
Oxidative Stress and Mitochondrial Damage in Dry Age-Related Macular Degeneration Like NFE2L2/PGC-1α -/- Mouse Model Evoke Complement Component C5a Independent of C3

 , ,
, ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics and Animal Experiments
2.2. Genotyping and Tissue Preparation
2.3. Immunohistochemical Staining
2.4. Confocal Imaging and Analysis
2.5. Statistics
3. Results
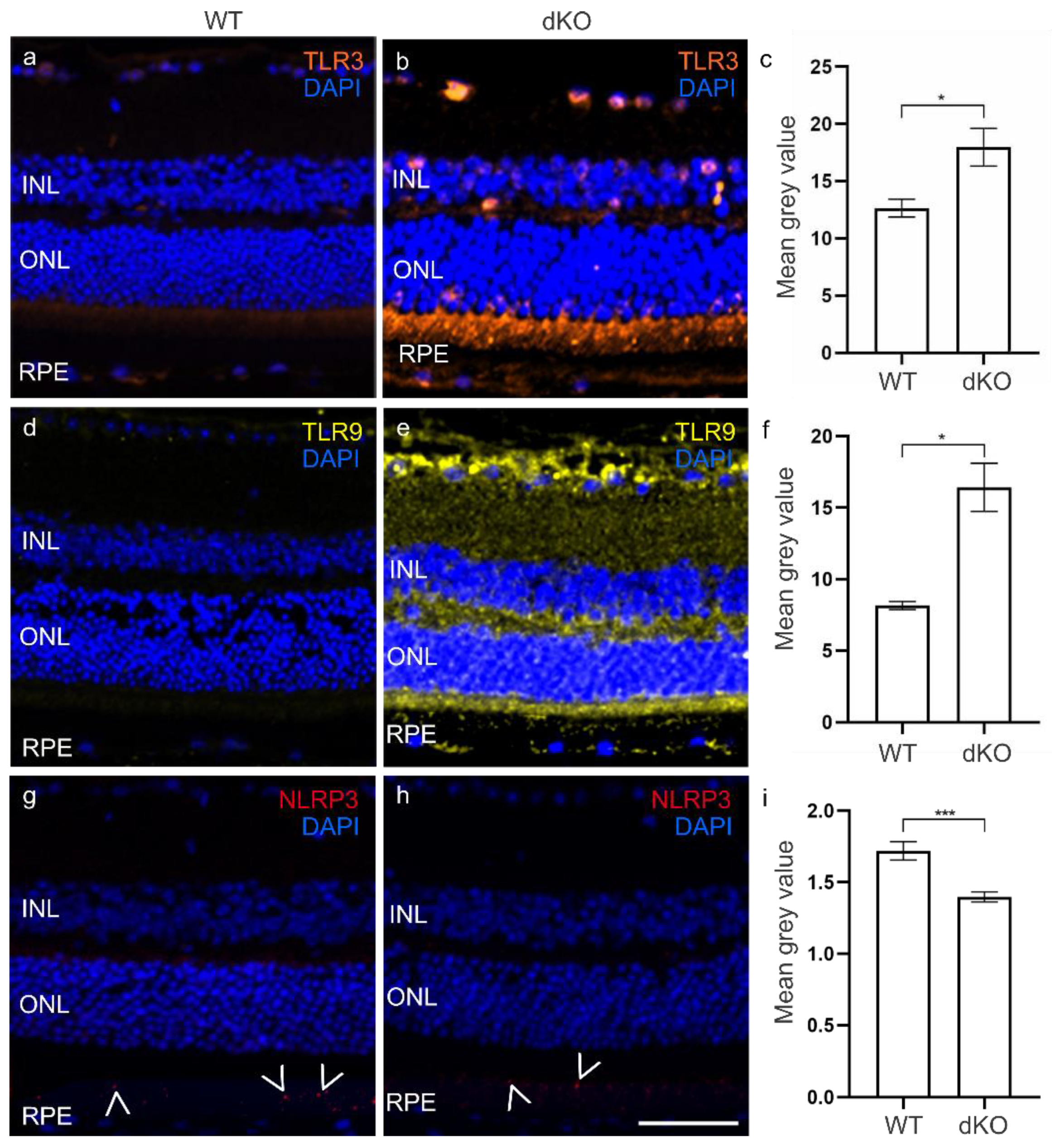
3.1. Elevated TLRs in the dKO Retina
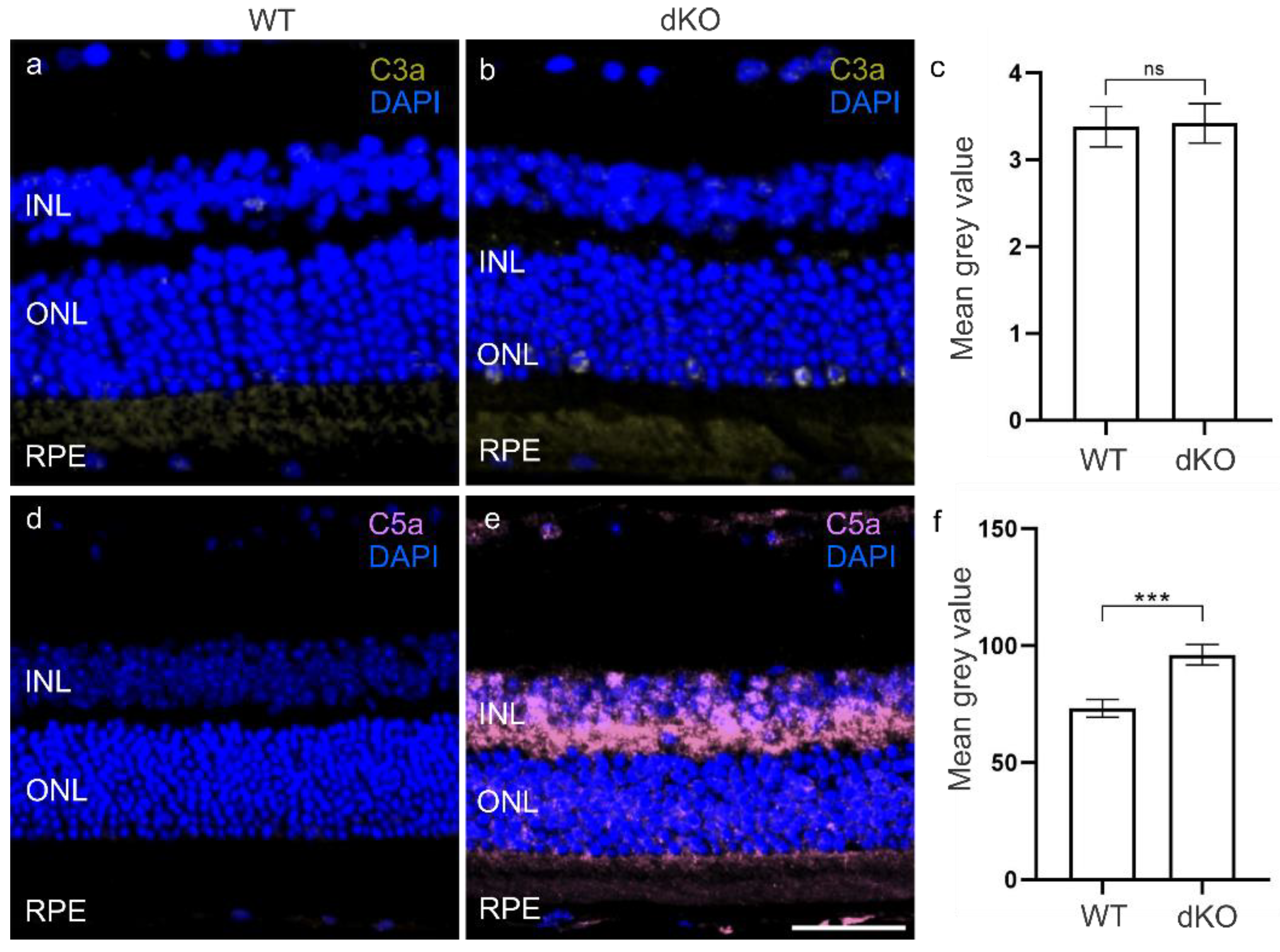
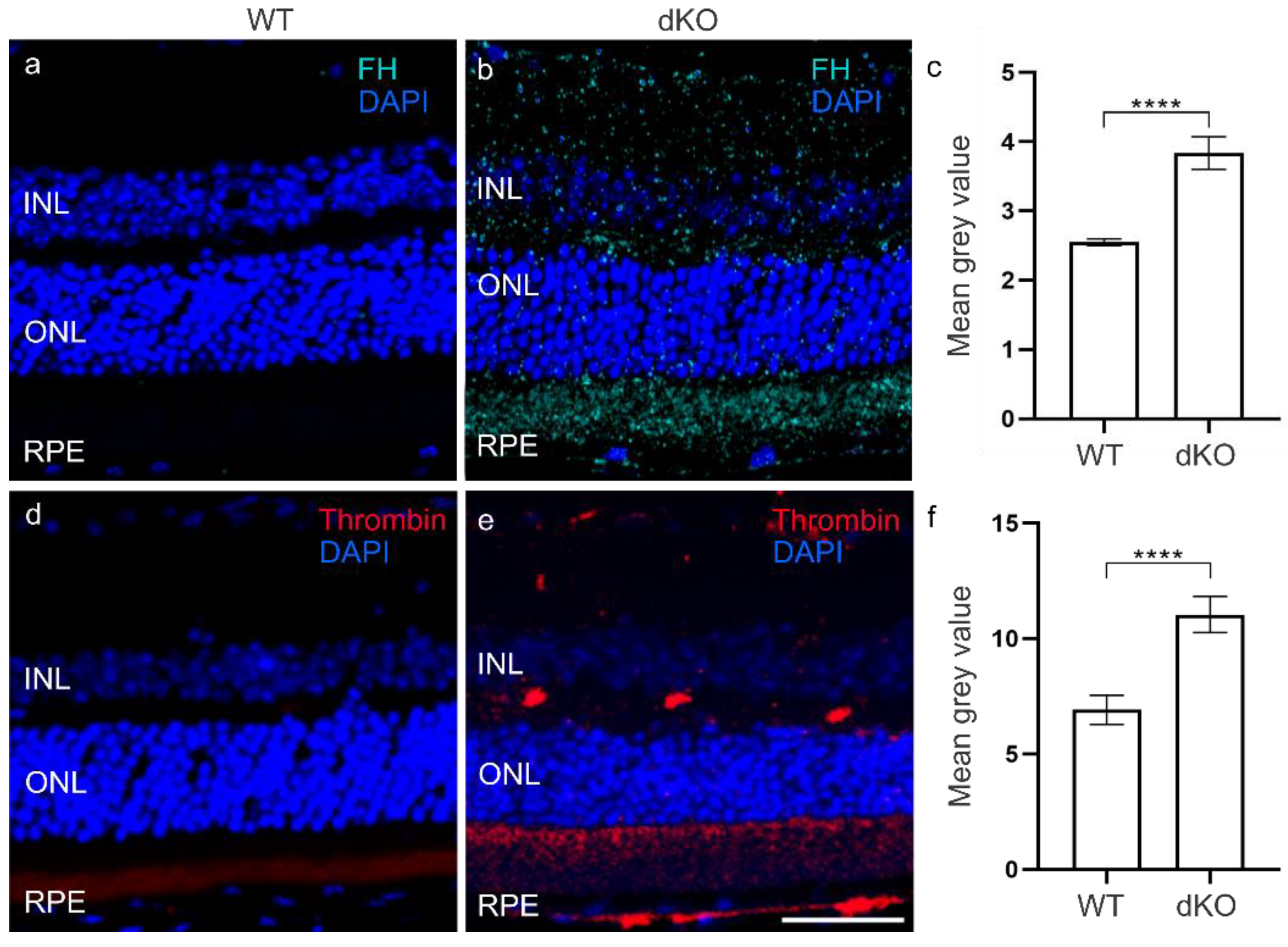
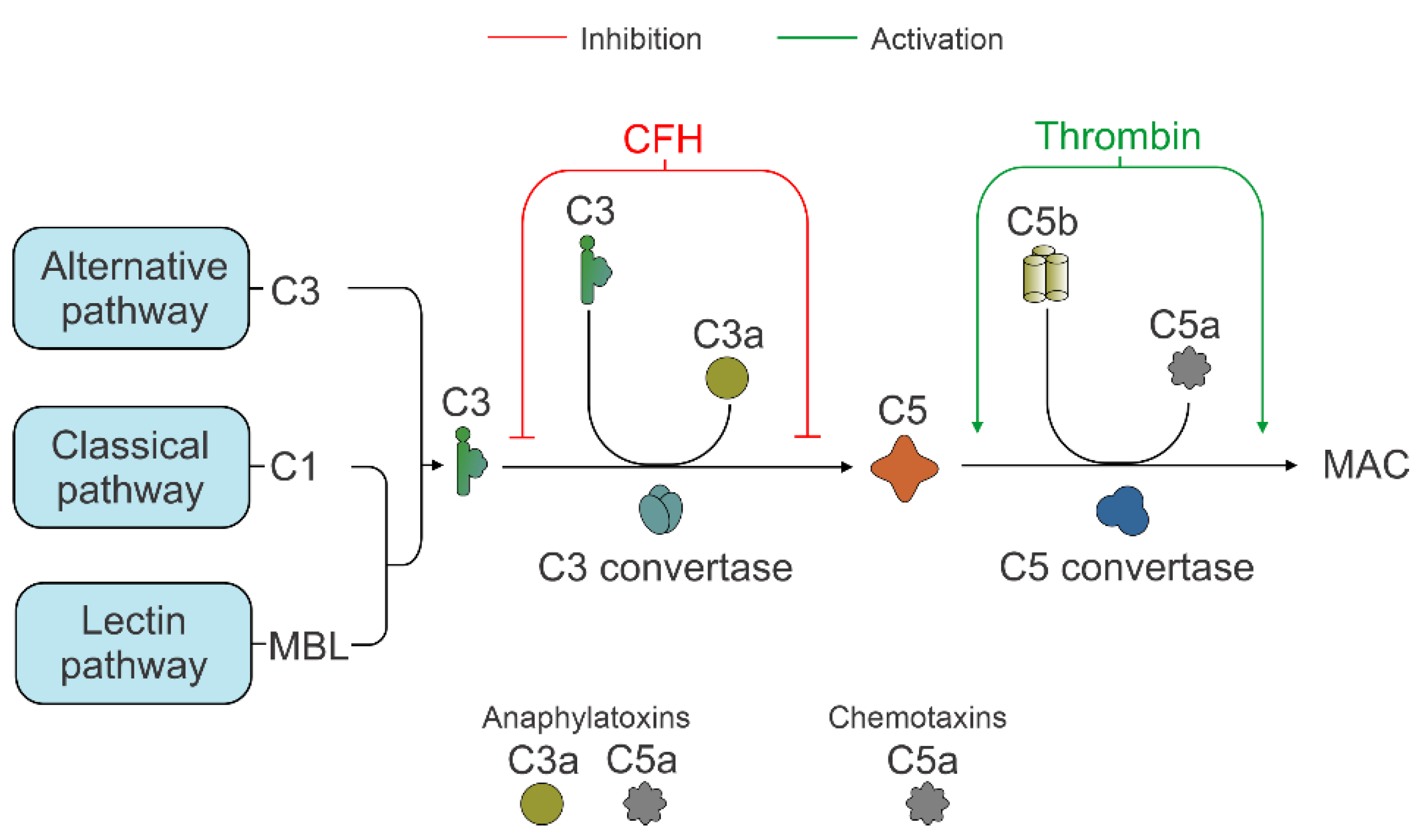
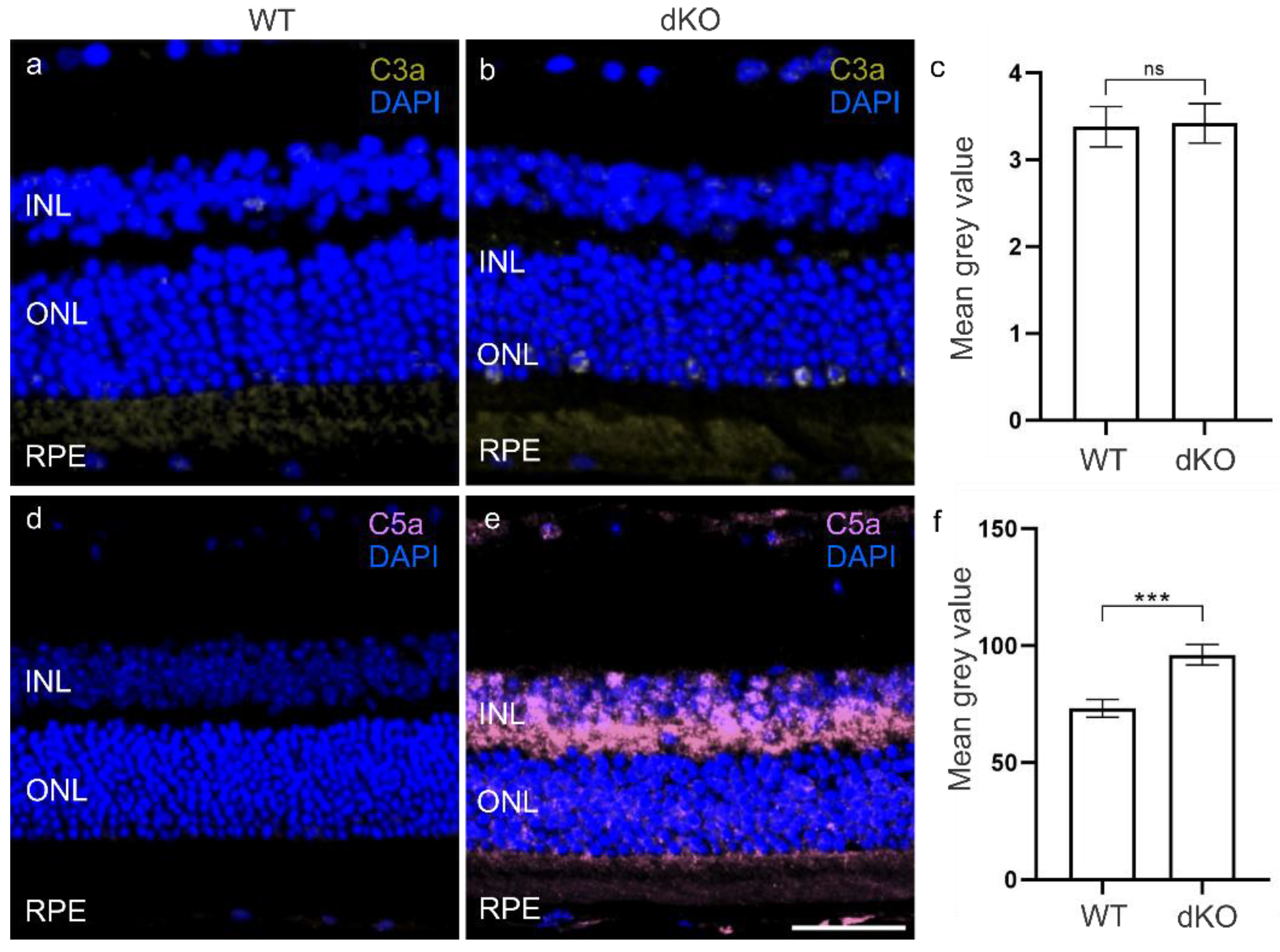
3.2. C3 Independent C5 Activation
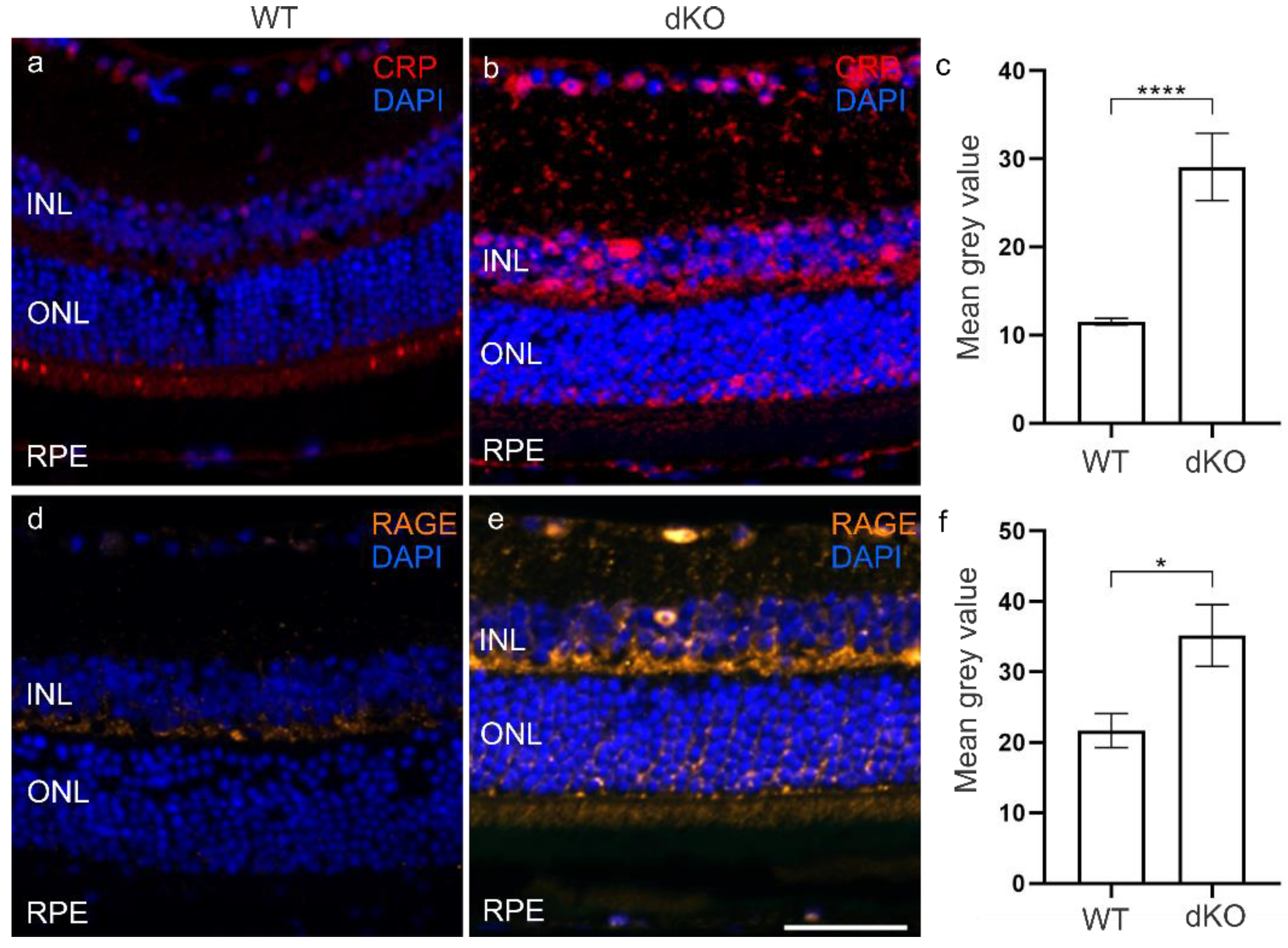
3.3. CRP and RAGE Coordinate under Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luu, J.; Palczewski, K. Human aging and disease: Lessons from age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 2866–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Hageman, G.S. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann. Med. 2006, 38, 450–471. [Google Scholar] [CrossRef]
- Al-Zamil, W.M.; Yassin, S.A. Recent developments in age-related macular degeneration: A review. Clin. Interv. Aging 2017, 12, 1313–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heesterbeek, T.J.; Lorés-Motta, L.; Hoyng, C.B.; Lechanteur, Y.T.E.; den Hollander, A.I. Risk factors for progression of age-related macular degeneration. Ophthalmic Physiol. Opt. 2020, 40, 140–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, R.; Harris, A.; Kheradiya, N.S.; Winston, D.M.; Ciulla, T.A.; Wirostko, B. Age-related macular degeneration and the aging eye. Clin. Interv. Aging 2008, 3, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Hyttinen, J.; Ryhanen, T.; Viiri, J.; Paimela, T.; Toropainen, E.; Sorri, I.; Salminen, A. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front. Biosci. 2010, 2, 1374–1384. [Google Scholar] [CrossRef] [Green Version]
- Younessi, P.; Yoonessi, A. Advanced glycation end-products and their receptor-mediated roles: Inflammation and oxidative stress. Iran. J. Med. Sci. 2011, 36, 154–166. [Google Scholar]
- Glenn, J.V.; Stitt, A.W. The role of advanced glycation end products in retinal ageing and disease. Biochim. Biophys. Acta 2009, 1790, 1109–1116. [Google Scholar] [CrossRef]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 2019, 10, 1007. [Google Scholar] [CrossRef]
- Wu, J.; Sun, X. Complement system and age-related macular degeneration: Drugs and challenges. Drug Des. Devel. Ther. 2019, 13, 2413–2425. [Google Scholar] [CrossRef]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.B.; Johnson, L.V.; Bowes Rickman, C. Complement factor H in AMD: Bridging genetic associations and pathobiology. Prog. Retin. Eye Res. 2018, 62, 38–57. [Google Scholar] [CrossRef] [PubMed]
- McHarg, S.; Clark, S.J.; Day, A.J.; Bishop, P.N. Age-related macular degeneration and the role of the complement system. Mol Immunol. 2015, 67, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Banevicius, M.; Vilkeviciute, A.; Kriauciuniene, L.; Liutkeviciene, R.; Deltuva, V.P. The Association between Variants of Receptor for Advanced Glycation End Products (RAGE) Gene Polymorphisms and Age-Related Macular Degeneration. Med. Sci. Monit. 2018, 24, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, J.E.; Chan, C.C.; Sen, H.N.; Ferris, F.L.; Nussenblatt, R.B. Inflammatory Mechanisms of Age-related Macular Degeneration. Int. Ophthalmol. Clin. 2015, 55, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Takeuchi, M.; Karasawa, Y.; Takayama, K.; Enoki, T. Comprehensive expression patterns of inflammatory cytokines in aqueous humor of patients with neovascular age-related macular degeneration. Sci. Rep. 2019, 9, 19447. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Romano, M.R.; Costagliola, C.; Semeraro, F.; Incorvaia, C.; D’Angelo, S.; Perri, P.; De Palma, P.; De Nadai, K.; Sebastiani, A. Mechanism of inflammation in age-related macular degeneration: An up-to-date on genetic landmarks. Mediat. Inflamm. 2013, 2013, 435607. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Salminen, A. Age-related macular degeneration: Activation of innate immunity system via pattern recognition receptors. J. Mol Med. 2009, 87, 117–123. [Google Scholar] [CrossRef]
- Witko-Sarsat, V.; Rieu, P.; Descamps-Latscha, B.; Lesavre, P.; Halbwachs-Mecarelli, L. Neutrophils: Molecules, functions and pathophysiological aspects. Lab. Investig. 2000, 80, 617–653. [Google Scholar] [CrossRef] [Green Version]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Puy, C.; Pang, J.; Reitsma, S.E.; Lorentz, C.U.; Tucker, E.I.; Gailani, D.; Gruber, A.; Lupu, F.; McCarty, O. Cross-Talk between the Complement Pathway and the Contact Activation System of Coagulation: Activated Factor XI Neutralizes Complement Factor, H. J. Immunol. 2021, 206, 1784–1792. [Google Scholar] [CrossRef]
- Kopp, A.; Hebecker, M.; Svobodová, E.; Józsi, M. Factor h: A complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2012, 2, 46–75. [Google Scholar] [CrossRef] [Green Version]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes-Gracia, K.; Raja, K.; Llanas-Cornejo, D.; Cobley, J.N.; Megson, I.L.; Chahwan, R.; Husi, H. Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel Plus 2020, 4, 27. [Google Scholar] [CrossRef]
- Rajendran, K.; Devarajan, N.; Ganesan, M.; Ragunathan, M. Obesity, Inflammation and Acute Myocardial Infarction—Expression of leptin, IL-6 and high sensitivity-CRP in Chennai based population. Thromb. J. 2012, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.L.; Hooper, W.C.; Jones, D.P.; Ashfaq, S.; Rhodes, S.D.; Weintraub, W.S.; Harrison, D.G.; Quyyumi, A.A.; Vaccarino, V. Association between novel oxidative stress markers and C-reactive protein among adults without clinical coronary heart disease. Atherosclerosis 2005, 178, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Cottone, S.; Mulè, G.; Nardi, E.; Vadalà, A.; Guarneri, M.; Briolotta, C.; Arsena, R.; Palermo, A.; Riccobene, R.; Cerasola, G. Relation of C-reactive protein to oxidative stress and to endothelial activation in essential hypertension. Am. J. Hypertens. 2006, 19, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Chirco, K.R.; Potempa, L.A. C-Reactive Protein As a Mediator of Complement Activation and Inflammatory Signaling in Age-Related Macular Degeneration. Front. Immunol. 2018, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Liu, W.; Huang, X.; Zhao, Y. Complement factor H levels are decreased and correlated with serum C-reactive protein in late-onset Alzheimer’s disease. Arq. Neuropsiquiatr. 2020, 78, 76–80. [Google Scholar] [CrossRef]
- Mold, C.; Kingzette, M.; Gewurz, H. C-reactive protein inhibits pneumococcal activation of the alternative pathway by increasing the interaction between factor H and C3b. J. Immunol. 1984, 133, 882–885. [Google Scholar] [PubMed]
- Jarva, H.; Jokiranta, T.S.; Hellwage, J.; Zipfel, P.F.; Meri, S. Regulation of complement activation by C-reactive protein: Targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J. Immunol. 1999, 163, 3957–3962. [Google Scholar] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Koskela, A.; Felszeghy, S.; Kivinen, N.; Salminen, A.; Kauppinen, A. Fatty acids and oxidized lipoproteins contribute to autophagy and innate immunity responses upon the degeneration of retinal pigment epithelium and development of age-related macular degeneration. Biochimie 2019, 159, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.K. Mitochondrial Reactive Oxygen Species: Double-Edged Weapon in Host Defense and Pathological Inflammation during Infection. Front. Immunol. 2020, 11, 1649. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The Role of Inflammation in Age-Related Macular Degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, W.T. Innate Immunity in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2021, 1256, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Stiles, T.; Douglas, C.; Ho, D.; Fan, W.; Du, H.; Xiao, X. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol. Sci. 2016, 3, 196–221. [Google Scholar] [CrossRef]
- Cavassani, K.A.; Ishii, M.; Wen, H.; Schaller, M.A.; Lincoln, P.M.; Lukacs, N.W.; Hogaboam, C.M.; Kunkel, S.L. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008, 205, 2609–2621. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.K.; Hackam, A.S. Toll-like receptor 3 (TLR3) protects retinal pigmented epithelium (RPE) cells from oxidative stress through a STAT3-dependent mechanism. Mol. Immunol. 2013, 54, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Borysiewicz, E.; Doppalapudi, S.; Kirschman, L.T.; Konat, G.W. TLR3 ligation protects human astrocytes against oxidative stress. J. Neuroimmunol. 2013, 255, 54–59. [Google Scholar] [CrossRef]
- Koarai, A.; Sugiura, H.; Yanagisawa, S.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Hirano, T.; Akamatsu, K.; Ichinose, M. Oxidative stress enhances toll-like receptor 3 response to double-stranded RNA in airway epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2010, 42, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, H.; Guo, D.; Hu, Y.; Fang, X.; Chen, Y.; Zhang, F. Oxidative stress and inflammation: Early predictive indicators of multiple recurrent coronary in-stent chronic total occlusions in elderly patients after coronary stenting. IUBMB Life 2020, 72, 1023–1033. [Google Scholar] [CrossRef]
- Wang, M.M.; Lu, M.; Zhang, C.L.; Wu, X.; Chen, J.X.; Lv, W.W.; Sun, T.; Qiu, H.; Huang, S.H. Oxidative stress modulates the expression of toll-like receptor 3 during respiratory syncytial virus infection in human lung epithelial A549 cells. Mol. Med. Rep. 2018, 18, 1867–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, 1077. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.C.; Xie, Q.M.; Xu, J.; Yan, X.B.; Fan, X.Y.; Wu, H.M. TLR9 mediates the activation of NLRP3 inflammasome and oxidative stress in murine allergic airway inflammation. Mol. Immunol. 2020, 125, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Foronjy, R.F.; Salathe, M.A.; Dabo, A.J.; Baumlin, N.; Cummins, N.; Eden, E.; Geraghty, P. TLR9 expression is required for the development of cigarette smoke-induced emphysema in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L154–L166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridevi Gurubaran, I.; Viiri, J.; Koskela, A.; Hyttinen, J.; Paterno, J.J.; Kis, G.; Antal, M.; Urtti, A.; Kauppinen, A.; Felszeghy, S.; et al. Mitophagy in the Retinal Pigment Epithelium of Dry Age-Related Macular Degeneration Investigated in the NFE2L2/PGC-1α-/- Mouse Model. Int. J. Mol. Sci. 2020, 21, 1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Xia, H.; Liang, Y.; Ye, Y.; Lu, Y.; Xu, X.; Duan, A.; He, J.; Chen, Z.; Wu, Y.; et al. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci. Rep. 2016, 6, 22579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celkova, L.; Doyle, S.L.; Campbell, M. NLRP3 Inflammasome and Pathobiology in AMD. J. Clin. Med. 2015, 4, 172–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Lambris, J.D. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010, 31, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Lambris, J.D. More than complementing Tolls: Complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol. Rev. 2016, 274, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Song, W.C. Crosstalk between complement and Toll-like receptors. Toxicol. Pathol. 2012, 40, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaprasad, S.; Chong, N.V. The complement system and age-related macular degeneration. Eye 2006, 20, 867–872. [Google Scholar] [CrossRef]
- Charbel Issa, P.; Chong, N.V.; Scholl, H.P. The significance of the complement system for the pathogenesis of age-related macular degeneration—current evidence and translation into clinical application. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Khandhadia, S.; Cipriani, V.; Yates, J.R.; Lotery, A.J. Age-related macular degeneration and the complement system. Immunobiology 2012, 217, 127–146. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur J. Pharmacol. 2016, 787, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [Green Version]
- Herbert, A.P.; Makou, E.; Chen, Z.A.; Kerr, H.; Richards, A.; Rappsilber, J.; Barlow, P.N. Complement Evasion Mediated by Enhancement of Captured Factor H: Implications for Protection of Self-Surfaces from Complement. J. Immunol. 2015, 195, 4986–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; El Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salvodelli, M.; Naud, M.C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 13873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Francis, P.J.; George, S.; Schultz, D.W.; Rosner, B.; Klein, M.L. Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA 2007, 297, 1793–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, V.P.; Pangburn, M.K.; Cortés, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Zwarthoff, S.A.; Berends, E.; Mol, S.; Ruyken, M.; Aerts, P.C.; Józsi, M.; De Haas, C.J.; Rooijakkers, S.H.; Gorham, R.D., Jr. Functional Characterization of Alternative and Classical Pathway C3/C5 Convertase Activity and Inhibition Using Purified Models. Front. Immunol. 2018, 9, 1691. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [Green Version]
- Krisinger, M.J.; Goebeler, V.; Lu, Z.; Meixner, S.C.; Myles, T.; Pryzdial, E.L.; Conway, E.M. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 2012, 120, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, H.; Imamura, T.; Matsushita, M.; Tanase, S.; Okada, H.; Ogawa, M.; Kambara, T. Thrombin generates monocyte chemotactic activity from complement factor H. Immunology 1993, 80, 140–145. [Google Scholar]
- Parrales, A.; López, E.; López-Colomé, A.M. Thrombin activation of PI3K/PDK1/Akt signaling promotes cyclin D1 upregulation and RPE cell proliferation. Biochim. Biophys. Acta 2011, 1813, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Palma-Nicolas, J.P.; López, E.; López-Colomé, A.M. PKC isoenzymes differentially modulate the effect of thrombin on MAPK-dependent RPE proliferation. Biosci. Rep. 2008, 28, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Hecquet, C.; Lefevre, G.; Valtink, M.; Engelmann, K.; Mascarelli, F. Activation and role of MAP kinase-dependent pathways in retinal pigment epithelial cells: ERK and RPE cell proliferation. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3091–3098. [Google Scholar]
- Goel, R.; Phillips-Mason, P.J.; Raben, D.M.; Baldassare, J.J. alpha-Thrombin induces rapid and sustained Akt phosphorylation by beta-arrestin1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. J. Biol. Chem. 2002, 277, 18640–18648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, E.; Majkic-Singh, N.; Zoric, L.; Radosavljevic, A.; Kosanovic-Jakovic, N. The role of CRP and inflammation in the pathogenesis of age-related macular degeneration. Biochem. Med. 2012, 22, 39–48. [Google Scholar] [CrossRef]
- Seddon, J.M.; Gensler, G.; Milton, R.C.; Klein, M.L.; Rifai, N. Association between C-reactive protein and age-related macular degeneration. JAMA 2004, 291, 704–710. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Wang, P.; Li, S.; Zeng, J.; Tu, X.; Yan, Q.; Xiao, Z.; Pan, M.; Zhu, F. Syncytin-1, an endogenous retroviral protein, triggers the activation of CRP via TLR3 signal cascade in glial cells. Brain Behav. Immun. 2018, 67, 324–334. [Google Scholar] [CrossRef]
- Lo Gullo, A.; Mandraffino, G.; Imbalzano, E.; Mamone, F.; Aragona, C.O.; D’Ascola, A.; Loddo, S.; Cinquegrani, A.; Alibrandi, A.; Mormina, E.; et al. Toll-like receptor 3 and interleukin 1β expression in CD34+ cells from patients with rheumatoid arthritis: Association with inflammation and vascular involvement. Clin. Exp. Rheumatol. 2014, 32, 922–929. [Google Scholar]
- McCausland, M.R.; Cruz-Lebrón, A.; Pilch-Cooper, H.A.; Howell, S.; Albert, J.M.; Park, Y.S.; Levine, A.D. Toll-like receptor distribution in colonic epithelium and lamina propria is disrupted in HIV viremic, immune success, and failure. AIDS 2020, 34, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; Baba, T.; Merges, C.; Juriasinghani, V.; McLeod, D.S.; Lutty, G.A. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br. J. Ophthalmol. 2011, 95, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Xu, X.Y.; Feng, C.H.; Wang, Y.; Li, Y.L.; Feng, B. Relationships among serum C-reactive protein, receptor for advanced glycation products, metabolic dysfunction, and cognitive impairments. BMC Neurol. 2013, 13, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Q.; Sun, Z.; Shao, C.; Cai, H.; Bao, Z.; Wang, L.; Li, L.; Jing, L.; Zhang, L.; Wang, Z. CML/RAGE Signal Bridges a Common Pathogenesis Between Atherosclerosis and Non-alcoholic Fatty Liver. Front. Med. 2020, 7, 583943. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, S.H.; Liu, S.M.; Szmitko, P.E.; He, X.Q.; Fedak, P.W.; Verma, S. C-Reactive protein upregulates receptor for advanced glycation end products expression in human endothelial cells. Hypertension 2006, 48, 504–511. [Google Scholar] [CrossRef]
- Mahajan, N.; Bahl, A.; Dhawan, V. C-reactive protein (CRP) up-regulates expression of receptor for advanced glycation end products (RAGE) and its inflammatory ligand EN-RAGE in THP-1 cells: Inhibitory effects of atorvastatin. Int. J. Cardiol. 2010, 142, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jin, J.; Song, M.; Dong, H.; Zhao, G.; Huang, L. C-reactive protein down-regulates endothelial nitric oxide synthase expression and promotes apoptosis in endothelial progenitor cells through receptor for advanced glycation end-products. Gene 2012, 496, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Paradela-Dobarro, B.; Raposeiras-Roubín, S.; Rodiño-Janeiro, B.K.; Grigorian-Shamagian, L.; García-Acuña, J.M.; Aguiar-Souto, P.; Jacquet-Hervet, M.; Reino-Maceiras, M.V.; González-Juanatey, J.R.; Alvarez, E. Statins modulate feedback regulation mechanisms between advanced glycation end-products and C-reactive protein: Evidence in patients with acute myocardial infarction. Eur. J. Pharm. Sci. 2013, 49, 512–518. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies Against | Isotope | Working Dilution | Supplier/Catalogue Number |
|---|---|---|---|
| C5a | Polyclonal | 1:250 | AB217027 |
| C3a | Monoclonal | 1:100 | NBP2-66994 |
| FH | Polyclonal | 1:50 | NBP2-90802 |
| C-reactive protein | Polyclonal | 1:100 | AB65842 |
| NLRP3 | Polyclonal | 1:100 | AB214185 |
| RAGE | Polyclonal | 1:100 | AB3611 |
| Thrombin | Monoclonal | 1:100 | SC271449 |
| TLR3 | Polyclonal | 1:100 | AB62566 |
| TLR9 | Polyclonal | 1:100 | AB37154 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sridevi Gurubaran, I.; Heloterä, H.; Marry, S.; Koskela, A.; Hyttinen, J.M.T.; Paterno, J.J.; Urtti, A.; Chen, M.; Xu, H.; Kauppinen, A.; et al. Oxidative Stress and Mitochondrial Damage in Dry Age-Related Macular Degeneration Like NFE2L2/PGC-1α -/- Mouse Model Evoke Complement Component C5a Independent of C3. Biology 2021, 10, 622. https://doi.org/10.3390/biology10070622
Sridevi Gurubaran I, Heloterä H, Marry S, Koskela A, Hyttinen JMT, Paterno JJ, Urtti A, Chen M, Xu H, Kauppinen A, et al. Oxidative Stress and Mitochondrial Damage in Dry Age-Related Macular Degeneration Like NFE2L2/PGC-1α -/- Mouse Model Evoke Complement Component C5a Independent of C3. Biology. 2021; 10(7):622. https://doi.org/10.3390/biology10070622
Chicago/Turabian StyleSridevi Gurubaran, Iswariyaraja, Hanna Heloterä, Stephen Marry, Ali Koskela, Juha M. T. Hyttinen, Jussi J. Paterno, Arto Urtti, Mei Chen, Heping Xu, Anu Kauppinen, and et al. 2021. "Oxidative Stress and Mitochondrial Damage in Dry Age-Related Macular Degeneration Like NFE2L2/PGC-1α -/- Mouse Model Evoke Complement Component C5a Independent of C3" Biology 10, no. 7: 622. https://doi.org/10.3390/biology10070622
APA StyleSridevi Gurubaran, I., Heloterä, H., Marry, S., Koskela, A., Hyttinen, J. M. T., Paterno, J. J., Urtti, A., Chen, M., Xu, H., Kauppinen, A., & Kaarniranta, K. (2021). Oxidative Stress and Mitochondrial Damage in Dry Age-Related Macular Degeneration Like NFE2L2/PGC-1α -/- Mouse Model Evoke Complement Component C5a Independent of C3. Biology, 10(7), 622. https://doi.org/10.3390/biology10070622