
Comparative Transcriptome Analysis of Different Actinidia arguta Fruit Parts Reveals Difference of Light Response during Fruit Coloration


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
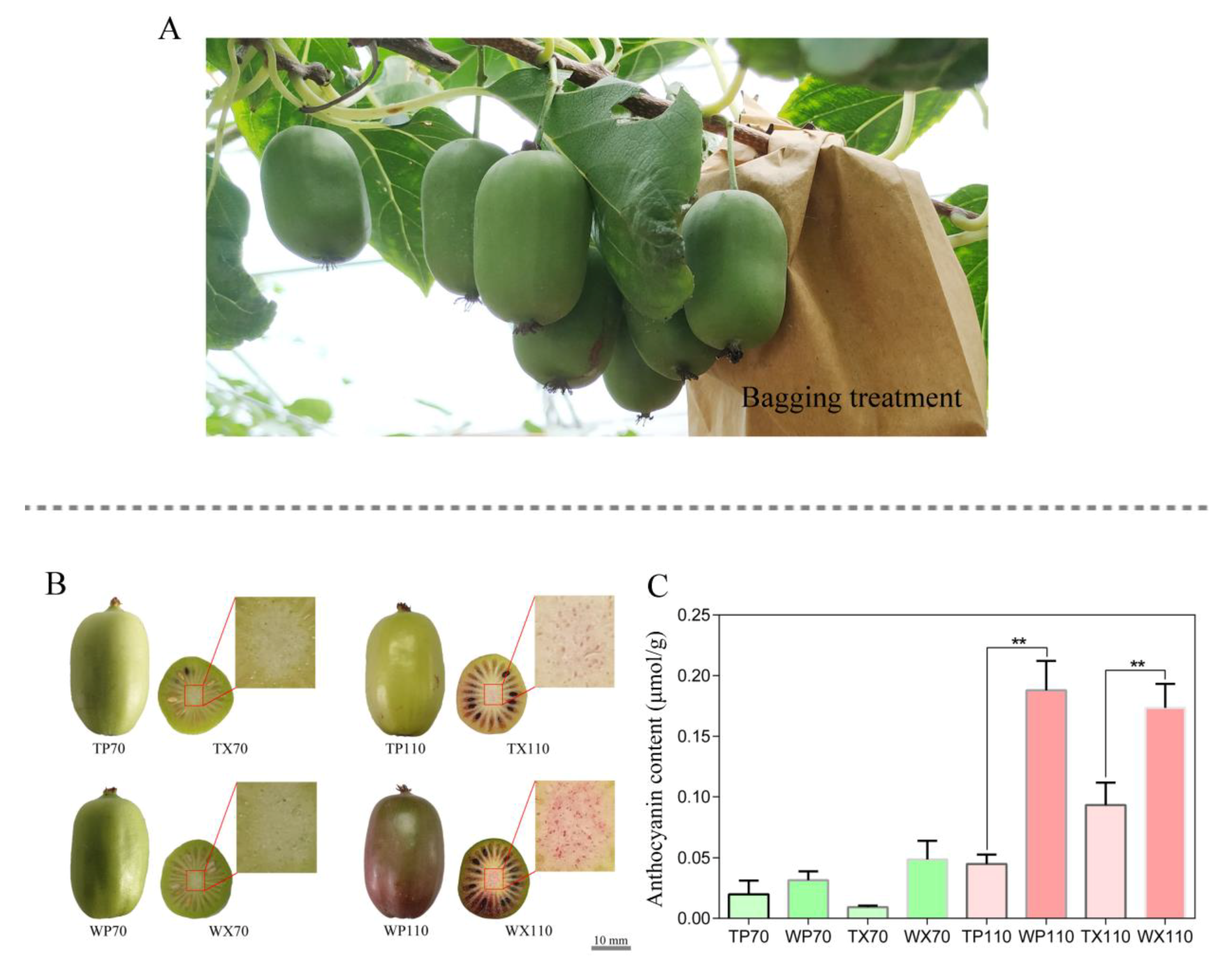
2.1. Bagging Treatment, Sample Preparation, and Anthocyanin Measurement
2.2. RNA Extraction, cDNA Synthesis, Library Construction, and Sequencing
2.3. Quality Control and Gene Functional Annotation
2.4. Quantification of Gene Expression and Differential Expression Analysis
2.5. GO and KEGG Enrichment Analysis
2.6. Vector Construction and Transient Overexpression of AaMYB308like in N. tabacum
2.7. qRT-PCR Analysis
3. Results
3.1. Changes in Phenotype of Peel and Core between Bagging and Non-Bagging Treatment
3.2. Transcriptome Assembly
3.3. Functional Annotation
3.4. Expression Analysis of DEGs among Different Pairwise
3.5. GO and KEGG Analysis of DEGs
3.6. The Possible MYB-Related Genes Controlling Coloration of Different Fruit Parts
3.7. Transient Overexpression of AaMYB308like in Nicotiana tabacum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, H.W.; Ferguson, A.R. Actinidia in China: Natural diversity, phylogeographical evolution, interspecific gene flow and kiwifruit cultivar improvement. Acta Hortic. 2007, 753, 31–40. [Google Scholar] [CrossRef]
- He, J.; Giusti, M.M. Anthocyanins: Natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef]
- Li, Y.K.; Cui, W.; Qi, X.J.; Lin, M.M.; Qiao, C.K.; Zhong, Y.P.; Hu, C.G.; Fang, J.B. MicroRNA858 negatively regulates anthocyanin biosynthesis by repressing AaMYBC1 expression in kiwifruit (Actinidia arguta). Plant. Sci. 2020, 296, 110476. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.H.; Chattopadhyay, S.; Wei, N.; Oyama, T.; Okada, K.; Batschauer, A.; Deng, X.W. Molecular interaction between COP1 and HY5 defines a regulatory switch for light control of Arabidopsis development. Mol. Cell 1998, 1, 213–222. [Google Scholar] [CrossRef]
- Seo, H.S.; Yang, J.Y.; Ishikawa, M.; Bolle, C.; Ballesteros, M.L.; Chua, N.H. LAF1 ubiquitination by COP1 controls photomorphogenesis and is stimulated by SPA1. Nature 2003, 423, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.; Heijde, M.; Heller, W.; Albert, A.; Seidlitz, H.K.; Ulm, R. Negative feedback regulation of UV-B-induced photomorphogenesis and stress acclimation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 101, 20132–20137. [Google Scholar] [CrossRef] [Green Version]
- Nagatani, A. Phytochrome: Structural basis for its functions. Curr. Opin. Plant Biol. 2010, 13, 565–570. [Google Scholar] [CrossRef]
- An, J.P.; Qu, F.J.; Yao, J.F.; Wang, X.N.; You, C.X.; Wang, X.F.; Hao, Y.J. The bZIP transcription factor MdHY5 regulates anthocyanin accumulation and nitrate assimilation in apple. Hortic. Res. 2017, 4, 17023. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Wang, Y.C.; Yu, L.; Jiang, H.Y.; Guo, Z.W.; Xu, H.F.; Jiang, S.H.; Fang, H.C.; Zhang, J.; Su, M.Y.; et al. MdWRKY11 participates in anthocyanin accumulation in red-fleshed apples by affecting MYB transcription factors and the photoresponse factor MdHY5. J. Agric. Food Chem. 2019, 67, 8783–8793. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, P.B.; Chen, G.Q.; Wu, J.; Liu, Z.C.; Lian, H.L. FvbHLH9 functions as a positive regulator of anthocyanin biosynthesis by forming a HY5-bHLH9 transcription complex in strawberry fruits. Plant Cell Physiol. 2020, 64, 826–837. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Chu, Y.J.; Corey, D.R. RNA sequencing: Platform selection, experimental design, and data interpretation. Nucleic Acid Ther. 2012, 22, 271–274. [Google Scholar] [CrossRef]
- Bai, S.; Sun, Y.; Qian, M.; Yang, F.; Ni, J.; Tao, R.; Li, L.; Shu, Q.; Zhang, D.; Teng, Y. Transcriptome analysis of bagging-treated red Chinese sand pear peels reveals light-responsive pathway functions in anthocyanin accumulation. Sci. Rep. 2017, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.B.; Zawora, C.; Li, Y.; Wu, J.; Liu, L.C.; Liu, Z.C.; Cai, R.; Lian, H.L. Transcriptome sequencing reveals role of light in promoting anthocyanin accumulation of strawberry fruit. Plant Growth Regul. 2018, 86, 121–132. [Google Scholar] [CrossRef]
- Yang, T.; Ma, H.Y.; Zhang, J.; Wu, T.; Song, T.T.; Tian, J.; Yao, Y.C. Systematic identification of long noncoding RNAs expressed during light-induced anthocyanin accumulation in apple fruit. Plant J. 2019, 100, 3. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Vandesompele, J.; Preter, D.P.; Pattyn, F.; Poppe, B.; Roy, N.V.; Paepe, A.D.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Takos, A.M.; Jaffé, F.W.; Jacob, S.R.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [Green Version]
- Albert, N.W.; Lewis, D.H.; Zhang, H.; Irving, L.J.; Jameson, P.E.; Davies, K.M. Light-induced vegetative anthocyanin pigmentation in Petunia. J. Exp. Bot. 2009, 60, 2191–2202. [Google Scholar] [CrossRef] [Green Version]
- Li, B.Q.; Xia, Y.X.; Wang, Y.Y.; Qin, G.Z.; Tian, S.P. Characterization of genes encoding key enzymes involved in anthocyanin metabolism of kiwifruit during storage period. Front. Plant Sci. 2017, 8, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Mao, K.; Zhao, C.; Zhao, X.Y.; Zhang, H.L.; Shu, H.R.; Hao, Y.J. MdCOP1 ubiquitin E3 ligases interact with MdMYB1 to regulate light-induced anthocyanin biosynthesis and red fruit coloration in apple. Plant Physiol 2012, 160, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.P.; Wang, X.F.; Li, Y.Y.; Song, L.Q.; Zhao, L.L.; You, C.X.; Hao, Y.J. EIN3-LIKE1, MYB1, and ETHYLENE RESPONSE FACTOR3 act in a regulatory loop that synergistically modulates ethylene biosynthesis and anthocyanin accumulation. Plant Physiol. 2018, 178, 808–823. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ding, Z.; Ruan, M.; Yu, X.; Peng, M.; Liu, Y. Kiwifruit R2R3-MYB transcription factors and contribution of the novel AcMYB75 to red kiwifruit anthocyanin biosynthesis. Sci. Rep. 2017, 7, 16861. [Google Scholar] [CrossRef] [Green Version]
- Theologis, A.; Ecker, J.R.; Palm, C.J.; Federspiel, N.A.; Kaul, S.; White, O.; Alonso, J.; Altafi, H.; Araujo, R.; Bowman, C.L.; et al. Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature 2000, 408, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.H.; Luo, Y.Q.; Zhang, W.F.; Jian, W.; Zhang, L.; Gao, X.L.; Hu, X.W.; Yuan, Y.J.; Wu, M.B.; Xu, X.; et al. A SlMYB75-centred transcriptional cascade regulates trichome formation and sesquiterpene accumulation in tomato. J. Exp. Bot. 2021, 72, 3806–3820. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Cao, H.; Yuan, S.; Liu, Y.; Lu, J.; Lu, W.; Li, N.; Wang, J.; Zou, J.; Tang, N.; et al. SlMYB75, an MYB-type transcription factor, promotes anthocyanin accumulation and enhances volatile aroma production in tomato fruits. Hortic. Res. 2019, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Qiu, Z.; Wang, X.; Van Giang, T.; Liu, X.; Wang, J.; Wang, X.; Gao, J.; Guo, Y.; Du, Y.; et al. A putative R3 MYB repressor is the candidate gene underlying atroviolacium, a locus for anthocyanin pigmentation in tomato fruit. J. Exp. Bot. 2017, 68, 5745–5758. [Google Scholar] [CrossRef] [PubMed]
- Kiferle, C.; Fantini, E.; Bassolino, L.; Povero, G.; Spelt, C.; Buti, S.; Giuliano, G.; Quattrocchio, F.; Koes, R.; Perata, P.; et al. Tomato R2R3-MYB proteins SlANT1 and SlAN2: Same protein activity, different roles. PLoS ONE 2015, 10, e0136365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Wang, J.R.; Wang, G.D.; Liang, X.Q.; Li, X.D.; Meng, Q.W. An R2R3-MYB gene, LeAN2, positively regulated the thermo-tolerance in transgenic tomato. J. Plant Physiol. 2015, 175, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Jing, C.; Chang, B.; Yan, J.; Liang, B.; Liu, L.; Yang, Y.; Zhao, Z. The effect of promoter methylation on MdMYB1 expression determines the level of anthocyanin accumulation in skins of two non-red apple cultivars. BMC Plant Biol. 2018, 18, 108. [Google Scholar] [CrossRef]
- An, J.P.; Liu, X.; Li, H.H.; You, C.X.; Wang, X.F.; Hao, Y.J. Apple RING E3 ligase MdMIEL1 inhibits anthocyanin accumulation by ubiquitinating and degrading MdMYB1 protein. Plant Cell Physiol. 2017, 58, 1953–1962. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Tuan, P.A.; Saito, T.; Honda, C.; Hatsuyama, Y.; Ito, A.; Moriguchi, T. Epigenetic regulation of MdMYB1 is associated with paper bagging-induced red pigmentation of apples. Planta 2016, 244, 573–586. [Google Scholar] [CrossRef]
- Hu, D.G.; Sun, C.H.; Ma, Q.J.; You, C.X.; Cheng, L.; Hao, Y.J. MdMYB1 regulates anthocyanin and malate accumulation by directly facilitating their transport into vacuoles in apples. Plant Physiol. 2016, 170, 1315–1330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.J.; Wang, L.X.; Chen, X.X.; Liu, Y.L.; Meng, R.; Wang, Y.J.; Zhao, Z.Y. A and MdMYB1 allele-specific markers controlling apple (Malus x domestica Borkh.) skin color and suitability for marker-assisted selection. Genet. Mol. Res. 2014, 13, 9103–9114. [Google Scholar] [CrossRef]
- Espley, R.V.; Brendolise, C.; Chagné, D.; Kutty-Amma, S.; Green, S.; Volz, R.; Putterill, J.; Schouten, H.J.; Gardiner, S.E.; Hellens, R.P.; et al. Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 2009, 21, 168–183. [Google Scholar] [CrossRef] [Green Version]
- Butelli, E.; Garcia-Lor, A.; Licciardello, C.; Las Casas, G.; Hill, L.; Recupero, G.R.; Keremane, M.L.; Ramadugu, C.; Krueger, R.; Xu, Q.; et al. Changes in anthocyanin production during domestication of Citrus. Plant Physiol. 2017, 173, 2225–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin-Wang, K.; Bolitho, K.; Grafton, K.; Kortstee, A.; Karunairetnam, S.; McGhie, T.K.; Espley, R.V.; Hellens, R.P.; Allan, A.C. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 2010, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Wang, Y.; Yang, S.; Xu, Y.; Chen, X. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta 2010, 232, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Yakushiji, H.; Kobayashi, S.; Goto-Yamamoto, N.; Tae Jeong, S.; Sueta, T.; Mitani, N.; Azuma, A. A skin color mutation of grapevine, from black-skinned Pinot Noir to white-skinned Pinot Blanc, is caused by deletion of the functional VvmybA1 allele. Biosci. Biotechnol. Biochem. 2006, 70, 1506–1508. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, H.; Yang, Y.; Li, M.; Zhang, Y.; Liu, J.; Dong, J.; Li, J.; Butelli, E.; Xue, Z.; et al. The control of red colour by a family of MYB transcription factors in octoploid strawberry (Fragaria × ananassa) fruits. Plant Biotechnol. J. 2020, 18, 1169–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampomah-Dwamena, C.; Thrimawithana, A.H.; Dejnoprat, S.; Lewis, D.; Espley, R.V.; Allan, A.C. A kiwifruit (Actinidia deliciosa) R2R3-MYB transcription factor modulates chlorophyll and carotenoid accumulation. New Phytol. 2019, 221, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Montefiori, M.; Brendolise, C.; Lin-Wang, K.; Davies, K.M.; Hellens, R.P.; Allan, A.C. AN1 and JAF13 Confer Distinct Target Specificity to the MYB/bHLH/WD40 Complex that Regulates the Anthocyanin Biosynthesis; 2014; Unpublished in NCBI. [Google Scholar]
- Liu, Y.; Zhou, B.; Qi, Y.; Chen, X.; Liu, C.; Liu, Z.; Ren, X. Expression differences of pigment structural genes and transcription factors explain flesh coloration in three contrasting kiwifruit cultivars. Front. Plant Sci. 2017, 8, 1507. [Google Scholar] [CrossRef]
- Wang, L.; Tang, W.; Hu, Y.; Zhang, Y.; Sun, J.; Guo, X.; Lu, H.; Yang, Y.; Fang, C.; Niu, X.; et al. A MYB/bHLH complex regulates tissue-specific anthocyanin biosynthesis in the inner pericarp of red-centered kiwifruit Actinidia chinensis cv. Hongyang. Plant J. 2019, 99, 359–378. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript Length | Total Number | Percentage | Unigene Length | Total Number | Percentage |
|---|---|---|---|---|---|
| 200–300 | 36,864 | 16.07% | 200–300 | 28,565 | 28.45% |
| 300–500 | 42,911 | 18.71% | 300–500 | 26,507 | 26.40% |
| 500–1000 | 53,495 | 23.32% | 500–1000 | 20,159 | 20.08% |
| 1000–2000 | 55,806 | 24.33% | 1000–2000 | 13,838 | 13.78% |
| >2000 | 40,321 | 17.58% | >2000 | 11,348 | 11.30% |
| Total number | 229,397 | Total number | 100,471 | ||
| Total length | 267,448,645 | Total length | 86,736,802 | ||
| N50 length | 1856 | N50 length | 1600 | ||
| Mean length | 1165.88 | Mean length | 863.77 | ||
| Database | Number of Annotated Unigenes | “Length” (300–1000) | “Length” (>1000) |
|---|---|---|---|
| COG | 10,167 | 2519 | 6358 |
| KEGG | 13,391 | 4343 | 7123 |
| KOG | 21,038 | 6921 | 10,791 |
| GO | 21,560 | 6918 | 11,001 |
| Swissprot | 22,123 | 6590 | 13,062 |
| Pfam | 22,300 | 6416 | 13,379 |
| eggNOG | 34,480 | 11,576 | 17,083 |
| NR | 34,561 | 11,396 | 17,548 |
| All_Annotated | 37,519 | 12,916 | 17,787 |
| Gene Name | Species | NCBI GenBank Accession | Length/bp | Identity with AaMYB308like | References |
|---|---|---|---|---|---|
| AtPAP1 | Arabidopsis thaliana | NM_104541.4 | 747 | 61.76% | [27] |
| AtMYB113 | Arabidopsis thaliana | NM_105308.2 | 741 | 56.52% | [27] |
| SlMYB75 | Solanum lycopersicum | NM_001279063.2 | 828 | 59.29% | [28,29,30,31,32] |
| MdMYB1 | Malus domestica | NM_001301116.1 | 1239 | 46.85% | [33,34,35,36,37] |
| MdMYB10 | Malus domestica | EU518249.2 | 732 | 59.43% | [38] |
| CsRUBY | Citrus sinensis | NM_001288889.1 | 789 | 45.68% | [39,40] |
| PpMYB10 | Prunus persica | EU155160.1 | 675 | 64.29% | [41] |
| PyMYB10 | Pyrus pyrifolia | GU253310.1 | 735 | 62.00% | [42] |
| VvMYBA1 | Vitis vinifera | B242302.1 | 753 | 50.00% | [43] |
| FaMYB10-1 | Fragaria ananassa | MG456859.1 | 702 | 65.69% | [44] |
| FaMYB10-2 | Fragaria ananassa | MG456860.1 | 540 | 65.69% | [44] |
| FvMYB10 | Fragaria vesca | EU155163.1 | 708 | 65.69% | [44] |
| AcMYB10 | Actinidia chinensis | MG581953.1 | 666 | 64.76% | [45] |
| AcMYB75 | Actinidia chinensis | KX349735.1 | 666 | 64.76% | [26] |
| AcMYB110 | Actinidia chinensis | KF311107.1 | 729 | 57.03% | [46] |
| AcMYBF110 | Actinidia chinensis | MH370827.1 | 666 | 64.76% | [47] |
| AcMYB123 | Actinidia chinensis | MH643775.1 | 801 | 68.50% | [48] |
| AaMYBC1 | Actinidia arguta | MN249175.1 | 798 | 69.83% | [3] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Abid, M.; Lin, M.; Wang, R.; Gu, H.; Li, Y.; Qi, X. Comparative Transcriptome Analysis of Different Actinidia arguta Fruit Parts Reveals Difference of Light Response during Fruit Coloration. Biology 2021, 10, 648. https://doi.org/10.3390/biology10070648
Huang H, Abid M, Lin M, Wang R, Gu H, Li Y, Qi X. Comparative Transcriptome Analysis of Different Actinidia arguta Fruit Parts Reveals Difference of Light Response during Fruit Coloration. Biology. 2021; 10(7):648. https://doi.org/10.3390/biology10070648
Chicago/Turabian StyleHuang, Hailei, Muhammad Abid, Miaomiao Lin, Ran Wang, Hong Gu, Yukuo Li, and Xiujuan Qi. 2021. "Comparative Transcriptome Analysis of Different Actinidia arguta Fruit Parts Reveals Difference of Light Response during Fruit Coloration" Biology 10, no. 7: 648. https://doi.org/10.3390/biology10070648
APA StyleHuang, H., Abid, M., Lin, M., Wang, R., Gu, H., Li, Y., & Qi, X. (2021). Comparative Transcriptome Analysis of Different Actinidia arguta Fruit Parts Reveals Difference of Light Response during Fruit Coloration. Biology, 10(7), 648. https://doi.org/10.3390/biology10070648