
Comprehensive CircRNA Profiling and Selection of Key CircRNAs Reveal the Potential Regulatory Roles of CircRNAs throughout Ovarian Development and Maturation in Cynoglossus semilaevis

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Strand-Specific Library Construction and Sequencing
2.3. Sequencing Assessment of CircRNA
2.4. CircRNA Identification
2.5. CircRNA Statistics
2.6. Functional Enrichment Analysis of Parental Genes
2.7. Analysis of Differentially Expressed CircRNAs
2.8. Database Annotation of CircRNAs
2.9. MiRNA Sponge Analysis and Integrated Analysis of CircRNA-miRNA-mRNA
2.10. RNase R Treatment and Sanger Sequencing
2.11. Quantitative Real-Time PCR Analysis of CircRNAs
3. Results
3.1. Expression Profiles of CircRNAs in the Brain, Ovary, Pituitary and Liver at Different Ovarian Stages
3.2. Identification of Differentially Expressed CircRNAs
3.3. Analysis between CircRNAs and Their Parental Genes
3.4. Delineation of GO and KEGG Pathway Analysis
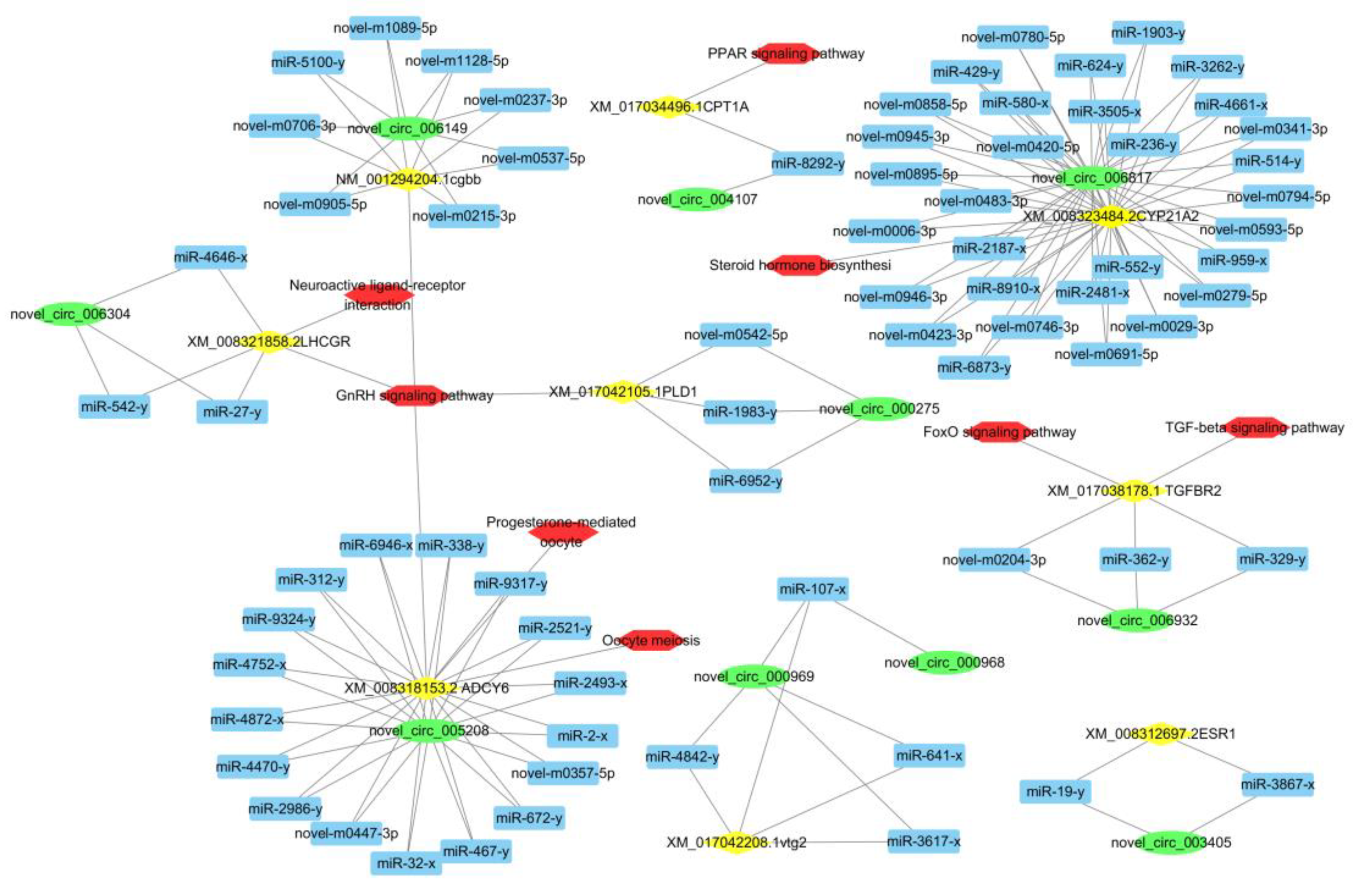
3.5. CeRNA Network
3.6. Identification of CircRNAs
3.7. Validation of Differentially Expressed CircRNAs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, Y.D.; Guo, X.W.; Wang, W.M. Identification and characterization of circular RNAs in zebrafish. FEBS Lett. 2017, 591, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.H.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.F.; Li, H.; Yang, J.M.; Hao, D.; Dong, D.; Huang, Y.Z.; Lan, X.Y.; Plath, M.; Lei, C.Z.; Lin, F.P.; et al. Circular RNA profiling reveals an abundant circLMO7 that regulates myoblasts differentiation and survival by sponging miR-378a-3p. Cell. Death Dis. 2017, 8, e3153. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.H.; Liu, Y.; Chen, S.; Zong, Z.H.; Du, Y.P.; Sheng, X.J.; Zhao, Y. circ-CSPP1 promotes proliferation, invasion and migration of ovarian cancer cells by acting as a miR-1236-3p sponge. Biomed. Pharmacother. 2019, 114, 108832. [Google Scholar] [CrossRef]
- Cai, Y.Q.; Lei, X.C.; Chen, Z.; Mo, Z.C. The roles of cirRNA in the development of germ cells. Acta Histochem. 2020, 122, 151506. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.C.; Xu, B.; Wu, J. Circular RNA expression profiles of mouse ovaries during postnatal development and the function of circular RNA epidermal growth factor receptor in granulosa cells. Metab. Clin. Exp. 2018, 85, 192–204. [Google Scholar] [CrossRef]
- Broer, S.L.; Broekmans, F.J.M.; Laven, J.S.E.; Fauser, B.C.J.M. Anti-Mullerian hormone: Ovarian reserve testing and its potential clinical implications. Hum. Reprod. Update 2014, 20, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, J.; Yuan, S.Z.; Zhou, S.; Yan, W.; Shen, W.; Chen, Y.; Xia, X.; Luo, A.Y.; Zhu, D.; et al. Circular RNA expression profiling of human granulosa cells during maternal aging reveals novel transcripts associated with assisted reproductive technology outcomes. PLoS ONE 2017, 12, e0177888. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Y.; Yin, J.; Yan, X.Y.; Chen, W.J.; Jiang, C.Y.; Hu, X.S.; Wang, X.Y.; Zhu, J.G.; Yu, Z.B.; et al. Profiles analysis reveals circular RNAs involving zebrafish physiological development. J. Cell. Physiol. 2019, 234, 15922–15933. [Google Scholar] [CrossRef]
- Xu, S.B.; Xiao, S.J.; Qiu, C.L.; Wang, Z.Y. Transcriptome-wide identification and functional investigation of circular RNA in the teleost large yellow croaker (Larimichthys crocea). Mar. Genom. 2017, 32, 71–78. [Google Scholar] [CrossRef]
- Li, J.H.; Lv, Y.; Liu, R.R.; Yu, Y.; Shan, C.M.; Bian, W.H.; Jiang, J.; Zhang, D.L.; Yang, C.; Sun, Y.Y. Identification and characterization of a conservative W chromosome-linked circRNA in half smooth tongue sole (Cynoglossus semilaevis) reveal its female-biased expression in immune organs. Fish. Shellfish. Immunol. 2018, 82, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Y.J.; Jiang, G.P.; Zhou, S.; Diao, J.; Liu, H.J.; Su, B.F.; Li, C. Identification of potential immune related circRNA–miRNA–mRNA regulatory network in intestine of Paralichthys olivaceus during Edwardsiella tarda infection. Front. Genet. 2019, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yuan, R.; Liang, Z.; Zhang, T.T.; Zhu, M.; Zhang, X.; Geng, W.; Fang, P.; Jiang, M.S.; Wang, Z.Z.; et al. Comprehensive analysis of circRNA expression pattern and circRNA–mRNA–miRNA network in Ctenopharyngodon idellus kidney (CIK) cells after grass carp reovirus (GCRV) infection. Aquaculture 2019, 512, 734349. [Google Scholar] [CrossRef]
- Shen, Y.D.; Liang, W.W.; Lin, Y.; Yang, H.Z.; Chen, X.H.; Feng, P.F.; Zhang, B.; Zhu, J.J.; Zhang, Y.D.; Luo, H.L. Single molecule real-time sequencing and RNA-seq unravel the role of long non-coding and circular RNA in the regulatory network during Nile tilapia (Oreochromis niloticus) infection with Streptococcus agalactiae. Fish. Shellfish. Immunol. 2020, 104, 640–653. [Google Scholar] [CrossRef]
- Pereiro, P.; Lama, R.; Moreira, R.; Valenzuela-Muñoz, V.; Gallardo-Escárate, C.; Novoa, B.; Figueras, A. Potential involvement of lncRNAs in the modulation of the transcriptome response to nodavirus challenge in European sea bass (Dicentrarchus labrax L.). Biology 2020, 9, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhong, Y.; Guo, S.Y.; Zhu, Y.F.; Guo, J.; Fu, Y.S.; Li, M.Y. CircRNA profiling reveals circ880 functions as miR-375-3p sponge in medaka gonads. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 38, 100797. [Google Scholar] [CrossRef]
- Tao, H.; Xiong, Q.; Zhang, F.; Zhang, N.; Liu, Y.; Suo, X.J.; Li, X.F.; Yang, Q.P.; Chen, M.X. Circular RNA profiling reveals chi_circ_0008219 function as microRNA sponges in pre-ovulatory ovarian follicles of goats (Capra hircus). Genomics 2018, 110, 257–266. [Google Scholar] [CrossRef]
- Qiao, J.; Wang, Z.B.; Feng, H.L.; Miao, Y.L.; Wang, Q.; Yu, Y.; Wei, Y.C.; Yan, J.; Wang, W.H.; Shen, W.; et al. The root of reduced fertility in aged women and possible therapentic options: Current status and future perspects. Mol. Asp. Med. 2014, 38, 54–85. [Google Scholar] [CrossRef]
- Mukherjee, A.; Koli, S.; Reddy, K.V.R. Regulatory non-coding transcripts in spermatogenesis: Shedding light on ‘dark matter’. Andrology 2014, 2, 360–369. [Google Scholar] [CrossRef]
- Shi, B.; Liu, X.Z.; Xu, Y.J.; Wang, S.S. Molecular characterization of three gonadotropin subunits and their expression patterns during ovarian maturation in Cynoglossus semilaevis. Int. J. Mol. Sci. 2015, 16, 2767–2793. [Google Scholar] [CrossRef]
- Shi, B.; Liu, X.Z.; Thomas, P.; Pang, Y.F.; Xu, Y.J.; Li, X.N.; Li, X.X. Identification and characterization of a progestin and adipoQ receptor (PAQR) structurally related to Paqr7 in the ovary of Cynoglossus semilaevis and its potential role in regulating oocyte maturation. Gen. Comp. Endocrinol. 2016, 237, 109–120. [Google Scholar] [CrossRef]
- Cheng, Z.F.; Deutscher, M.P. Purification and characterization of the Escherichia coli Exoribonuclease RNase R: Comparison with RNase II. J. Biol. Chem. 2002, 277, 21624–21629. [Google Scholar] [CrossRef]
- Suzuki, H.; Zuo, Y.H.; Wang, J.H.; Zhang, M.Q.; Malhotra, A.; Mayeda, A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006, 34, e63. [Google Scholar] [CrossRef]
- Yang, Y.B.; Gao, X.Y.; Zhang, M.L.; Yan, S.; Sun, C.J.; Xiao, F.Z.; Huang, N.; Yang, X.S.; Zhao, K.; Zhou, H.K.; et al. Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef]
- Liu, K.S.; Pan, F.; Mao, X.D.; Liu, C.; Chen, Y.J. Biological functions of circular RNAs and their roles in occurrence of reproduction and gynecological diseases. Am. J. Transl. Res. 2019, 11, 1–15. [Google Scholar]
- Zhao, W.; Cheng, Y.H.; Zhang, C.; You, Q.B.; Shen, X.J.; Guo, W.; Jiang, Y.Q. Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci. Rep. 2017, 7, 5636. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.F.; Yang, Z.; Jia, R.B.; Ge, S.F. The novel roles of circRNAs in human cancer. Mol. Cancer. 2019, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.J.; Zhou, Z.J.; Niu, Y.J.; Sun, X.Y.; Deng, Z.P. Identification and functional characterization of tomato circRNAs derived from genes involved in fruit pigment accumulation. Sci. Rep. 2017, 7, 8594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, H.D.; Wang, Z.; Li, T.; Guo, J.L.; Adu-Asiamah, P.; Leng, Q.Y.; An, L.L.; Liu, M.Q.; Zhang, X.Q.; et al. Identification and characterization of circular RNAs in chicken hepatocytes. Growth. Horm. IGF. Res. 2019, 46–47, 16–23. [Google Scholar] [CrossRef]
- Chen, X.; Shi, W.; Chen, C. Differential circular RNAs expression in ovary during oviposition in honey bees. Genomics 2019, 111, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.N.; Zou, Q.; Lv, D.J.; Raza, M.A.; Wang, X.; Li, P.L.; Chen, Y.; Xi, X.Y.; Wen, A.X.; Zhu, L.; et al. Comprehensive transcriptional profiling of porcine brain aging. Gene 2019, 693, 1–9. [Google Scholar] [CrossRef]
- Cai, H.C.; Li, Y.M.; Li, H.M.; Niringiyumukiza, J.D.; Zhang, M.D.; Chen, L.; Chen, G.; Xiang, W.P. Identification and characterization of human ovary-derived circular RNAs and their potential roles in ovarian aging. Aging 2018, 10, 2511–2534. [Google Scholar] [CrossRef]
- Dong, W.W.; Li, H.M.; Qing, X.R.; Huang, D.H.; Li, H.G. Identification and characterization of human testis derived circular RNAs and their existence in seminal plasma. Sci. Rep. 2016, 6, 39080. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wei, X.F.; Yang, J.M.; Dong, D.; Hao, D.; Huang, Y.Z.; Lan, X.Y.; Plath, M.; Lei, C.Z.; Ma, Y.; et al. CircFGFR4 promotes differentiation of myoblasts via binding miR-107 to relieve its inhibition of Wnt3a. Mol. Ther.-Nucl. Acids. 2018, 11, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, X.X.; Du, Q.Y.; Wu, N.; Liu, X.C.; Chen, Y.Q.; Wang, X.J. The circRNA circP4HB promotes NSCLC aggressiveness and metastasis by sponging miR-133a-5p. Biochem. Biophys. Res. Commun. 2019, 513, 904–911. [Google Scholar] [CrossRef]
- Guo, T.Y.; Huang, L.; Yao, W.; Du, X.; Li, Q.Q.; Ma, M.L.; Li, Q.F.; Liu, H.L.; Zhang, J.B.; Pan, Z.X. The potential biological functions of circular RNAs during the initiation of atresia in pig follicles. Domest. Anim. Endocrin. 2020, 72, 106401. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartk, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell. 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.R.; Yang, T.T.; Xiao, J.J. Circular RNAs: Promising biomarkers for human diseases. EBioMedicine 2018, 34, 267–274. [Google Scholar] [CrossRef]
- Chen, W.; Schuman, E. Circular RNAs in brain and other tissues: A functional enigma. Trends. Neurosci. 2016, 39, 597–604. [Google Scholar] [CrossRef]
- Hentze, M.W.; Preiss, T. Circular RNAs: Splicing’s enigma variations. EMBO J. 2013, 32, 923–925. [Google Scholar] [CrossRef]
- Sheng, R.; Li, X.D.; Wang, Z.L.; Wang, X.P. Circular RNAs and their emerging roles as diagnostic and prognostic biomarkers in ovarian cancer. Cancer. Lett. 2020, 473, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Long, B.; Zhang, W.; Yu, M.; Wang, S.; Cao, D.Y.; Yang, J.X.; Shen, K.; Huang, Y.N.; Lang, J.H. Circular RNA profiling reveals circEXOC6B and circN4BP2L2 as novel prognostic biomarkers in epithelial ovarian cancer. Int. J. Oncol. 2018, 53, 2637–2646. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.P.; Bao, C.Y.; Li, S.Y.; Guo, W.J.; Zhao, J.; Chen, D.; Gu, J.R.; He, X.H.; Huang, S.L. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell. Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Y.; Chen, Q.Q.; Yao, T.; Li, T.W.; Ying, S.; Hu, Y.R.; Guo, J.M. Hsa_circ_0005986 inhibits carcinogenesis by acting as a miR-129-5p sponge and is used as a novel biomarker for hepatocellular carcinoma. Oncotarget 2017, 8, 43878–43888. [Google Scholar] [CrossRef]
- Meng, S.J.; Zhou, H.C.; Feng, Z.Y.; Xu, Z.H.; Tang, Y.; Li, P.Y.; Wu, M.H. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 2017, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.L.; Wang, X.Y.; Wei, S.Z.; Chen, Y.; Chen, Y.; Fan, X.B.; Han, S.H.; Wu, G.Q. hsa_circ_0013958: A circular RNA and potential novel biomarker for lung adenocarcinoma. FEBS J. 2017, 284, 2170–2182. [Google Scholar] [CrossRef]
- Shao, J.J.; Wang, L.Q.; Liu, X.Y.; Yang, M.; Chen, H.M.; Wu, B.; Liu, C. Identification and characterization of circular RNAs in Ganoderma lucidum. Sci. Rep. 2019, 9, 16522. [Google Scholar] [CrossRef]
- Reading, B.J.; Hiramatsu, N.; Sawaguchi, S.; Matsubara, T.; Hara, A.; Lively, M.O.; Sullivan, C.V. Conserved and variant molecular and functional features of multiple egg yolk precursor proteins (vitellogenins) in white perch (Morone americana) and other teleosts. Mar. Biotechnol. 2009, 11, 169–187. [Google Scholar] [CrossRef]
- Couse, J.F.; Curtis, S.W.; Washburn, T.F.; Lindzey, J.; Golding, T.S.; Lubahn, D.B.; Smithies, O.; Korach, K.S. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol. Endocrinol. 1995, 9, 1441–1454. [Google Scholar] [CrossRef]
- Lubahn, D.B.; Moyer, J.S.; Golding, T.S.; Couse, J.F.; Korach, K.S.; Smithies, O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 11162–11166. [Google Scholar] [CrossRef]
- Li, W.G.; Zhang, J.R.; Mu, W.J.; Wen, H.S. Cloning, characterization and expression of estrogen receptor beta in the male half-smooth tongue sole, Cynoglossus semilaevis. Fish. Physiol. Biochem. 2013, 39, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Wu, R.S.S.; Ge, W. Luteinizing hormone receptor (lhcgr) as a marker gene for characterizing estrogenic endocrine-disrupting chemicals in zebrafish ovarian follicle cells. Gen. Comp. Endocrinol. 2013, 192, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wohlres-Viana, S.; Arashiro, E.K.N.; Minare, T.P.; Fernandes, C.A.C.; Grazia, J.G.V.; Siqueira, L.G.B.; Machado, M.A.; Viana, J.H.M. Differential expression of LHCGR and its isoforms is associated to the variability in superovulation responses of Gir cattle. Theriogenology 2019, 126, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.Y.; Zhang, J.B.; Yao, W.; Du, X.; Li, Q.Q.; Huang, L.; Ma, M.L.; Li, Q.F.; Liu, H.L.; Pan, Z.X. CircINHA resists granulosa cell apoptosis by upregulating CTGF as a ceRNA of miR-10a-5p in pig ovarian follicles. Biochim. Biophys. Acta Gene. Regul. Mech. 2019, 1862, 194420. [Google Scholar] [CrossRef]
- Shen, M.M.; Li, T.T.; Zhang, G.X.; Wu, P.F.; Chen, F.X.; Lou, Q.H.; Chen, L.; Yin, X.M.; Zhang, T.; Wang, J.Y. Dynamic expression and functional analysis of circRNA in granulosa cells during follicular development in chicken. BMC Genom. 2019, 20, 96. [Google Scholar] [CrossRef]
- Liu, Z.L.; Rudd, M.D.; Hernandez-Gonzalez, I.; Gonzalez-Robayna, I.; Fan, H.-Y.; Zeleznik, A.J.; Richards, J.S. FSH and FOXO1 regulate genes in the sterol/steroid and lipid biosynthetic pathways in granulosa cells. Mol. Endocrinol. 2009, 23, 649–661. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | CircRNA | mRNA | Number of Targets |
|---|---|---|---|
| 3186 | 11,276 | 4887 | 1,841,040 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Shi, B.; Wang, C.; Shao, C.; Liu, X.; Zhang, D. Comprehensive CircRNA Profiling and Selection of Key CircRNAs Reveal the Potential Regulatory Roles of CircRNAs throughout Ovarian Development and Maturation in Cynoglossus semilaevis. Biology 2021, 10, 830. https://doi.org/10.3390/biology10090830
Li J, Shi B, Wang C, Shao C, Liu X, Zhang D. Comprehensive CircRNA Profiling and Selection of Key CircRNAs Reveal the Potential Regulatory Roles of CircRNAs throughout Ovarian Development and Maturation in Cynoglossus semilaevis. Biology. 2021; 10(9):830. https://doi.org/10.3390/biology10090830
Chicago/Turabian StyleLi, Jing, Bao Shi, Chongnv Wang, Changwei Shao, Xuezhou Liu, and Daiqiang Zhang. 2021. "Comprehensive CircRNA Profiling and Selection of Key CircRNAs Reveal the Potential Regulatory Roles of CircRNAs throughout Ovarian Development and Maturation in Cynoglossus semilaevis" Biology 10, no. 9: 830. https://doi.org/10.3390/biology10090830
APA StyleLi, J., Shi, B., Wang, C., Shao, C., Liu, X., & Zhang, D. (2021). Comprehensive CircRNA Profiling and Selection of Key CircRNAs Reveal the Potential Regulatory Roles of CircRNAs throughout Ovarian Development and Maturation in Cynoglossus semilaevis. Biology, 10(9), 830. https://doi.org/10.3390/biology10090830