, B.; Semanjski, M.; Orlovetskie, N.; Bhattacharya, S.; Alon, S.; Argaman, L.; Jarrous, N.; Zhang, Y.; Macek, B.; Sinai, L. Arginine dephosphorylation propels spore germination in bacteria. Proc. Natl. Acad. Sci. USA 2019, 116, 14228–14237. [Google Scholar] [CrossRef] [Green Version]
- Osherov, N.; May, G. Conidial germination in Aspergillus nidulans requires RAS signaling and protein synthesis. Genetics 2000, 155, 647–656. [Google Scholar] [CrossRef]
- Sinai, L.; Rosenberg, A.; Smith, Y.; Segev, E.; Ben-Yehuda, S. The molecular timeline of a reviving bacterial spore. Mol. Cell 2015, 57, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, M.; Noben, J.-P.; Van Dijck, P.; Valcke, R. Fire blight host-pathogen interaction: Proteome profiles of Erwinia amylovora infecting apple rootstocks. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Djouiai, B.; Thwaite, J.E.; Laws, T.R.; Commichau, F.M.; Setlow, B.; Setlow, P.; Moeller, R. Role of DNA repair and protective components in Bacillus subtilis spore resistance to inactivation by 400-nm-wavelength blue light. Appl. Environ. Microbiol. 2018, 84, e01604–e01618. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huber, N.; Moeller, R.; Stülke, J.; Dubovcova, B.; Akepsimaidis, G.; Meneses, N.; Drissner, D.; Mathys, A. Role of DNA repair in Bacillus subtilis spore resistance to high energy and low energy electron beam treatments. Food Microbiol. 2020, 87, 103353. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, S.; Leveau, J.H.; Stergiopoulos, I. Repeated exposure of Aspergillus niger spores to the antifungal bacterium Collimonas fungivorans Ter331 selects for delayed spore germination. Appl. Environ. Microbiol. 2021, 87, e00233-21. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified | Differentially Expressed (Gladiator vs. Iwa) | |
|---|---|---|
| RNA | 7650 | 1377 |
| Protein | 117 | 42 |
| Categories | GO Terms | adj p-Value |
|---|---|---|
| Biological process | Peptide metabolic process | 0.0032 |
| Organonitrogen compound metabolic process | 0.0032 | |
| Cellular amide metabolic process | 0.0032 | |
| Translation | 0.014 | |
| Peptide biosynthetic process | 0.014 | |
| Amide biosynthetic process | 0.014 | |
| Molecular function | Structural constituent of ribosome | 0.00004 |
| Structural molecule activity | 0.00009 | |
| Pathway | Ribosome | 0.00048 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. In Planta Transcriptome and Proteome Profiles of Spongospora subterranea in Resistant and Susceptible Host Environments Illuminates Regulatory Principles Underlying Host–Pathogen Interaction. Biology 2021, 10, 840. https://doi.org/10.3390/biology10090840
Balotf S, Wilson R, Tegg RS, Nichols DS, Wilson CR. In Planta Transcriptome and Proteome Profiles of Spongospora subterranea in Resistant and Susceptible Host Environments Illuminates Regulatory Principles Underlying Host–Pathogen Interaction. Biology. 2021; 10(9):840. https://doi.org/10.3390/biology10090840
Chicago/Turabian StyleBalotf, Sadegh, Richard Wilson, Robert S. Tegg, David S. Nichols, and Calum R. Wilson. 2021. "In Planta Transcriptome and Proteome Profiles of Spongospora subterranea in Resistant and Susceptible Host Environments Illuminates Regulatory Principles Underlying Host–Pathogen Interaction" Biology 10, no. 9: 840. https://doi.org/10.3390/biology10090840
APA StyleBalotf, S., Wilson, R., Tegg, R. S., Nichols, D. S., & Wilson, C. R. (2021). In Planta Transcriptome and Proteome Profiles of Spongospora subterranea in Resistant and Susceptible Host Environments Illuminates Regulatory Principles Underlying Host–Pathogen Interaction. Biology, 10(9), 840. https://doi.org/10.3390/biology10090840