
Mutual Interactions between Brain States and Alzheimer’s Disease Pathology: A Focus on Gamma and Slow Oscillations

Abstract
:Simple Summary
Abstract
1. Introduction
2. Gamma Oscillations and AD
2.1. Mechanisms of Gamma Oscillations
2.2. Gamma Oscillations and AD in Humans
2.3. Gamma Oscillations and AD in Mouse Models
2.4. Neuromodulation of Gamma Oscillations for AD
3. Slow Oscillations and AD
3.1. Mechanisms of Slow Oscillations
3.2. Slow Oscillations and AD in Humans
3.3. Slow Oscillations and AD in Mouse Models
3.4. Neuromodulation of Slow Oscillations for AD
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buzsáki, G.; Draguhn, A. Neuronal Oscillations in Cortical Networks. Science 2004, 304, 1926–1929. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.; Thiele, A. Cortical state and attention. Nat. Rev. Neurosci. 2011, 12, 509–523. [Google Scholar] [CrossRef]
- Buzsáki, G.; Logothetis, N.; Singer, W. Scaling Brain Size, Keeping Timing: Evolutionary Preservation of Brain Rhythms. Neuron 2013, 80, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Berger, H. Über das Elektrenkephalogramm des Menschen. Eur. Arch. Psychiatry Clin. Neurosci. 1929, 87, 527–570. [Google Scholar] [CrossRef]
- Buzsáki, G.; Wang, X.-J. Mechanisms of Gamma Oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef] [Green Version]
- Engel, A.K.; Fries, P.; Singer, W. Dynamic predictions: Oscillations and synchrony in top–down processing. Nat. Rev. Neurosci. 2001, 2, 704–716. [Google Scholar] [CrossRef]
- Fries, P. Neuronal Gamma-Band Synchronization as a Fundamental Process in Cortical Computation. Annu. Rev. Neurosci. 2009, 32, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Fries, P. Rhythms for Cognition: Communication through Coherence. Neuron 2015, 88, 220–235. [Google Scholar] [CrossRef] [Green Version]
- Singer, W.; Gray, C.M. Visual Feature Integration and the Temporal Correlation Hypothesis. Annu. Rev. Neurosci. 1995, 18, 555–586. [Google Scholar] [CrossRef]
- Varela, F.; Lachaux, J.-P.; Rodriguez, E.; Martinerie, J. The brainweb: Phase synchronization and large-scale integration. Nat. Rev. Neurosci. 2001, 2, 229–239. [Google Scholar] [CrossRef]
- Wang, X.-J. Neurophysiological and Computational Principles of Cortical Rhythms in Cognition. Physiol. Rev. 2010, 90, 1195–1268. [Google Scholar] [CrossRef]
- Miller, E.K.; Lundqvist, M.; Bastos, A.M. Working Memory 2.0. Neuron 2018, 100, 463–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamantidis, A.R.; Herrera, C.G.; Gent, T.C. Oscillating circuitries in the sleeping brain. Nat. Rev. Neurosci. 2019, 20, 746–762. [Google Scholar] [CrossRef]
- Brown, R.; Basheer, R.; McKenna, J.; Strecker, R.E.; McCarley, R. Control of Sleep and Wakefulness. Physiol. Rev. 2012, 92, 1087–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzsáki, G. Theta Oscillations in the Hippocampus. Neuron 2002, 33, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Buzsáki, G. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 2015, 25, 1073–1188. [Google Scholar] [CrossRef] [PubMed]
- Callaway, C.; Lydic, R.; Baghdoyan, H.A.; Hobson, J.A. Pontogeniculooccipital waves: Spontaneous visual system activity during rapid eye movement sleep. Cell. Mol. Neurobiol. 1987, 7, 105–149. [Google Scholar] [CrossRef]
- Colgin, L.L. Rhythms of the hippocampal network. Nat. Rev. Neurosci. 2016, 17, 239–249. [Google Scholar] [CrossRef]
- Datta, S. Cellular basis of pontine ponto-geniculo-occipital wave generation and modulation. Cell. Mol. Neurobiol. 1997, 17, 341–365. [Google Scholar] [CrossRef]
- Steriade, M.; Nunez, A.; Amzica, F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: Depolarizing and hyperpolarizing components. J. Neurosci. 1993, 13, 3252–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunematsu, T.; Patel, A.A.; Onken, A.; Sakata, S. State-dependent brainstem ensemble dynamics and their interactions with hippocampus across sleep states. eLife 2020, 9, 9. [Google Scholar] [CrossRef]
- Mestre, H.; Mori, Y.; Nedergaard, M. The Brain’s Glymphatic System: Current Controversies. Trends Neurosci. 2020, 43, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Buzsáki, G. Two-stage model of memory trace formation: A role for “noisy” brain states. Neuroscience 1989, 31, 551–570. [Google Scholar] [CrossRef]
- Fernández-Ruiz, A.; Oliva, A.; De Oliveira, E.F.; Rocha-Almeida, F.; Tingley, D.; Buzsáki, G. Long-duration hippocampal sharp wave ripples improve memory. Science 2019, 364, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gulati, T.; Ganguly, K. Competing Roles of Slow Oscillations and Delta Waves in Memory Consolidation versus Forgetting. Cell 2019, 179, 514–526. [Google Scholar] [CrossRef]
- Born, J.; Rasch, B.; Gais, S. Sleep to Remember. Neuroscience 2006, 12, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Stickgold, R.; Hobson, J.A.; Fosse, R.; Fosse, M. Sleep, Learning, and Dreams: Off-line Memory Reprocessing. Science 2001, 294, 1052–1057. [Google Scholar] [CrossRef] [Green Version]
- Todorova, R.; Zugaro, M. Isolated cortical computations during delta waves support memory consolidation. Science 2019, 366, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Berger, H. Über das Elektrenkephalogramm des Menschen. Dritte Mitteilung. Eur. Arch. Psychiatry Clin. Neurosci. 1931, 94, 16–60. [Google Scholar] [CrossRef]
- Jeong, J. EEG dynamics in patients with Alzheimer’s disease. Clin. Neurophysiol. 2004, 115, 1490–1505. [Google Scholar] [CrossRef] [PubMed]
- Mably, A.J.; Colgin, L.L. Gamma oscillations in cognitive disorders. Curr. Opin. Neurobiol. 2018, 52, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Berényi, A. Oscillotherapeutics—Time-targeted interventions in epilepsy and beyond. Neurosci. Res. 2020, 152, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef]
- Rojas, D.C.; Wilson, L.B. γ-band abnormalities as markers of autism spectrum disorders. Biomark. Med. 2014, 8, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.M.; Wallace, M.T. Dysfunction of sensory oscillations in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2016, 68, 848–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskaran, A.; Milev, R.; McIntyre, R.S. The neurobiology of the EEG biomarker as a predictor of treatment response in depression. Neuropharmacology 2012, 63, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.J.; Watson, B.O. Gamma oscillations as a biomarker for major depression: An emerging topic. Transl. Psychiatry 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Osipova, D.; Pekkonen, E.; Ahveninen, J. Enhanced magnetic auditory steady-state response in early Alzheimer’s disease. Clin. Neurophysiol. 2006, 117, 1990–1995. [Google Scholar] [CrossRef]
- Stam, C.J.; van Cappellen van Walsum, A.M.; Pijnenburg, Y.A.L.; Berendse, H.W.; de Munck, J.C.; Scheltens, P.; Van Dijk, B.W. Generalized Synchronization of MEG Recordings in Alzheimer’s Disease: Evidence for Involvement of the Gamma Band. J. Clin. Neurophysiol. 2002, 19, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.A.; Vuurman, E.F.P.M.; Verhey, F.R.J.; Van Kranen-Mastenbroek, V.H.J.M.; Riedel, W. Increased EEG gamma band activity in Alzheimer’s disease and mild cognitive impairment. J. Neural Transm. 2008, 115, 1301–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, D.E.; Kamerow, D.B. Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? JAMA 1989, 262, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Kaskie, E.R.; Graziano, B.; Ferrarelli, F. Schizophrenia and sleep disorders: Links, risks, and management challenges. Nat. Sci. Sleep 2017, 9, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoach, D.S.; Stickgold, R. Abnormal Sleep Spindles, Memory Consolidation, and Schizophrenia. Annu. Rev. Clin. Psychol. 2019, 15, 451–479. [Google Scholar] [CrossRef] [PubMed]
- Dolsen, M.R.; Harvey, A.G. Life-time history of insomnia and hypersomnia symptoms as correlates of alcohol, cocaine and heroin use and relapse among adults seeking substance use treatment in the United States from 1991 to 1994. Addiction 2017, 112, 1104–1111. [Google Scholar] [CrossRef]
- Kent, B.A.; Feldman, H.H.; Nygaard, H.B. Sleep and its regulation: An emerging pathogenic and treatment frontier in Alzheimer’s disease. Prog. Neurobiol. 2021, 197, 101902. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Gerashchenko, D.; Timofeev, I.; Bacskai, B.J.; Kastanenka, K.V. Slow Wave Sleep Is a Promising Intervention Target for Alzheimer’s Disease. Front. Neurosci. 2020, 14, 705. [Google Scholar] [CrossRef]
- Wang, C.; Holtzman, D.M. Bidirectional relationship between sleep and Alzheimer’s disease: Role of amyloid, tau, and other factors. Neuropsychopharmacology 2020, 45, 104–120. [Google Scholar] [CrossRef]
- Mander, B.A. Local Sleep and Alzheimer’s Disease Pathophysiology. Front. Neurosci. 2020, 14, 525970. [Google Scholar] [CrossRef]
- Krames, E.; Peckham, P.H.; Rezai, A. Neuromodulation: Comprehensive Textbook of Principles, Technologies, and Terapies, 2nd ed.; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Benabid, A.; Pollak, P.; Hoffmann, D.; Gervason, C.; Hommel, M.; Perret, J.; De Rougemont, J.; Gao, D. Long-term suppression of tremor by chronic stimulation of the ventral intermediate thalamic nucleus. Lancet 1991, 337, 403–406. [Google Scholar] [CrossRef]
- Scangos, K.W.; Makhoul, G.S.; Sugrue, L.P.; Chang, E.F.; Krystal, A.D. State-dependent responses to intracranial brain stimulation in a patient with depression. Nat. Med. 2021, 27, 229–231. [Google Scholar] [CrossRef]
- Mayberg, H.S.; Lozano, A.M.; Voon, V.; McNeely, H.E.; Seminowicz, D.; Hamani, C.; Schwalb, J.M.; Kennedy, S. Deep Brain Stimulation for Treatment-Resistant Depression. Neuron 2005, 45, 651–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, S.; Nguyen, J.A.; Viswanathan, V.; Reinhart, R.M.G. High-frequency neuromodulation improves obsessive–compulsive behavior. Nat. Med. 2021, 27, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.D.; Malone, D.A.; Friehs, G.M.; Rezai, A.R.; Kubu, C.S.; Malloy, P.F.; Salloway, S.P.; Okun, M.; Goodman, W.K.; Rasmussen, S.A. Three-Year Outcomes in Deep Brain Stimulation for Highly Resistant Obsessive–Compulsive Disorder. Neuropsychopharmacology 2006, 31, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Mirzadeh, Z.; Bari, A.; Lozano, A.M. The rationale for deep brain stimulation in Alzheimer’s disease. J. Neural Transm. 2015, 123, 775–783. [Google Scholar] [CrossRef]
- Kuhn, J.; Hardenacke, K.; Lenartz, D.; Gruendler, T.; Ullsperger, M.; Bartsch, C.; Mai, J.K.; Zilles, K.; Bauer, A.; Matusch, A.; et al. Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer’s dementia. Mol. Psychiatry 2014, 20, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Mellin, J.M.; Alagapan, S.; Alexander, M.L.; Gilmore, J.H.; Jarskog, L.F.; Fröhlich, F. Targeting reduced neural oscillations in patients with schizophrenia by transcranial alternating current stimulation. NeuroImage 2019, 186, 126–136. [Google Scholar] [CrossRef]
- Brunelin, J.; Mondino, M.; Gassab, L.; Haesebaert, F.; Gaha, L.; Suaud-Chagny, M.-F.; Saoud, M.; Mechri, A.; Poulet, E. Examining Transcranial Direct-Current Stimulation (tDCS) as a Treatment for Hallucinations in Schizophrenia. Am. J. Psychiatry 2012, 169, 719–724. [Google Scholar] [CrossRef]
- Farzan, F.; Barr, M.S.; Sun, Y.; Fitzgerald, P.B.; Daskalakis, Z.J. Transcranial magnetic stimulation on the modulation of gamma oscillations in schizophrenia. Ann. New York Acad. Sci. 2012, 1265, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.D.; Buckner, R.L.; White, M.P.; Greicius, M.D.; Pascual-Leone, A. Efficacy of Transcranial Magnetic Stimulation Targets for Depression Is Related to Intrinsic Functional Connectivity with the Subgenual Cingulate. Biol. Psychiatry 2012, 72, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Terraneo, A.; Leggio, L.; Saladini, M.; Ermani, M.; Bonci, A.; Gallimberti, L. Transcranial magnetic stimulation of dorsolateral prefrontal cortex reduces cocaine use: A pilot study. Eur. Neuropsychopharmacol. 2016, 26, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diana, M.; Raij, T.; Melis, M.; Nummenmaa, A.; Leggio, L.; Bonci, A. Rehabilitating the addicted brain with transcranial magnetic stimulation. Nat. Rev. Neurosci. 2017, 18, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Holczer, A.; Németh, V.L.; Vékony, T.; Vécsei, L.; Klivenyi, P.; Must, A. Non-invasive Brain Stimulation in Alzheimer’s Disease and Mild Cognitive Impairment—A State-of-the-Art Review on Methodological Characteristics and Stimulation Parameters. Front. Hum. Neurosci. 2020, 14, 179. [Google Scholar] [CrossRef]
- Freitas, C.; Mondragón-Llorca, H.; Pascual-Leone, A. Noninvasive brain stimulation in Alzheimer’s disease: Systematic review and perspectives for the future. Exp. Gerontol. 2011, 46, 611–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bréchet, L.; Michel, C.M.; Schacter, D.L.; Pascual-Leone, A. Improving autobiographical memory in Alzheimer’s disease by transcranial alternating current stimulation. Curr. Opin. Behav. Sci. 2021, 40, 64–71. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Long, J.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Musiek, E.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ’wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Sweeney, M.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Belloy, M.; Napolioni, V.; Greicius, M.D. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 2019, 101, 820–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef]
- Golde, T.E.; DeKosky, S.T.; Galasko, D. Alzheimer’s disease: The right drug, the right time. Science 2018, 362, 1250–1251. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dementia: Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Sierksma, A.; Escott-Price, V.; De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 2020, 370, 61–66. [Google Scholar] [CrossRef]
- Pérez, M.; Hernández, F.; Avila, J. Protein Biomarkers for the Diagnosis of Alzheimer’s Disease at Different Stages of Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 6749. [Google Scholar] [CrossRef]
- Adaikkan, C.; Tsai, L.-H. Gamma Entrainment: Impact on Neurocircuits, Glia, and Therapeutic Opportunities. Trends Neurosci. 2020, 43, 24–41. [Google Scholar] [CrossRef]
- Kastanenka, K.V.; Hou, S.S.; Shakerdge, N.; Logan, R.; Feng, D.; Wegmann, S.; Chopra, V.; Hawkes, J.M.; Chen, X.; Bacskai, B.J. Optogenetic Restoration of Disrupted Slow Oscillations Halts Amyloid Deposition and Restores Calcium Homeostasis in an Animal Model of Alzheimer’s Disease. PLoS ONE 2017, 12, e0170275. [Google Scholar] [CrossRef]
- Etter, G.; Van Der Veldt, S.; Manseau, F.; Zarrinkoub, I.; Trillaud-Doppia, E.; Williams, S. Optogenetic gamma stimulation rescues memory impairments in an Alzheimer’s disease mouse model. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nat. Cell Biol. 2016, 540, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorell, A.J.; Paulson, A.; Suk, H.-J.; Abdurrob, F.; Drummond, G.T.; Guan, W.; Young, J.Z.; Kim, D.N.-W.; Kritskiy, O.; Barker, S.J.; et al. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019, 177, 256–271.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaikkan, C.; Middleton, S.; Marco, A.; Pao, P.-C.; Mathys, H.; Kim, D.N.-W.; Gao, F.; Young, J.Z.; Suk, H.-J.; Boyden, E.S.; et al. Gamma Entrainment Binds Higher-Order Brain Regions and Offers Neuroprotection. Neuron 2019, 102, 929–943.e8. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Kekuš, M.; Adelsberger, H.; Noda, T.; Förstl, H.; Nelken, I.; Konnerth, A. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat. Neurosci. 2015, 18, 1623–1630. [Google Scholar] [CrossRef]
- Isla, A.G.; Balleza-Tapia, H.; Fisahn, A. Efficacy of preclinical pharmacological interventions against alterations of neuronal network oscillations in Alzheimer’s disease: A systematic review. Exp. Neurol. 2021, 343, 113743. [Google Scholar] [CrossRef]
- Jasper, H.H.; Andrews, H.L. Brain potentials and voluntary muscle activity in man. J. Neurophysiol. 1938, 1, 87–100. [Google Scholar] [CrossRef]
- Bressler, S.L.; Freeman, W.J. Frequency analysis of olfactory system EEG in cat, rabbit, and rat. Electroencephalogr. Clin. Neurophysiol. 1980, 50, 19–24. [Google Scholar] [CrossRef]
- Freeman, W.J. Mass Action in the Nervous System; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Nakazono, T.; Lam, T.N.; Patel, A.Y.; Kitazawa, M.; Saito, T.; Saido, T.C.; Igarashi, K.M. Impaired In Vivo Gamma Oscillations in the Medial Entorhinal Cortex of Knock-in Alzheimer Model. Front. Syst. Neurosci. 2017, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, J.J.; Buzsaki, G. Gamma Oscillations in the Entorhinal Cortex of the Freely Behaving Rat. J. Neurosci. 1998, 18, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Charpak, S.; Paré, D.; Llinás, R. The Entorhinal Cortex Entrains Fast CA1 Hippocampal Oscillations in the Anaesthetized Guinea-pig: Role of the Monosynaptic Component of the Perforant Path. Eur. J. Neurosci. 1995, 7, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.P.; Paz, R.; Paré, D. Gamma Oscillations Coordinate Amygdalo-Rhinal Interactions during Learning. J. Neurosci. 2007, 27, 9369–9379. [Google Scholar] [CrossRef]
- Harris, K.; Csicsvari, J.; Hirase, H.; Dragoi, G.; Buzsaki, G. Organization of cell assemblies in the hippocampus. Nat. Cell Biol. 2003, 424, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G.; Leung, L.-W.S.; Vanderwolf, C.H. Cellular bases of hippocampal EEG in the behaving rat. Brain Res. Rev. 1983, 6, 139–171. [Google Scholar] [CrossRef]
- Wang, X.-J.; Buzsaki, G. Gamma Oscillation by Synaptic Inhibition in a Hippocampal Interneuronal Network Model. J. Neurosci. 1996, 16, 6402–6413. [Google Scholar] [CrossRef]
- Cohen, M.X.; Axmacher, N.; Lenartz, D.; Elger, C.E.; Sturm, V.; Schlaepfer, T. Good Vibrations: Cross-frequency Coupling in the Human Nucleus Accumbens during Reward Processing. J. Cogn. Neurosci. 2009, 21, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.T.; Popa, D.; Pare, D. Coherent gamma oscillations couple the amygdala and striatum during learning. Nat. Neurosci. 2009, 12, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beshel, J.; Kopell, N.; Kay, L.M. Olfactory Bulb Gamma Oscillations Are Enhanced with Task Demands. J. Neurosci. 2007, 27, 8358–8365. [Google Scholar] [CrossRef]
- Eeckman, F.H.; Freeman, W.J. Correlations between unit firing and EEG in the rat olfactory system. Brain Res. 1990, 528, 238–244. [Google Scholar] [CrossRef]
- Yague, J.G.; Tsunematsu, T.; Sakata, S. Distinct Temporal Coordination of Spontaneous Population Activity between Basal Forebrain and Auditory Cortex. Front. Neural Circuits 2017, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, J.; Klaassen, A.-L.; Arato, J.; Vyssotski, A.L.; Harvey, M.; Rainer, G. Basal forebrain contributes to default mode network regulation. Proc. Natl. Acad. Sci. USA 2018, 115, 1352–1357. [Google Scholar] [CrossRef] [Green Version]
- Minlebaev, M.; Colonnese, M.; Tsintsadze, T.; Sirota, A.; Khazipov, R. Early Gamma Oscillations Synchronize Developing Thalamus and Cortex. Science 2011, 334, 226–229. [Google Scholar] [CrossRef]
- Jensen, O.; Kaiser, J.; Lachaux, J.-P. Human gamma-frequency oscillations associated with attention and memory. Trends Neurosci. 2007, 30, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.W.; Rizzuto, D.S.; Caplan, J.B.; Madsen, J.R.; Lisman, J.; Aschenbrenner-Scheibe, R.; Schulze-Bonhage, A.; Kahana, M.J. Gamma Oscillations Correlate with Working Memory Load in Humans. Cereb. Cortex 2003, 13, 1369–1374. [Google Scholar] [CrossRef] [Green Version]
- Osipova, D.; Takashima, A.; Oostenveld, R.; Fernandez, G.; Maris, E.; Jensen, O. Theta and Gamma Oscillations Predict Encoding and Retrieval of Declarative Memory. J. Neurosci. 2006, 26, 7523–7531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiesinga, P.; Sejnowski, T.J. Cortical Enlightenment: Are Attentional Gamma Oscillations Driven by ING or PING? Neuron 2009, 63, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Whittington, M.; Traub, R.; Kopell, N.; Ermentrout, B.; Buhl, E. Inhibition-based rhythms: Experimental and mathematical observations on network dynamics. Int. J. Psychophysiol. 2000, 38, 315–336. [Google Scholar] [CrossRef] [Green Version]
- Traub, R.D.; Whittington, M.A.; Stanford, I.M.; Jefferys, J.G.R. A mechanism for generation of long-range synchronous fast oscillations in the cortex. Nat. Cell Biol. 1996, 383, 621–624. [Google Scholar] [CrossRef]
- Lytton, W.W.; Sejnowski, T.J. Simulations of cortical pyramidal neurons synchronized by inhibitory interneurons. J. Neurophysiol. 1991, 66, 1059–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traub, R.D.; Whittington, M.A.; Colling, S.B.; Buzsaki, G.; Jefferys, J.G. Analysis of gamma rhythms in the rat hippocampus in vitro and in vivo. J. Physiol. 1996, 493, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Whittington, M.A.; Traub, R.D.; Jefferys, J. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nat. Cell Biol. 1995, 373, 612–615. [Google Scholar] [CrossRef]
- Cardin, J.A.; Carlen, M.; Meletis, K.; Knoblich, U.; Zhang, F.; Deisseroth, K.; Tsai, L.H.; Moore, C.I. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009, 459, 663–667. [Google Scholar] [CrossRef] [Green Version]
- Sohal, V.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nat. Cell Biol. 2009, 459, 698–702. [Google Scholar] [CrossRef] [Green Version]
- Jefferys, J.G.; Traub, R.D.; Whittington, M.A. Neuronal networks for induced ‘40 Hz’ rhythms. Trends Neurosci. 1996, 19, 202–208. [Google Scholar] [CrossRef]
- Wilson, H.R.; Cowan, J.D. Excitatory and Inhibitory Interactions in Localized Populations of Model Neurons. Biophys. J. 1972, 12, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Tiesinga, P.; Fellous, J.-M.; Sejnowski, T.J. Regulation of spike timing in visual cortical circuits. Nat. Rev. Neurosci. 2008, 9, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Fisahn, A.; Pike, F.G.; Buhl, E.H.; Paulsen, O. Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nat. Cell Biol. 1998, 394, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Galter, D.; Papadia, D.; Fisahn, A. Histamine induces KCNQ channel-dependent gamma oscillations in rat hippocampus via activation of the H1 receptor. Neuropharmacology 2017, 118, 13–25. [Google Scholar] [CrossRef]
- Gray, C.M.; McCormick, D.A. Chattering Cells: Superficial Pyramidal Neurons Contributing to the Generation of Synchronous Oscillations in the Visual Cortex. Science 1996, 274, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Ribary, U.; Ioannides, A.A.; Singh, K.; Hasson, R.; Bolton, J.P.; Lado, F.; Mogilner, A.; Llinas, R. Magnetic field tomography of coherent thalamocortical 40-Hz oscillations in humans. Proc. Natl. Acad. Sci. USA 1991, 88, 11037–11041. [Google Scholar] [CrossRef] [Green Version]
- Başar, E.; Emek-Savaş, D.D.; Güntekin, B.; Yener, G.G. Delay of cognitive gamma responses in Alzheimer’s disease. NeuroImage: Clin. 2016, 11, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.; Vuurman, E.; van Kranen-Mastenbroek, V.; Verhey, F.; Riedel, W. 40-Hz steady state response in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2011, 32, 24–30. [Google Scholar] [CrossRef]
- Rossini, P.; Del Percio, C.; Pasqualetti, P.; Cassetta, E.; Binetti, G.; Forno, G.D.; Ferreri, F.; Frisoni, G.B.; Chiovenda, P.; Miniussi, C.; et al. Conversion from mild cognitive impairment to Alzheimer’s disease is predicted by sources and coherence of brain electroencephalography rhythms. Neuroscience 2006, 143, 793–803. [Google Scholar] [CrossRef]
- Başar, E.; Femir, B.; Emek-Savaş, D.D.; Güntekin, B.; Yener, G.G. Increased long distance event-related gamma band connectivity in Alzheimer’s disease. NeuroImage: Clin. 2017, 14, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, Y.; Wang, X.; Yang, H.; Yu, X.; Wang, H. Enhanced Gamma Activity and Cross-Frequency Interaction of Resting-State Electroencephalographic Oscillations in Patients with Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, T.; Prichep, L.; Dierks, T.; Hubl, D.; Wahlund, L.; John, E.; Jelic, V. Decreased EEG synchronization in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2005, 26, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaubert, S.; Raimondo, F.; Houot, M.; Corsi, M.-C.; Naccache, L.; Sitt, J.D.; Hermann, B.; Oudiette, D.; Gagliardi, G.; Habert, M.-O.; et al. EEG evidence of compensatory mechanisms in preclinical Alzheimer’s disease. Brain 2019, 142, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Knopman, D.S.; Gunter, J.L.; Graff-Radford, J.; Vemuri, P.; Boeve, B.F.; Petersen, R.C.; Weiner, M.W.; Jack, C.R. Cascading network failure across the Alzheimer’s disease spectrum. Brain 2016, 139, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Nebes, R.; Snitz, B.; Cohen, A.; Mathis, C.; Price, J.; Weissfeld, L.; Klunk, W.; Aizenstein, H.J. Regional amyloid burden and intrinsic connectivity networks in cognitively normal elderly subjects. Brain 2014, 137, 3327–3338. [Google Scholar] [CrossRef] [Green Version]
- Mormino, E.C.; Smiljic, A.; Hayenga, A.O.; Onami, S.H.; Greicius, M.D.; Rabinovici, G.D.; Janabi, M.; Baker, S.L.; Yen, I.V.; Madison, C.M.; et al. Relationships between Beta-Amyloid and Functional Connectivity in Different Components of the Default Mode Network in Aging. Cereb. Cortex 2011, 21, 2399–2407. [Google Scholar] [CrossRef]
- Klein, A.S.; Donoso, J.R.; Kempter, R.; Schmitz, D.; Beed, P. Early Cortical Changes in Gamma Oscillations in Alzheimer’s Disease. Front. Syst. Neurosci. 2016, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Verret, L.; Mann, E.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Losa, M.; Tracy, T.E.; Ma, K.; Verret, L.; Clemente-Perez, A.; Khan, A.S.; Cobos, I.; Ho, K.; Gan, L.; Mucke, L.; et al. Nav1.1-Overexpressing Interneuron Transplants Restore Brain Rhythms and Cognition in a Mouse Model of Alzheimer’s Disease. Neuron 2018, 98, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Goutagny, R.; Gu, N.; Cavanagh, C.; Jackson, J.; Chabot, J.-G.; Quirion, R.; Krantic, S.; Williams, S. Alterations in hippocampal network oscillations and theta-gamma coupling arise before Aβ overproduction in a mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2013, 37, 1896–1902. [Google Scholar] [CrossRef]
- Hamm, V.; Héraud, C.; Bott, J.-B.; Herbeaux, K.; Strittmatter, C.; Mathis, C.; Goutagny, R. Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer’s disease. Sci. Adv. 2017, 3, e1601068. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.A.; Gillespie, A.K.; Yoon, S.Y.; Frank, L.; Huang, Y. Early Hippocampal Sharp-Wave Ripple Deficits Predict Later Learning and Memory Impairments in an Alzheimer’s Disease Mouse Model. Cell Rep. 2019, 29, 2123–2133.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, A.K.; Jones, E.A.; Lin, Y.-H.; Karlsson, M.P.; Kay, K.; Yoon, S.Y.; Tong, L.M.; Nova, P.; Carr, J.S.; Frank, L.; et al. Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron 2016, 90, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Bégard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhong, W.; Brankačk, J.; Weyer, S.W.; Müller, U.C.; Tort, A.B.L.; Draguhn, A. Impaired theta-gamma coupling in APP-deficient mice. Sci. Rep. 2016, 6, 21948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, A.; Petrache, A.L.; Shi, J.; Ali, A.B. Preserved Calretinin Interneurons in an App Model of Alzheimer’s Disease Disrupt Hippocampal Inhibition via Upregulated P2Y1 Purinoreceptors. Cereb. Cortex 2019, 30, 1272–1290. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Possenti, A.; Freer, R.; Nakano, Y.; Villegas, N.C.H.; Tang, M.; Cauhy, P.V.M.; Lassus, B.A.; Chen, S.; Fowler, S.L.; et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 2018, 22, 47–56. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Zalocusky, K.A.; Najm, R.; Taubes, A.L.; Hao, Y.; Yoon, S.Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Bennett, D.A.; Bant, J.; et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef]
- Roussarie, J.-P.; Yao, V.; Rodriguez, P.R.; Oughtred, R.; Rust, J.; Plautz, Z.; Kasturia, S.; Albornoz, C.; Wang, W.; Schmidt, E.F.; et al. Selective Neuronal Vulnerability in Alzheimer’s Disease: A Network-Based Analysis. Neuron 2020, 107, 821–835.e12. [Google Scholar] [CrossRef]
- Ahnaou, A.; Moechars, D.; Raeymaekers, L.; Biermans, R.; Manyakov, N.V.; Bottelbergs, A.; Wintmolders, C.; Van Kolen, K.; Van De Casteele, T.; Kemp, J.A.; et al. Emergence of early alterations in network oscillations and functional connectivity in a tau seeding mouse model of Alzheimer’s disease pathology. Sci. Rep. 2017, 7, 14189. [Google Scholar] [CrossRef] [Green Version]
- Hermann, D.; Both, M.; Ebert, U.; Gross, G.; Schoemaker, H.; Draguhn, A.; Wicke, K.; Nimmrich, V. Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein–overexpressing mice without altering behaviorally-correlated sharp wave–ripple complexes. Neuroscience 2009, 162, 1081–1090. [Google Scholar] [CrossRef]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2020, 6. [Google Scholar] [CrossRef]
- Wilson, C.A.; Fouda, S.; Sakata, S. Effects of optogenetic stimulation of basal forebrain parvalbumin neurons on Alzheimer’s disease pathology. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Bobola, M.; Chen, L.; Ezeokeke, C.; Olmstead, T.; Nguyen, C.; Sahota, A.; Williams, R.; Mourad, P. Transcranial focused ultrasound, pulsed at 40 Hz, activates microglia acutely and reduces Aβ load chronically, as demonstrated in vivo. Brain Stimul. 2020, 13, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-S.; Park, H.-S.; Kim, C.-J.; Kang, H.-S.; Kim, D.-H.; Baek, S.-S.; Kim, T.-W. Physical exercise during exposure to 40-Hz light flicker improves cognitive functions in the 3xTg mouse model of Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Yao, Y.; Ying, Y.; Deng, Q.; Zhang, W.; Zhu, H.; Lin, Z.; Zhang, S.; Ma, J.; Zhao, Y. Non-invasive 40-Hz Light Flicker Ameliorates Alzheimer’s-Associated Rhythm Disorder via Regulating Central Circadian Clock in Mice. Front. Physiol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Freund, T.F.; Meskenaite, V. gamma-Aminobutyric acid-containing basal forebrain neurons innervate inhibitory interneurons in the neocortex. Proc. Natl. Acad. Sci. USA 1992, 89, 738–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klunk, W.; Bacskai, B.J.; Mathis, C.A.; Kajdasz, S.T.; McLellan, M.E.; Frosch, M.P.; Debnath, M.L.; Holt, D.P.; Wang, Y.; Hyman, B.T. Imaging Aβ Plaques in Living Transgenic Mice with Multiphoton Microscopy and Methoxy-X04, a Systemically Administered Congo Red Derivative. J. Neuropathol. Exp. Neurol. 2002, 61, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Steriade, M.; Nuñez, A.; Amzica, F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J. Neurosci. 1993, 13, 3266–3283. [Google Scholar] [CrossRef]
- Steriade, M.; Contreras, D.; Dossi, R.C.; Nunez, A. The slow (<1 Hz) oscillation in reticular thalamic and thalamocortical neurons: Scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J. Neurosci 1993, 13, 3284–3299. [Google Scholar] [CrossRef] [Green Version]
- Sakata, S.; Harris, K.D. Laminar-dependent effects of cortical state on auditory cortical spontaneous activity. Front. Neural Circuits 2012, 6, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, S.; Harris, K.D. Laminar Structure of Spontaneous and Sensory-Evoked Population Activity in Auditory Cortex. Neuron 2009, 64, 404–418. [Google Scholar] [CrossRef] [Green Version]
- Luczak, A.; Bartho, P.; Marguet, S.L.; Buzsaki, G.; Harris, K.D. Sequential structure of neocortical spontaneous activity in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Chauvette, S.; Volgushev, M.; Timofeev, I. Origin of Active States in Local Neocortical Networks during Slow Sleep Oscillation. Cereb. Cortex 2010, 20, 2660–2674. [Google Scholar] [CrossRef] [Green Version]
- Timofeev, I.; Grenier, F.; Bazhenov, M.; Sejnowski, T.; Steriade, M. Origin of Slow Cortical Oscillations in Deafferented Cortical Slabs. Cereb. Cortex 2000, 10, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isomura, Y.; Sirota, A.; Özen, S.; Montgomery, S.; Mizuseki, K.; Henze, D.A.; Buzsáki, G. Integration and Segregation of Activity in Entorhinal-Hippocampal Subregions by Neocortical Slow Oscillations. Neuron 2006, 52, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.W.; Cope, D.W.; Blethyn, K.L.; Crunelli, V. Cellular Mechanisms of the Slow (<1 Hz) oscillation in thalamocortical neurons in vitro. Neuron 2002, 33, 947–958. [Google Scholar] [CrossRef] [Green Version]
- David, F.; Schmiedt, J.T.; Taylor, H.L.; Orban, G.; Di Giovanni, G.; Uebele, V.N.; Renger, J.J.; Lambert, R.C.; LeResche, N.; Crunelli, V. Essential thalamic contribution to slow waves of natural sleep. J. Neurosci. 2013, 33, 19599–19610. [Google Scholar] [CrossRef] [Green Version]
- Blethyn, K.L.; Hughes, S.W.; Tóth, T.I.; Cope, D.W.; Crunelli, V. Neuronal Basis of the Slow (<1 Hz) oscillation in neurons of the nucleus reticularis thalami in vitro. J. Neurosci 2006, 26, 2474–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.J. Chapter 18 The generation of natural firing patterns in neostriatal neurons. Prog. Brain Res. 1993, 99, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Groves, P.M. Spontaneous firing patterns of identified spiny neurons in the rat neostriatum. Brain Res. 1981, 220, 67–80. [Google Scholar] [CrossRef]
- Mena-Segovia, J.; Sims, H.M.; Magill, P.J.; Bolam, J.P. Cholinergic brainstem neurons modulate cortical gamma activity during slow oscillations. J. Physiol. 2008, 586, 2947–2960. [Google Scholar] [CrossRef]
- Narikiyo, K.; Mizuguchi, R.; Ajima, A.; Shiozaki, M.; Hamanaka, H.; Johansen, J.P.; Mori, K.; Yoshihara, Y. The claustrum coordinates cortical slow-wave activity. Nat. Neurosci. 2020, 23, 741–753. [Google Scholar] [CrossRef]
- Timofeev, I.; Chauvette, S. Sleep slow oscillation and plasticity. Curr. Opin. Neurobiol. 2017, 44, 116–126. [Google Scholar] [CrossRef]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-β Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [Green Version]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef]
- Gagnon, J.; Lafreniere, A.; Rauchs, G.; Petit, D.; Carrier, J. Sleep in normal aging, Alzheimer’s disease, and mild cognitive impairment. In Handbook of Sleep Research; Dringenberg, H.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 30, pp. 677–692. [Google Scholar]
- Ning, S.; Jorfi, M. Beyond the sleep-amyloid interactions in Alzheimer’s disease pathogenesis. J. Neurophysiol. 2019, 122, 1–4. [Google Scholar] [CrossRef]
- Sanchez-Vives, M.V. Origin and dynamics of cortical slow oscillations. Curr. Opin. Physiol. 2020, 15, 217–223. [Google Scholar] [CrossRef]
- Noya, S.B.; Colameo, D.; Brüning, F.; Spinnler, A.; Mircsof, D.; Opitz, L.; Mann, M.; Tyagarajan, S.K.; Robles, M.S.; Brown, S.A. The forebrain synaptic transcriptome is organized by clocks but its proteome is driven by sleep. Science 2019, 366, eaav2642. [Google Scholar] [CrossRef]
- Brüning, F.; Noya, S.B.; Bange, T.; Koutsouli, S.; Rudolph, J.D.; Tyagarajan, S.K.; Cox, J.; Mann, M.; Brown, S.A.; Robles, M.S. Sleep-wake cycles drive daily dynamics of synaptic phosphorylation. Science 2019, 366, eaav3617. [Google Scholar] [CrossRef]
- Borbély, A.A. A two process model of sleep regulation. Hum. Neurobiol. 1982, 1, 195–204. [Google Scholar]
- Timofeev, I.; Steriade, M. Low-frequency rhythms in the thalamus of intact-cortex and decorticated cats. J. Neurophysiol. 1996, 76, 4152–4168. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vives, M.; McCormick, D.A. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 2000, 3, 1027–1034. [Google Scholar] [CrossRef]
- Beltramo, R.; D’Urso, G.; Maschio, M.D.; Farisello, P.; Bovetti, S.; Clovis, Y.; Lassi, G.; Tucci, V.; Tonelli, D.D.P.; Fellin, T. Layer-specific excitatory circuits differentially control recurrent network dynamics in the neocortex. Nat. Neurosci. 2013, 16, 227–234. [Google Scholar] [CrossRef]
- Levenstein, D.; Buzsáki, G.; Rinzel, J. NREM sleep in the rodent neocortex and hippocampus reflects excitable dynamics. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.; Tononi, G. Modeling Sleep and Wakefulness in the Thalamocortical System. J. Neurophysiol. 2005, 93, 1671–1698. [Google Scholar] [CrossRef]
- Destexhe, A. Self-sustained asynchronous irregular states and Up–Down states in thalamic, cortical and thalamocortical networks of nonlinear integrate-and-fire neurons. J. Comput. Neurosci. 2009, 27, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Bazhenov, M.; Timofeev, I.; Steriade, M.; Sejnowski, T.J. Model of Thalamocortical Slow-Wave Sleep Oscillations and Transitions to Activated States. J. Neurosci. 2002, 22, 8691–8704. [Google Scholar] [CrossRef] [Green Version]
- Compte, A.; Sanchez-Vives, M.; McCormick, D.A.; Wang, X.-J. Cellular and Network Mechanisms of Slow Oscillatory Activity (<1 Hz) and wave propagations in a cortical network model. J. Neurophysiol. 2003, 89, 2707–2725. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, F.; Bazhenov, M.; Timofeev, I.; Steriade, M.; Sejnowski, T.J. Slow State Transitions of Sustained Neural Oscillations by Activity-Dependent Modulation of Intrinsic Excitability. J. Neurosci. 2006, 26, 6153–6162. [Google Scholar] [CrossRef]
- Shu, Y.; Hasenstaub, A.; McCormick, D.A. Turning on and off recurrent balanced cortical activity. Nat. Cell Biol. 2003, 423, 288–293. [Google Scholar] [CrossRef]
- Zucca, S.; D’Urso, G.; Pasquale, V.; Vecchia, D.; Pica, G.; Bovetti, S.; Moretti, C.; Varani, S.; Mazon, M.M.; Chiappalone, M.; et al. An inhibitory gate for state transition in cortex. eLife 2017, 6, e26177. [Google Scholar] [CrossRef]
- Valero, M.; Viney, T.J.; Machold, R.; Mederos, S.; Zutshi, I.; Schuman, B.; Senzai, Y.; Rudy, B.; Buzsáki, G. Sleep down state-active ID2/Nkx2.1 interneurons in the neocortex. Nat. Neurosci. 2021, 24, 401–411. [Google Scholar] [CrossRef]
- Parga, N.; Abbott, L.F. Network model of spontaneous activity exhibiting synchronous transitions between up and down states. Front. Behav. Neurosci. 2007, 1, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jercog, D.; Roxin, A.; Barthó, P.; Luczak, A.; Compte, A.; De La Rocha, J. UP-DOWN cortical dynamics reflect state transitions in a bistable network. eLife 2017, 6, e22425. [Google Scholar] [CrossRef]
- Zucca, S.; Pasquale, V.; Roig, P.L.D.L.; Panzeri, S.; Fellin, T. Thalamic Drive of Cortical Parvalbumin-Positive Interneurons during Down States in Anesthetized Mice. Curr. Biol. 2019, 29, 1481–1490.e6. [Google Scholar] [CrossRef]
- Bojarskaite, L.; Bjørnstad, D.M.; Pettersen, K.H.; Cunen, C.; Hermansen, G.H.; Åbjørsbråten, K.S.; Chambers, A.R.; Sprengel, R.; Vervaeke, K.; Tang, W.; et al. Astrocytic Ca2+ signaling is reduced during sleep and is involved in the regulation of slow wave sleep. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Buskila, Y.; Bellot-Saez, A.; Morley, J.W. Generating Brain Waves, the Power of Astrocytes. Front. Neurosci. 2019, 13, 1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, Z.; Héja, L.; Szalay, G.; Kékesi, O.; Füredi, A.; Szebényi, K.; Dobolyi, Á.; Orbán, T.I.; Kolacsek, O.; Tompa, T.; et al. Extensive astrocyte synchronization advances neuronal coupling in slow wave activity in vivo. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hirase, H.; Qian, L.; Barthó, P.; Buzsaki, G. Calcium Dynamics of Cortical Astrocytic Networks In Vivo. PLoS Biol. 2004, 2, e96. [Google Scholar] [CrossRef] [Green Version]
- Amzica, F.; Steriade, M. Neuronal and Glial Membrane Potentials during Sleep and Paroxysmal Oscillations in the Neocortex. J. Neurosci. 2000, 20, 6648–6665. [Google Scholar] [CrossRef] [Green Version]
- Timofeev, I.; Schoch, S.; LeBourgeois, M.K.; Huber, R.; Riedner, B.A.; Kurth, S. Spatio-temporal properties of sleep slow waves and implications for development. Curr. Opin. Physiol. 2020, 15, 172–182. [Google Scholar] [CrossRef]
- Billeh, Y.N.; Cai, B.; Gratiy, S.L.; Dai, K.; Iyer, R.; Gouwens, N.W.; Abbasi-Asl, R.; Jia, X.; Siegle, J.H.; Olsen, S.R.; et al. Systematic Integration of Structural and Functional Data into Multi-scale Models of Mouse Primary Visual Cortex. Neuron 2020, 106, 388–403.e18. [Google Scholar] [CrossRef]
- Markram, H.; Muller, E.; Ramaswamy, S.; Reimann, M.W.; Abdellah, M.; Sanchez, C.A.; Ailamaki, A.; Alonso-Nanclares, L.; Antille, N.; Arsever, S.; et al. Reconstruction and Simulation of Neocortical Microcircuitry. Cell 2015, 163, 456–492. [Google Scholar] [CrossRef]
- Nir, Y.; Staba, R.J.; Andrillon, T.; Vyazovskiy, V.; Cirelli, C.; Fried, I.; Tononi, G. Regional Slow Waves and Spindles in Human Sleep. Neuron 2011, 70, 153–169. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.; Riedner, B.A.; Huber, R.; Massimini, M.; Ferrarelli, F.; Tononi, G. Source modeling sleep slow waves. Proc. Natl. Acad. Sci. USA 2009, 106, 1608–1613. [Google Scholar] [CrossRef] [Green Version]
- Massimini, M.; Huber, R.; Ferrarelli, F.; Hill, S.; Tononi, G. The Sleep Slow Oscillation as a Traveling Wave. J. Neurosci. 2004, 24, 6862–6870. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.L.; Hu, T.; Spadola, C.; Burgess, A.; Li, T.; Cadet, T. Treatment of Sleep Disturbance May Reduce the Risk of Future Probable Alzheimer’s Disease. J. Aging Health 2019, 31, 322–342. [Google Scholar] [CrossRef] [PubMed]
- Keage, H.A.D.; Banks, S.; Yang, K.L.; Morgan, K.; Brayne, C.; Matthews, F. What sleep characteristics predict cognitive decline in the elderly? Sleep Med. 2012, 13, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.S.P.; Kowgier, M.; Yu, L.; Buchman, A.S.; Bennett, D.A. Sleep Fragmentation and the Risk of Incident Alzheimer’s Disease and Cognitive Decline in Older Persons. Sleep 2013, 36, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovsky, D.V.; McPhillips, M.V.; Li, J.; Brody, A.; Caffeé, L.; Hodgson, N.A. Sleep disruption and quality of life in persons with dementia: A state-of-the-art review. Geriatr. Nurs. 2018, 39, 640–645. [Google Scholar] [CrossRef]
- Potvin, O.; Lorrain, D.; Forget, H.; Dubé, M.; Grenier, S.; Préville, M.; Hudon, C. Sleep Quality and 1-Year Incident Cognitive Impairment in Community-Dwelling Older Adults. Sleep 2012, 35, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Sindi, S.; Kåreholt, I.; Johansson, L.; Skoog, J.; Sjöberg, L.; Wang, H.-X.; Johansson, B.; Fratiglioni, L.; Soininen, H.; Solomon, A.; et al. Sleep disturbances and dementia risk: A multicenter study. Alzheimer’s Dement. 2018, 14, 1235–1242. [Google Scholar] [CrossRef]
- Yaffe, K.; Nettiksimmons, J.; Yesavage, J.; Byers, A. Sleep Quality and Risk of Dementia Among Older Male Veterans. Am. J. Geriatr. Psychiatry 2015, 23, 651–654. [Google Scholar] [CrossRef]
- Bliwise, D.; Tinklenberg, J.; Yesavage, J.; Davies, H.; Pursley, A.; Petta, D.; Widrow, L.; Guilleminault, C.; Zarcone, V.; Dement, W. REM latency in Alzheimer’s disease. Biol. Psychiatry 1989, 25, 320–328. [Google Scholar] [CrossRef]
- Liguori, C.; Placidi, F.; Izzi, F.; Spanetta, M.; Mercuri, N.B.; Di Pucchio, A. Sleep dysregulation, memory impairment, and CSF biomarkers during different levels of neurocognitive functioning in Alzheimer’s disease course. Alzheimer’s Res. Ther. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Liguori, C.; Romigi, A.; Nuccetelli, M.; Zannino, S.; Sancesario, G.; Martorana, A.; Albanese, M.; Mercuri, N.B.; Izzi, F.; Bernardini, S.; et al. Orexinergic System Dysregulation, Sleep Impairment, and Cognitive Decline in Alzheimer Disease. JAMA Neurol. 2014, 71, 1498–1505. [Google Scholar] [CrossRef]
- Liu, S.; Pan, J.; Tang, K.; Lei, Q.; He, L.; Meng, Y.; Cai, X.; Li, Z. Sleep spindles, K-complexes, limb movements and sleep stage proportions may be biomarkers for amnestic mild cognitive impairment and Alzheimer’s disease. Sleep Breath. 2019, 24, 637–651. [Google Scholar] [CrossRef]
- Loewenstein, R.J.; Weingartner, H.; Gillin, J.; Kaye, W.; Ebert, M.; Mendelson, W.B. Disturbances of sleep and cognitive functioning in patients with dementia. Neurobiol. Aging 1982, 3, 371–377. [Google Scholar] [CrossRef]
- Vitiello, M.V.; Prinz, P.N.; Williams, D.E.; Frommlet, M.S.; Ries, R.K. Sleep Disturbances in Patients With Mild-Stage Alzheimer’s Disease. J. Gerontol. 1990, 45, M131–M138. [Google Scholar] [CrossRef]
- Prinz, P.N.; Vitaliano, P.P.; Vitiello, M.V.; Bokan, J.; Raskind, M.; Peskind, E.; Gerber, C. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol. Aging 1982, 3, 361–370. [Google Scholar] [CrossRef]
- Prinz, P.N.; Peskind, E.R.; Vitaliano, P.P.; Raskind, M.A.; Eisdorfer, C.; Zemcuznikov, H.N.; Gerber, C.J. Changes in the Sleep and Waking EEGs of Nondemented and Demented Elderly Subjects. J. Am. Geriatr. Soc. 1982, 30, 86–92. [Google Scholar] [CrossRef]
- Moe, K.E.; Vitiello, M.V.; Larsen, L.H.; Prinz, P.N. Sleep/wake patterns In Alzheimer’s disease: Relationships with cognition and function. J. Sleep Res. 1995, 4, 15–20. [Google Scholar] [CrossRef]
- Mander, A.B.; Rao, V.; Lu, B.; Saletin, J.; Lindquist, J.R.; Ancoli-Israel, S.; Jagust, W.; Walker, M.P. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat. Neurosci. 2013, 16, 357–364. [Google Scholar] [CrossRef]
- Mander, B.A.; Marks, S.M.; Vogel, J.W.; Rao, V.; Lu, B.; Saletin, J.M.; Ancoli-Israel, S.; Jagust, W.J.; Walker, M.P. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci. 2015, 18, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Lucey, B.P.; McCullough, A.; Landsness, E.C.; Toedebusch, C.D.; McLeland, J.S.; Zaza, A.M.; Fagan, A.M.; McCue, L.; Xiong, C.; Morris, J.C.; et al. Reduced non–rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaau6550. [Google Scholar] [CrossRef]
- Castano-Prat, P.; Perez-Mendez, L.; Perez-Zabalza, M.; Sanfeliu, C.; Giménez-Llort, L.; Sanchez-Vives, M.V. Altered slow (<1 Hz) and fast (beta and gamma) neocortical oscillations in the 3xTg-AD mouse model of Alzheimer's disease under anes-thesia. Neurobiol. Aging 2019, 79, 142–151. [Google Scholar] [CrossRef]
- Kent, B.; Strittmatter, S.; Nygaard, H.B. Sleep and EEG Power Spectral Analysis in Three Transgenic Mouse Models of Alzheimer’s Disease: APP/PS1, 3xTgAD, and Tg2576. J. Alzheimer’s Dis. 2018, 64, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.H.; Huang, Y.; Bero, A.W.; Kasten, T.; Stewart, F.R.; Bateman, R.; Holtzman, D.M. Disruption of the Sleep-Wake Cycle and Diurnal Fluctuation of -Amyloid in Mice with Alzheimer’s Disease Pathology. Sci. Transl. Med. 2012, 4, 150ra122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Veasey, S.C.; Wood, M.A.; Leng, L.Z.; Kaminski, C.; Leight, S.; Abel, T.; Lee, V.M.-Y.; Trojanowski, J.Q. Impaired Rapid Eye Movement Sleep in the Tg2576 APP Murine Model of Alzheimer’s Disease with Injury to Pedunculopontine Cholinergic Neurons. Am. J. Pathol. 2005, 167, 1361–1369. [Google Scholar] [CrossRef]
- Holth, J.K.; Mahan, T.E.; Robinson, G.O.; da Rocha, A.S.; Holtzman, D.M. Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann. Clin. Transl. Neurol. 2017, 4, 180–190. [Google Scholar] [CrossRef]
- Holton, C.M.; Hanley, N.; Shanks, E.; Oxley, P.; McCarthy, A.; Eastwood, B.J.; Murray, T.K.; Nickerson, A.; Wafford, K.A. Longitudinal changes in EEG power, sleep cycles and behaviour in a tau model of neurodegeneration. Alzheimer’s Res. Ther. 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Jyoti, A.; Plano, A.; Riedel, G.; Platt, B. Progressive age-related changes in sleep and EEG profiles in the PLB1Triple mouse model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2768–2784. [Google Scholar] [CrossRef]
- Koss, D.J.; Robinson, L.; Drever, B.D.; Plucinska, K.; Stoppelkamp, S.; Veselcic, P.; Riedel, G.; Platt, B. Mutant Tau knock-in mice display frontotemporal dementia relevant behaviour and histopathology. Neurobiol. Dis. 2016, 91, 105–123. [Google Scholar] [CrossRef] [Green Version]
- Kastanenka, K.V.; Calvo-Rodriguez, M.; Hou, S.S.; Zhou, H.; Takeda, S.; Arbel-Ornath, M.; Lariviere, A.; Lee, Y.F.; Kim, A.; Hawkes, J.M.; et al. Frequency-dependent exacerbation of Alzheimer’s disease neuropathophysiology. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Keskin, A.D.; Kekuš, M.; Adelsberger, H.; Neumann, U.; Shimshek, D.R.; Song, B.; Nelken, I.; Nelken, I.; Sakmann, B.; Konnerth, A.; et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl. Acad. Sci. USA 2017, 114, 8631–8636. [Google Scholar] [CrossRef] [Green Version]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [Green Version]
- Bero, A.W.; Yan, P.; Roh, J.H.; Cirrito, J.R.; Stewart, F.R.; Raichle, M.E.; Lee, J.-M.; Holtzman, D.M. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 2011, 14, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.A.; Barrett, G.M.; Duff, K.E.; Hussaini, S.A. Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Aβ and tau pathology in the hippocampus. PLoS Biol. 2020, 18, e3000851. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tanei, Z.-I.; Hashimoto, T.; Wakabayashi, T.; Okuno, H.; Naka, Y.; Yizhar, O.; Fenno, L.E.; Fukayama, M.; Bito, H.; et al. Chronic Optogenetic Activation Augments Aβ Pathology in a Mouse Model of Alzheimer Disease. Cell Rep. 2015, 11, 859–865. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Grutzendler, J. Attenuation of β-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity. J. Neurosci. 2016, 36, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.-E.; Lucey, B.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [Green Version]
- Götz, J.; Bodea, L.-G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M.; McCormick, D.; Sejnowski, T.J. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993, 262, 679–685. [Google Scholar] [CrossRef]
- Romanella, S.; Roe, D.; Tatti, E.; Cappon, D.; Paciorek, R.; Testani, E.; Rossi, A.; Rossi, S.; Santarnecchi, E. The Sleep Side of Aging and Alzheimer’s Disease. Sleep Med. 2021, 77, 209–225. [Google Scholar] [CrossRef]
- Rasch, B.; Born, J. About Sleep’s Role in Memory. Physiol. Rev. 2013, 93, 681–766. [Google Scholar] [CrossRef]
- Héricé, C.; Patel, A.A.; Sakata, S. Circuit mechanisms and computational models of REM sleep. Neurosci. Res. 2019, 140, 77–92. [Google Scholar] [CrossRef]
- Karashima, A.; Nakao, M.; Honda, K.; Iwasaki, N.; Katayama, N.; Yamamoto, M. Theta wave amplitude and frequency are differentially correlated with pontine waves and rapid eye movements during REM sleep in rats. Neurosci. Res. 2004, 50, 283–289. [Google Scholar] [CrossRef]
- Karashima, A.; Nakamura, K.; Sato, N.; Nakao, M.; Katayama, N.; Yamamoto, M. Phase-locking of spontaneous and elicited ponto–geniculo–occipital waves is associated with acceleration of hippocampal theta waves during rapid eye movement sleep in cats. Brain Res. 2002, 958, 347–358. [Google Scholar] [CrossRef]
- Ramirez-Villegas, J.F.; Besserve, M.; Murayama, Y.; Evrard, H.C.; Oeltermann, A.; Logothetis, N.K. Coupling of hippocampal theta and ripples with pontogeniculooccipital waves. Nat. Cell Biol. 2021, 589, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; McAlinden, N.; Mathieson, K.; Sakata, S. Simultaneous Electrophysiology and Fiber Photometry in Freely Behaving Mice. Front. Neurosci. 2020, 14, 148. [Google Scholar] [CrossRef] [Green Version]
- Mufson, E.J.; Mash, D.C.; Hersh, L.B. Neurofibrillary tangles in cholinergic pedunculopontine neurons in Alzheimer’s disease. Ann. Neurol. 1988, 24, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Parvizi, J.; Van Hoesen, G.W.; Damasio, A. The selective vulnerability of brainstem nuclei to Alzheimer’s disease. Ann. Neurol. 2001, 49, 53–66. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Albin, R.L. The cholinergic system and Parkinson disease. Behav. Brain Res. 2011, 221, 564–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Y.-Y.; Lei, X.; Yu, J. Sleep spindle abnormalities related to Alzheimer’s disease: A systematic mini-review. Sleep Med. 2020, 75, 37–44. [Google Scholar] [CrossRef]
- Ngo, H.-V.V.; Martinetz, T.; Born, J.; Mölle, M. Auditory Closed-Loop Stimulation of the Sleep Slow Oscillation Enhances Memory. Neuron 2013, 78, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Arzi, A.; Shedlesky, L.; Ben-Shaul, M.; Nasser, K.; Oksenberg, A.; Hairston, I.S.; Sobel, N. Humans can learn new information during sleep. Nat. Neurosci. 2012, 15, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Ball, W.A.; Morrison, A.R.; Ross, R.J. The effects of tones on PGO waves in slow wave sleep and paradoxical sleep. Exp. Neurol. 1989, 104, 251–256. [Google Scholar] [CrossRef]
- Buzsáki, G. Rhythms of the Brain; Oxford University Press (OUP): New York, NY, USA, 2006. [Google Scholar]
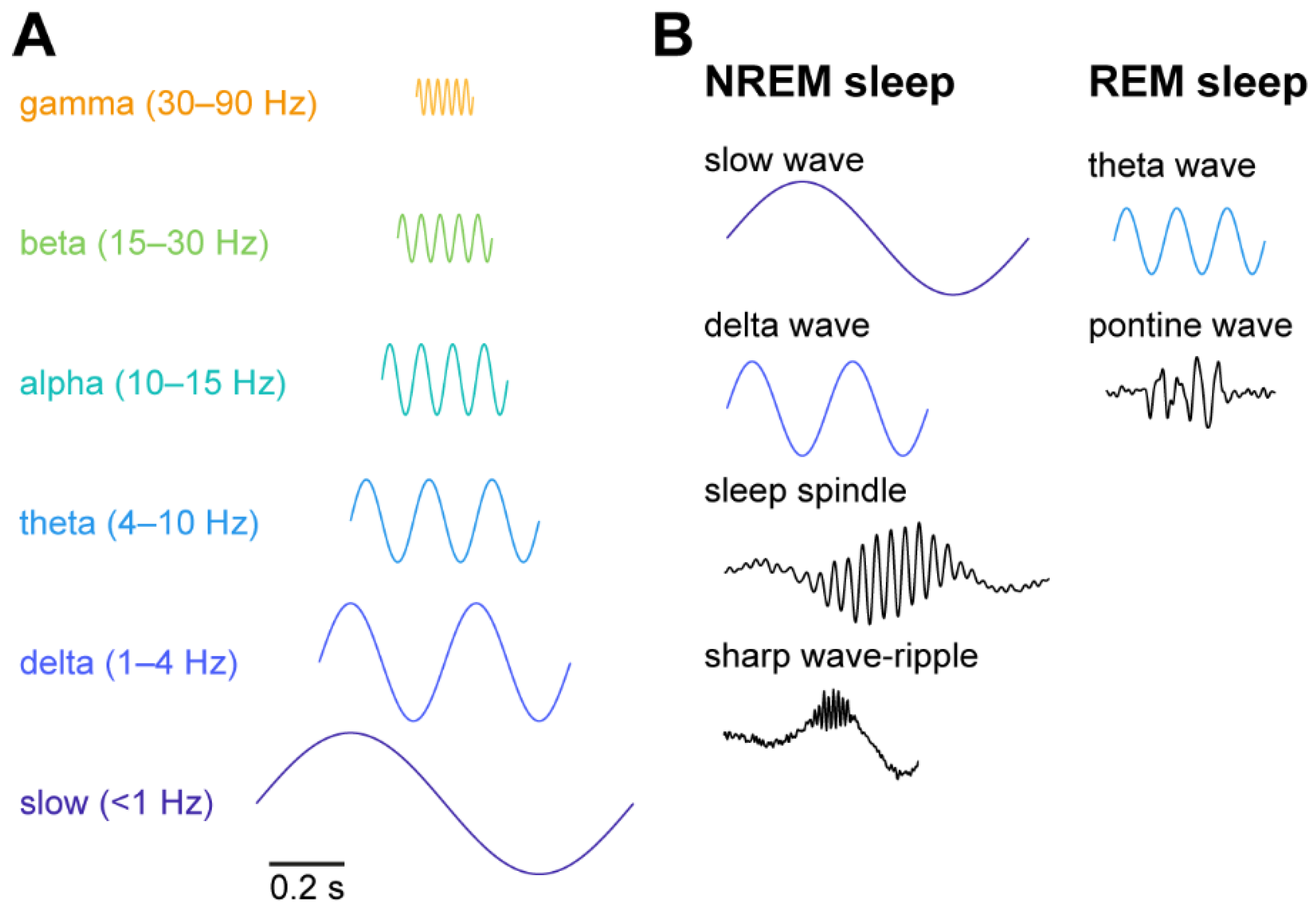
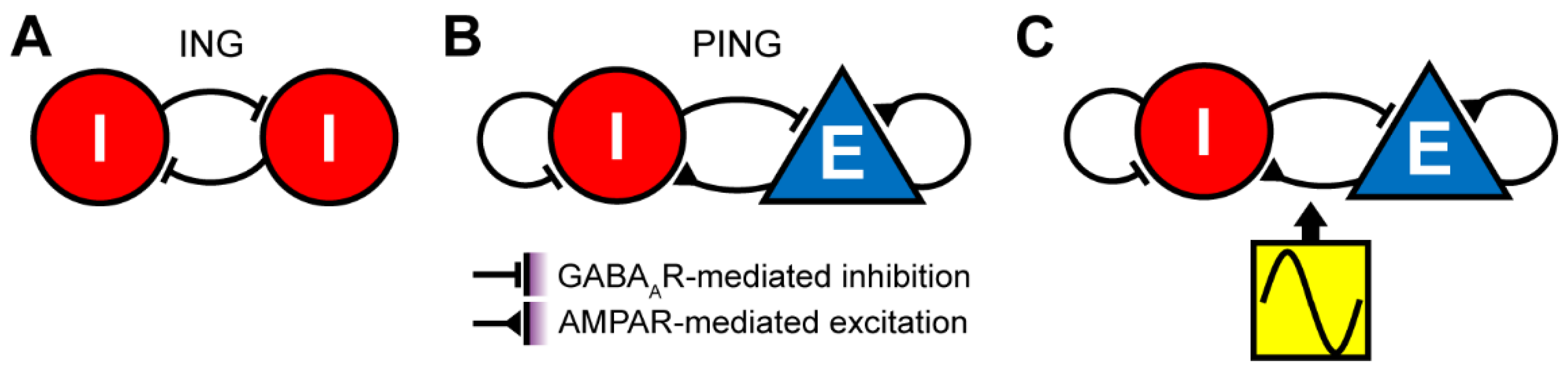
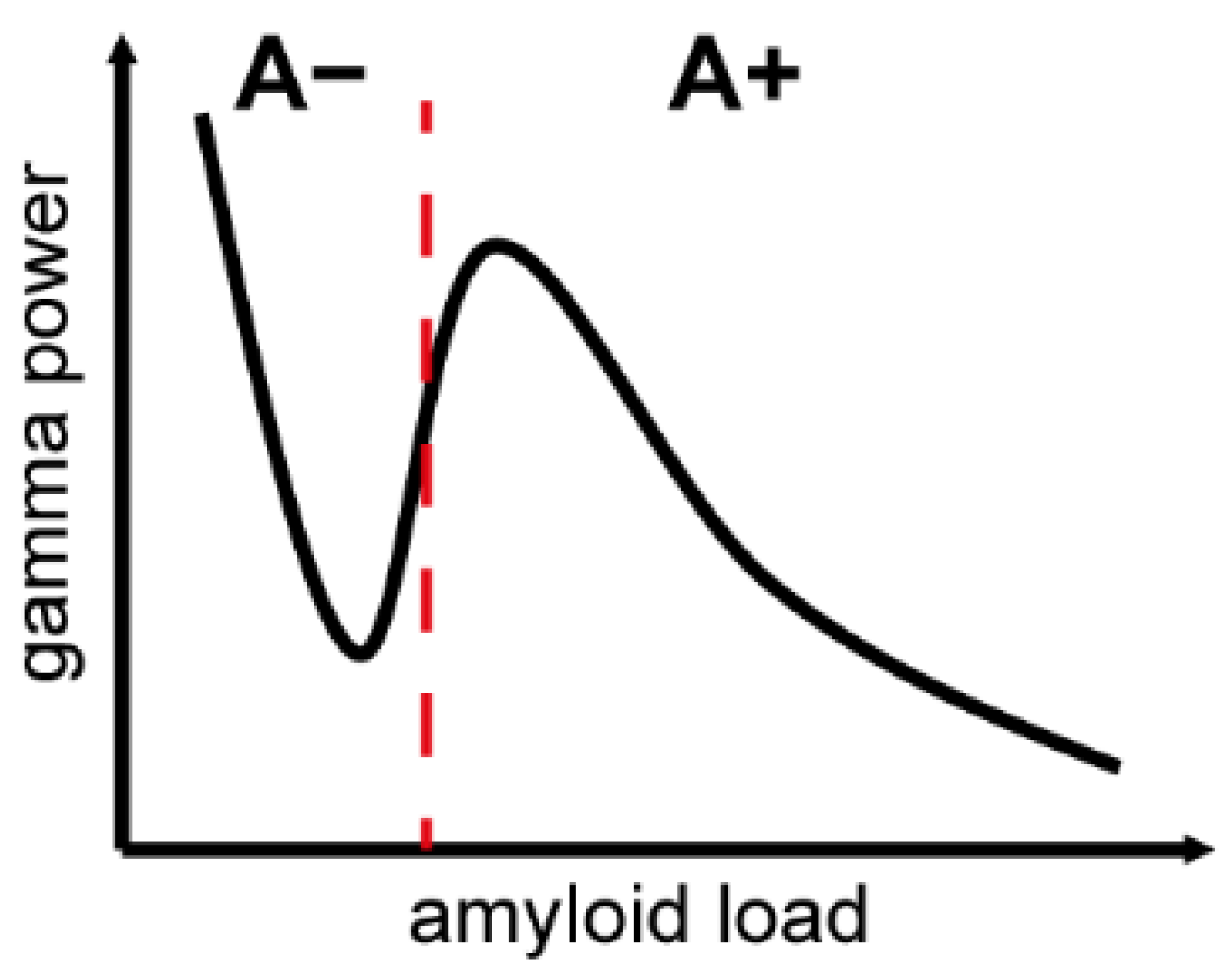
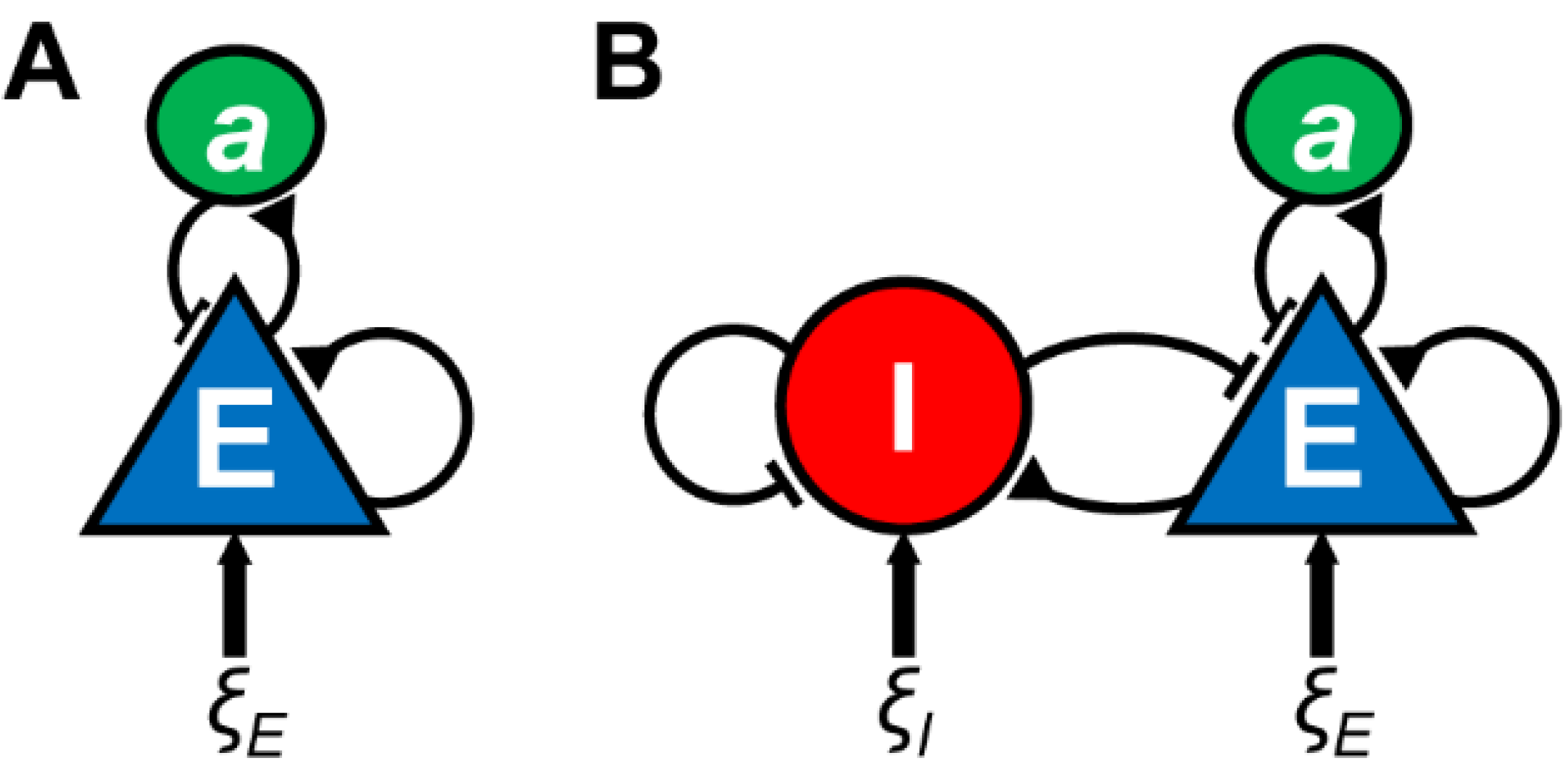

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Age (Months) | Sex | Preparation | Frequency Band (Hz) | Changes in γ Oscillations | Reference |
|---|---|---|---|---|---|---|
| APP/PS1 | 4–5 | NA | EC slices | 20–60 | Reduced γ frequency in LEC No effect in MEC | [139] |
| J20 | 4–7 | M/F | in vivo cEEG | 20–80 | Reduced γ power | [140] |
| 7–8 | M/F | in vivo cEEG | 30–90 | Reduced γ power | [141] | |
| 5xFAD | 3 | M | in vivo LFP in CA1 | 20–50 | Reduced γ power during SWRs | [89] |
| TgCRND8 | 1 | NA | HC slices | θ: 3–12 low γ: 25–85 high γ: 120–250 | No change in γ power Disrupted θ–γ coupling | [142] |
| 1 | M | in vivo HC LFP | low γ: 25–45 high γ: 60–100 | Reduced γ power | [143] | |
| APOE4 | 5–17 | F | in vivo HC LFP | 30–50 | Reduced γ power | [144] |
| 4–5 | F | in vivo HC LFP | 30–50 | Reduced γ power during SWRs | [145] | |
| 3R tau overexpression | 7 | M | HC slices | 50–90 | Reduced γ power and peak frequency | [146] |
| Induction Method | Stimulation Protocol | Duration | Model | Sex | Age (Months) | Modulated AD Phenotype | Reference | |
|---|---|---|---|---|---|---|---|---|
| Invasive | Optogenetic | 1 ms pulses, 40 Hz, CA1 | 1 h | 5xFAD::PV-Cre, AAV5-EF1α-DIO-ChR2-eYFP | M | 3 | Reduced Aβ Reduced inflammation | [89] |
| 12 ms pulses, 40 Hz, Medial Septum | 10 min | PVJ20, AAVdj-EF1α-DIO-ChETA-eYFP | M/F | NA | Improved spatial memory | [88] | ||
| 40 Hz, Basal Forebrain | 1 h/d for 3 days | 5xFAD::PV-Cre::Ai32 | M/F | 4–6 | Increased Aβ | [157] | ||
| Non-Invasive | Visual | 12.5 ms on, 12.5 ms off, 40 Hz flicker | 1 h/day for 7 days | 5xFAD | M | 6 | Reduced Aβ | [89] |
| APP/PS1 | M/F | 5 | Reduced Aβ | |||||
| TauP301S | M | 4 | Reduced tauopathy | |||||
| 40 Hz flicker | 1 h/day for 30 days | APP/PS1 | F | 8 | Reduced Aβ Reduced tauopathy Increased sleep regulation | [160] | ||
| 12.5 ms on, 12.5 ms off, 40 Hz flicker | 1 h/day for 22 days | TauP301S | M | 7.5–8 | Reduced neuronal damage Reduced inflammation Reduced tauopathy Improved spatial memory | [91] | ||
| 1 h/day for 6 weeks | CK-p25 | M/F | 6–9 | Reduced neuronal damage Reduced inflammation Improved spatial memory | ||||
| Auditory | 1 ms 10 kHz tones, 40 Hz, 60 dB | 1 h/day for 7 days | 5xFAD | NA | 6 | Reduced Aβ Reduced inflammation Improved memory | [90] | |
| APP/PS1 | NA | 6–9 | Reduced Aβ Reduced inflammation | |||||
| TauP301S | NA | 2 | Reduced tauopathy | |||||
| Combined Auditory and Visual | 10 s on, 10 s off | 1 h/day for 7 days | 5xFAD | NA | 6 | Reduced Aβ Reduced inflammation | ||
| Visual and Exercise | 40 Hz light flicker and 30–50 min exercise | Daily, 6 days a week for 12 weeks | 3xTg | M | 12–15 | Reduced Aβ Reduced tauopathy Reduced neuronal damage Improved spatial memory | [159] | |
| Transcranial Focused Ultrasound | 400 μs pulses, 5 s on 5 s off, 40 Hz, Hippocampus | 1 h/day for 5 days | 5xFAD | M | 6 | Increased microglia/Aβ Co-localisation | [158] |
| Mouse Model | Age (Months) | Sex | Frequency Band (Hz) | Changes in Oscillations | Reference |
|---|---|---|---|---|---|
| 3xTg-AD | 7, 20 | M/F | <1 | Increased frequency at 7 months Decreased frequency at 20 months More irregular at 20 months | [233] |
| 3xTg-AD | 18 | M/F | 0.1–4 | No change | [234] |
| APP/PS1 | 8–10 | M/F | 0.1–4 | Decreased power during NREM | |
| Tg2576 | 12 | M/F | 0.1–4 | Decreased power during W | |
| Tg2576 | 2, 6, 12 | NA | 0.5–4 | Decreased power during NREM at 6–12 months | [236] |
| APP/PS1 | 3, 6, 9 | NA | >1 | Shorter NREM at 9 months | [235] |
| P301S | 3–12 | M | 1–4 | Increased power during NREM at 6–9 months Decreased power during W and NREM at 11 months | [237] |
| rTg4510 | 5–10 | M | 0.1–4 | Decreased power during NREM from 6 months | [238] |
| PLB1triple | 5–21 | M/F | 0.5–5 | Decreased power during REM at 9 months Decreased power during W at 21 months | [239] |
| PLB2tau | 6 | F | 1.5–5 | Increased power during REM Decreased power during NREM | [240] |
| Induction Method | Protocol | Duration | Model | Sex | Age (Months) | Modulated AD Phenotype | Reference | |
|---|---|---|---|---|---|---|---|---|
| Invasive | Optogenetic | 400 ms pulses, 0.6 Hz, Anterior Cortex | 24 h/day for 1 month | APP/PS1, AAV5-CamKIIα-hChR2(H134R)-mCherry | M/F | 4–7 | Reduced Aβ Reduced calcium overload Restored GABA levels | [87] |
| 400 ms pulses, 1.2 Hz, Anterior Cortex | 24 h/day for 1 month | APP/PS1, AAV5-CamKIIα-hChR2(H134R)-mCherry | M/F | 3–9 | Increased Aβ Increased calcium overload Decreased spine densityNo change in GABA levels | [241] | ||
| Non-Invasive | BACE Inhibitor (oral) | Administration of 0.25 g/kg NB-360 in food pellets | 6 weeks ad lib | APP23xPS45 | F | 6–8 | Reduced Aβ Reduced calcium overload Improved spatial memory | [242] |
| GABA-A Agonist (i.p.) | Administration of 0.05 mg/kg clonazepam | Once/day for 5 days | APP23xPS45 | M/F | 6–8 | Improved spatial memory | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byron, N.; Semenova, A.; Sakata, S. Mutual Interactions between Brain States and Alzheimer’s Disease Pathology: A Focus on Gamma and Slow Oscillations. Biology 2021, 10, 707. https://doi.org/10.3390/biology10080707
Byron N, Semenova A, Sakata S. Mutual Interactions between Brain States and Alzheimer’s Disease Pathology: A Focus on Gamma and Slow Oscillations. Biology. 2021; 10(8):707. https://doi.org/10.3390/biology10080707
Chicago/Turabian StyleByron, Nicole, Anna Semenova, and Shuzo Sakata. 2021. "Mutual Interactions between Brain States and Alzheimer’s Disease Pathology: A Focus on Gamma and Slow Oscillations" Biology 10, no. 8: 707. https://doi.org/10.3390/biology10080707
APA StyleByron, N., Semenova, A., & Sakata, S. (2021). Mutual Interactions between Brain States and Alzheimer’s Disease Pathology: A Focus on Gamma and Slow Oscillations. Biology, 10(8), 707. https://doi.org/10.3390/biology10080707