
In Silico Studies on Sennidines—Natural Dianthrones from Senna


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
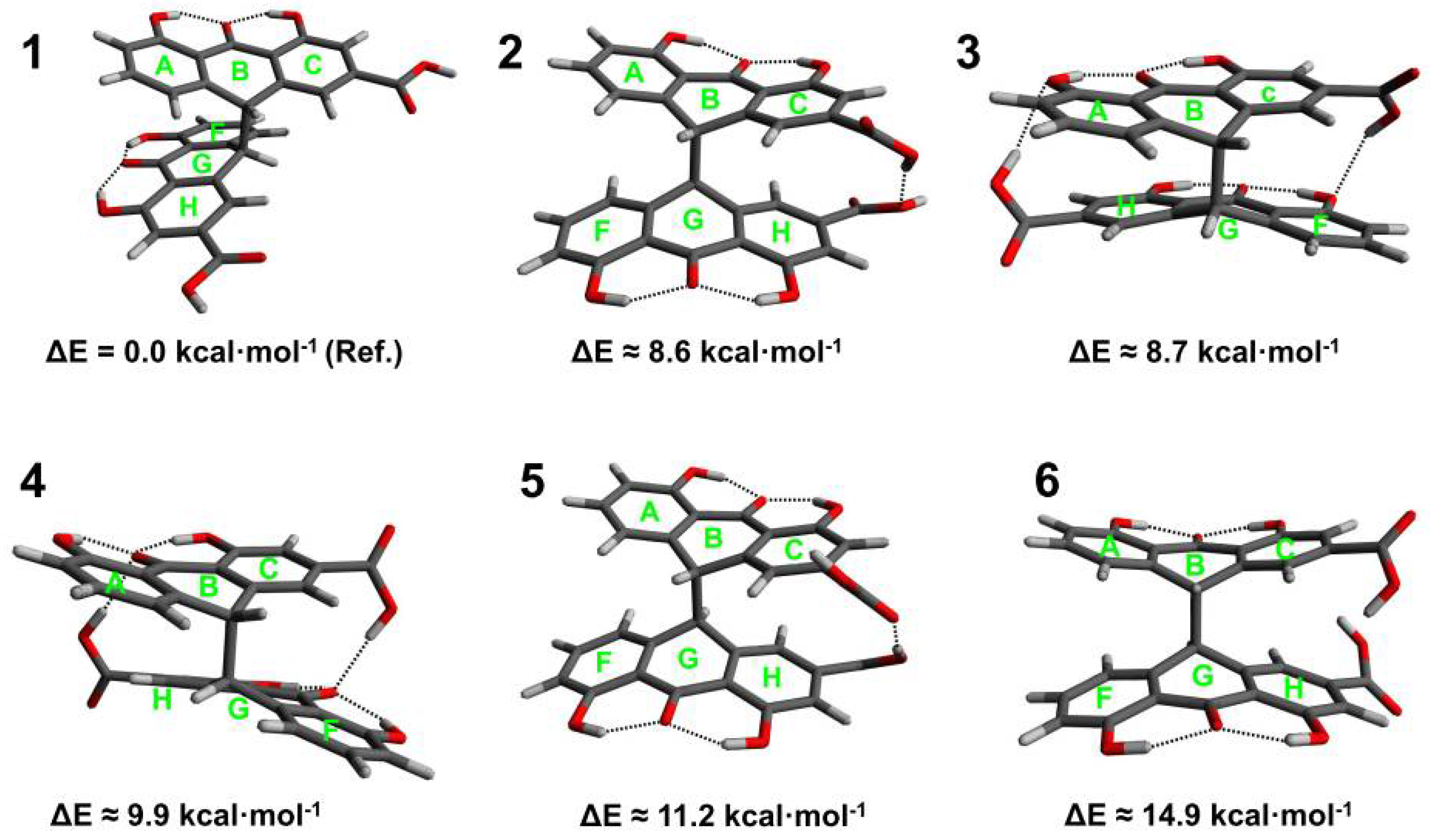
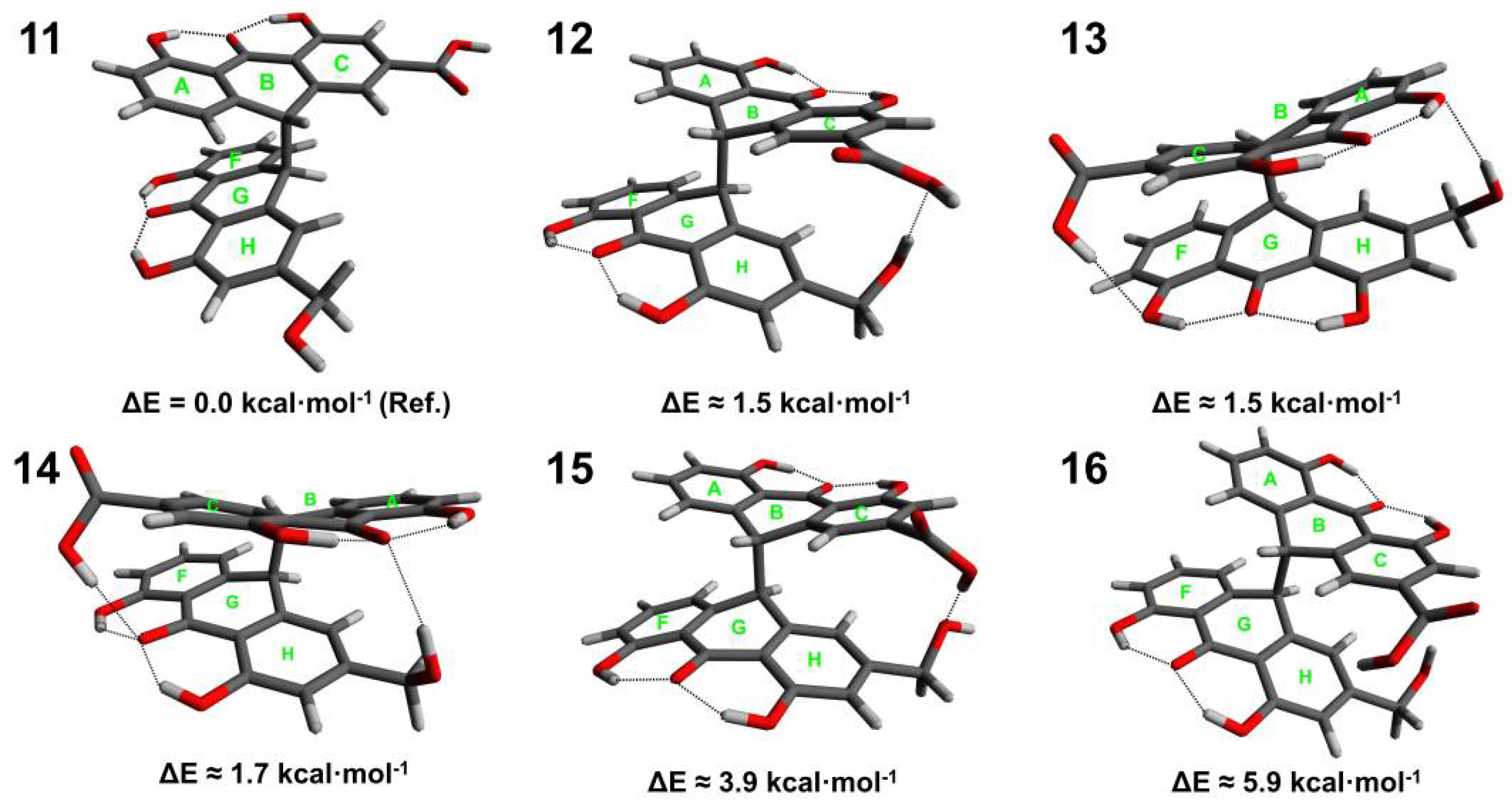
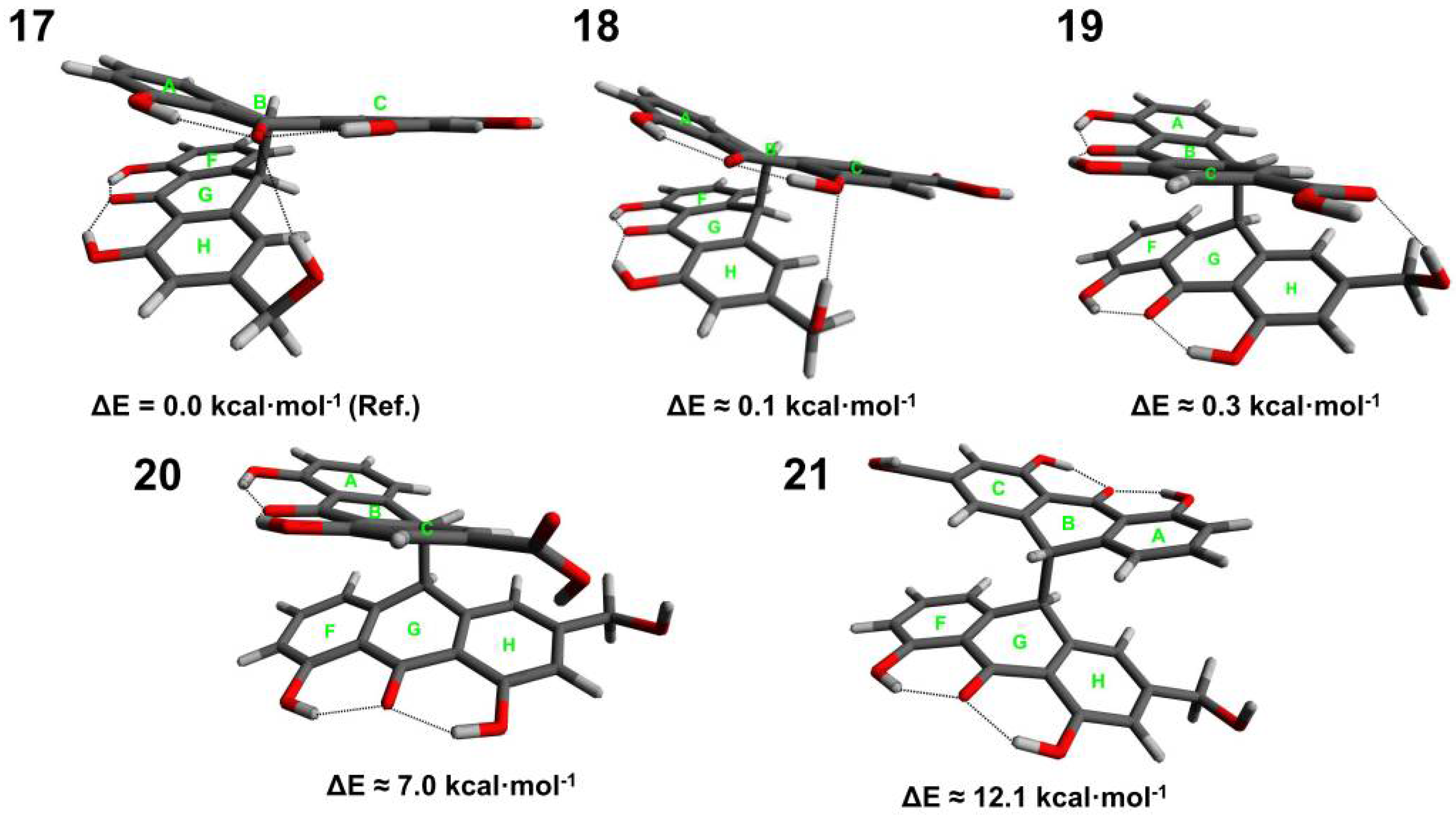
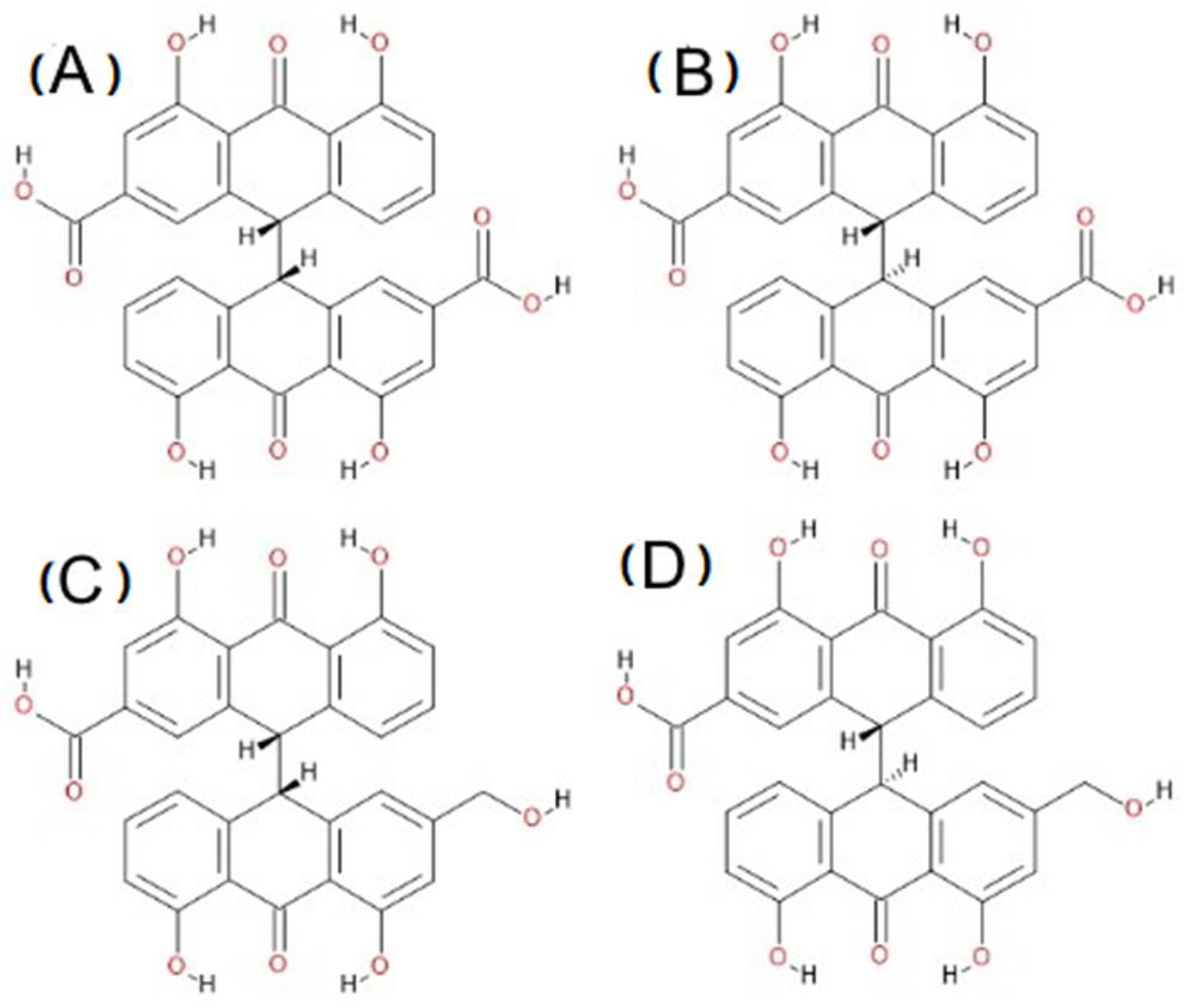
3.1. Conformational Analysis of Sennidines
3.2. Analysis of Geometry of Sennidin Structures
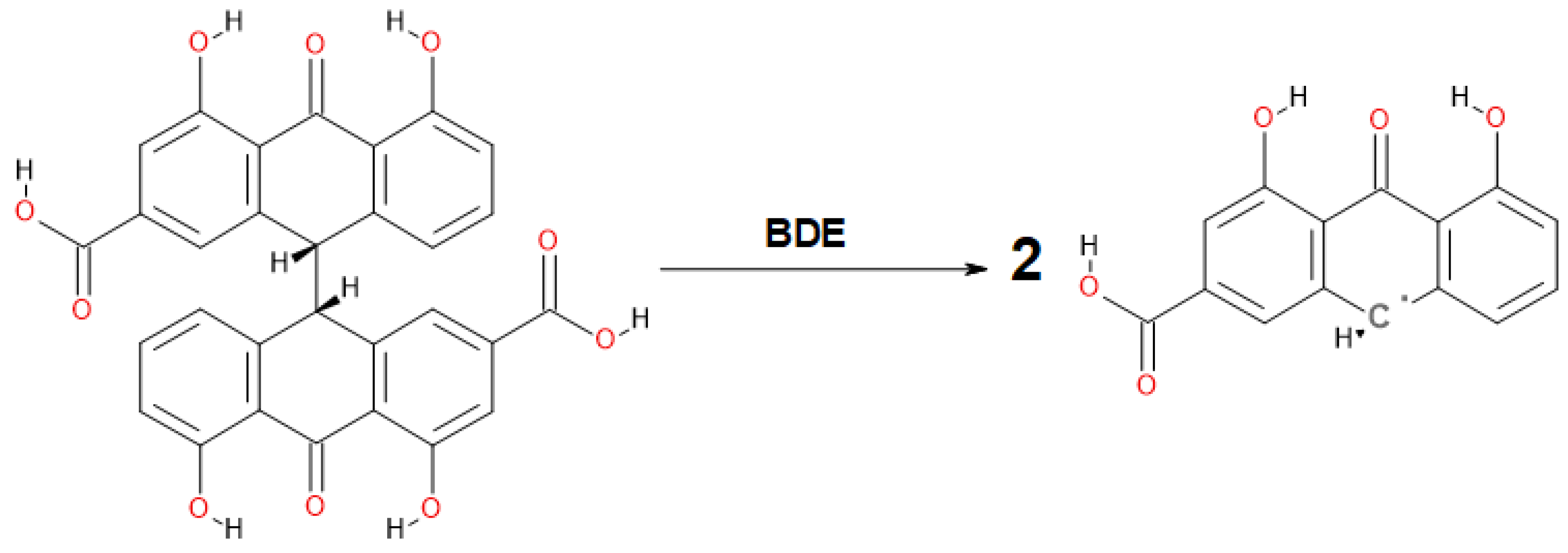
3.3. Analysis of Intramolecular Interactions in Sennidin Derivatives
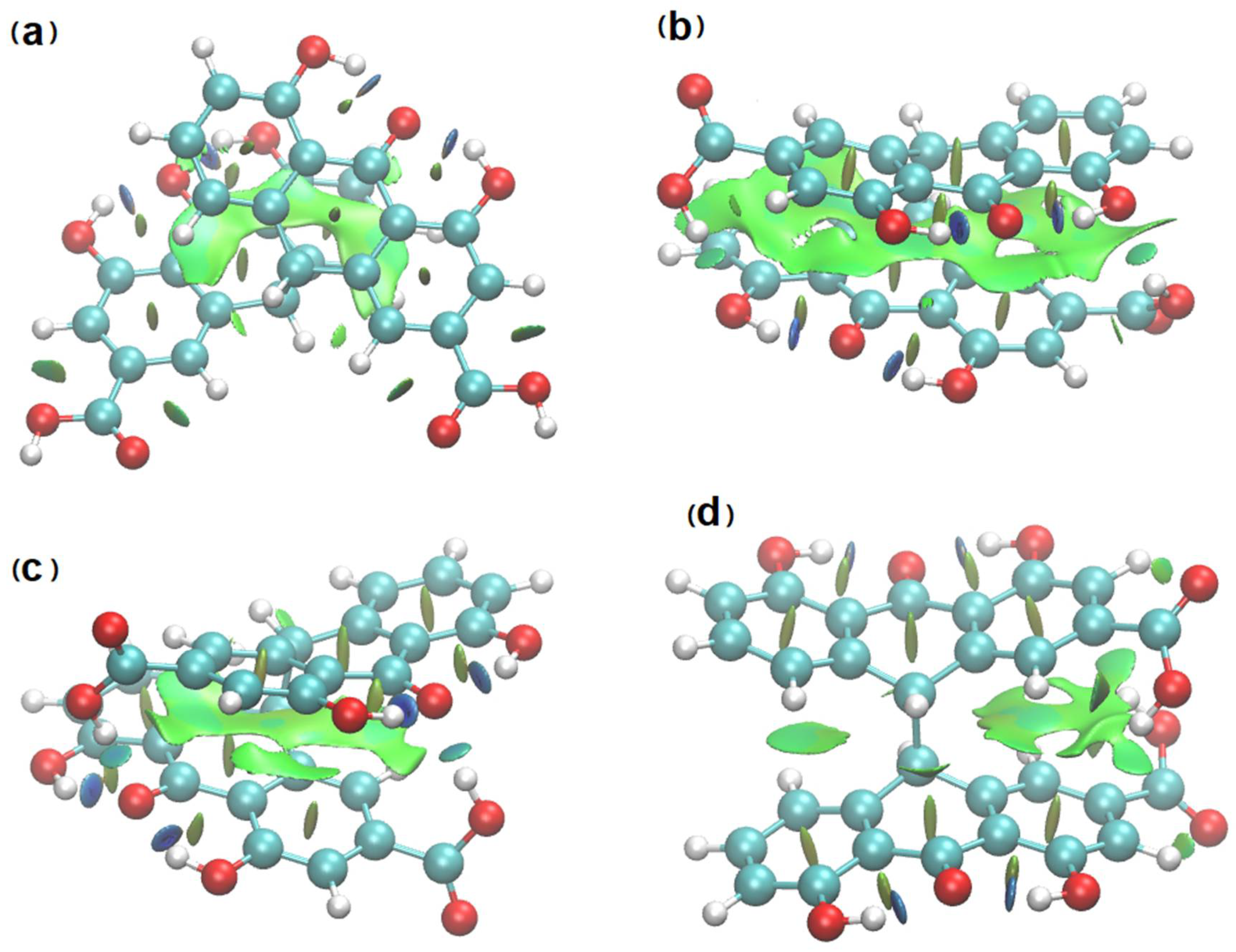
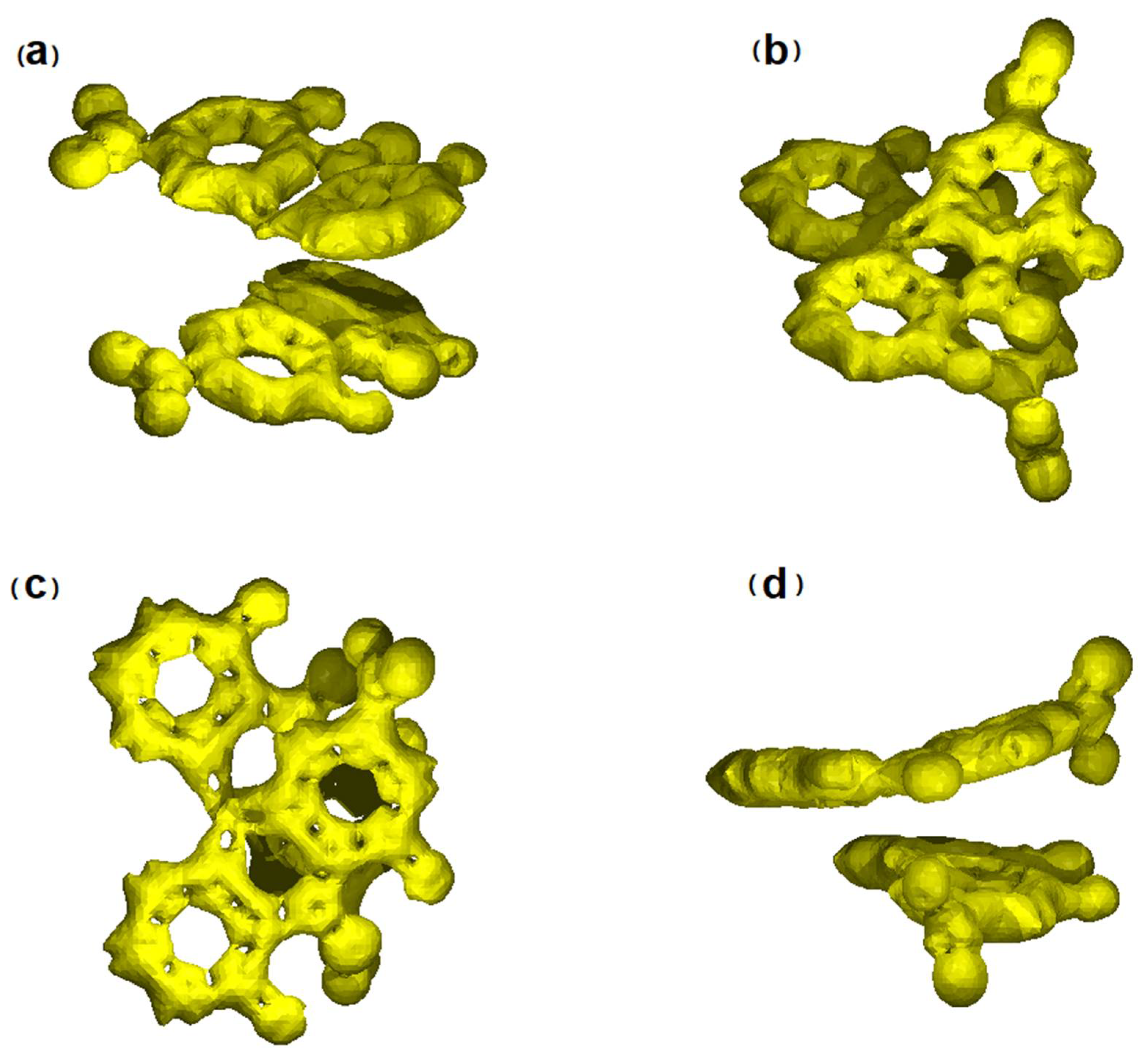
3.4. NCI Analysis of Sennidin Derivatives
3.5. Delocalization of Electrons in the Sennidin Molecule
3.6. Spectroscopic Properties of Sennidines
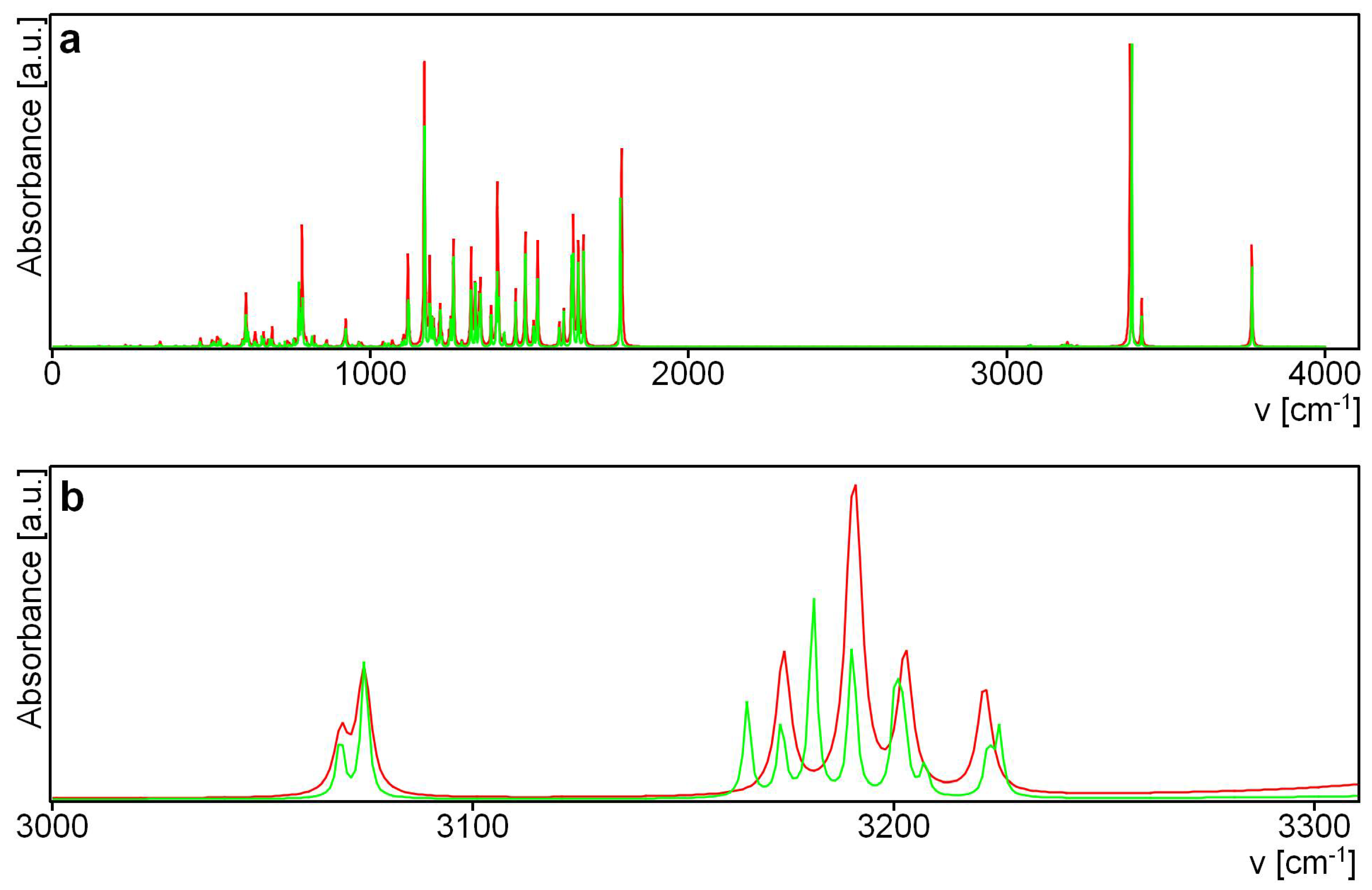
3.6.1. IR Spectra
3.6.2. UV Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Szymanski, S.; Majerz, I. Aromaticity and Electron Density of Hypericin. J. Nat. Prod. 2019, 82, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Goppel, M.; Franz, G. Stability control of senna leaves and senna extracts. Planta Med. 2004, 70, 432–436. [Google Scholar]
- Abe, D.; Saito, T.; Sekiya, K. Sennidin stimulates glucose incorporation in rat adipocytes. Life Sci. 2006, 79, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Franz, G. The senna drug and its chemistry. Pharmacology 1993, 47, 2–6. [Google Scholar] [CrossRef]
- Monkheang, P.; Sudmoon, R.; Tanee, T.; Noikotr, K.; Bletter, N.; Chaveerach, A. Species diversity, usages, molecular markers and barcode of medicinal Senna species (Fabaceae, Caesalpinioideae) in Thailand. J. Med. Plant Res. 2011, 5, 6173–6181. [Google Scholar] [CrossRef]
- Khan, S.; Mirza, K.J.; Al-Qurainy, F.; Abdin, M.Z. Authentication of the medicinal plant Senna angustifolia by RAPD profiling. Saudi J. Biol. Sci. 2011, 18, 287–292. [Google Scholar] [CrossRef]
- Marciniak, C.M.; Toledo, S.; Lee, J.; Jesselson, M.; Bateman, J.; Grover, B.; Tierny, J. Lubiprostone vs Senna in postoperative orthopedic surgery patients with opioid-induced constipation: A double-blind, active-comparator trial. World J. Gastroenterol. 2014, 20, 16323–16333. [Google Scholar] [CrossRef]
- Fairbairn, J.W.; Moss, M.J.R. The relative purgative activities of 1,8-dihydroxyanthracene derivatives. J. Pharm. Pharmacol. 1970, 22, 584–593. [Google Scholar] [CrossRef]
- Akao, T.; Mibu, K.; Hattori, M.; Namba, T.; Kobashi, K. Enzymatic reduction of sennidin and sennoside in Peptostreptococcus intermedius. J. Pharmacobiodyn. 1985, 8, 800–807. [Google Scholar] [CrossRef]
- Hattori, M.; Akao, T.; Kobashi, K.; Namba, T. Cleavages of the O- and C-Glucosyl bonds of anthrone and 10,10′-bianthrone derivatives by human intestinal bacteria. Pharmacology 1993, 47, 125–133. [Google Scholar] [CrossRef]
- Fan, M.; Peng, C.; Peng, Y.; Zhang, M.; Li, X. Analysis of Metabolites of Anthraquinones by Human Fecal Bacteria Using UPLC-Q-TOF-HRMS/MS. Chromatographia 2016, 79, 1593–1604. [Google Scholar] [CrossRef]
- Hietala, P.; Marvola, M.; Parviainen, T.; Lainonen, H. Laxative Potency and Acute Toxicity of Some Anthraquinone Derivatives, Senna Extracts and Fractions of Senna Extracts. Pharmacol. Toxicol. 1987, 61, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Schörkhuber, M.; Richter, M.; Dutter, A.; Sontag, G.; Marian, B. Effect of anthraquinone-laxatives on the proliferation and urokinase secretion of normal, premalignant and malignant colonic epithelial cells. Eur. J. Cancer 1998, 34, 1091–1098. [Google Scholar] [CrossRef]
- Wald, A. Is chronic use of stimulant laxatives harmful to the colon? J. Clin. Gastroenterol. 2003, 36, 386–389. [Google Scholar] [CrossRef]
- Morales, M.A.; Hernández, D.; Bustamante, S.; Bachiller, I.; Rojas, A. Is Senna Laxative Use Associated to Cathartic Colon, Genotoxicity, or Carcinogenicity? J. Toxicol. 2009, 1–8. [Google Scholar] [CrossRef]
- Ji, Y.; Hou, X.; Jiang, C.; Liu, W.; Gao, M.; Li, Y.; Wang, J.; Wang, Q.; Sun, Z.; Jiang, X.; et al. Necrosis targeted combinational theragnostic approach using radioiodinated Sennidin A in rodent tumor models. Oncotarget 2014, 5, 2934–2946. [Google Scholar] [CrossRef]
- Jiang, C.; Gao, M.; Li, Y.; Huang, D.; Yao, N.; Ji, Y.; Liu, X.; Zhang, D.; Wang, X.; Yin, Z.; et al. Exploring diagnostic potentials of radioiodinated sennidin A in rat model of reperfused myocardial infarction. Int. J. Pharm. 2015, 495, 31–40. [Google Scholar] [CrossRef]
- Li, L.; Zhang, D.; Yang, S.; Song, S.; Li, J.; Wang, Q.; Wang, C.; Feng, Y.; Ni, Y.; Zhang, J.; et al. Effects of Glycosylation on Biodistribution and Imaging Quality of Necrotic Myocardium of Iodine-131-Labeled Sennidins. Mol. Imaging Biol. 2016, 18, 877–886. [Google Scholar] [CrossRef]
- Shah, S.A.; Ravishankara, M.N.; Nirmal, A.; Shishoo, C.J.; Rathod, I.S.; Suhagia, B.N. Estimation of Individual Sennosides in Plant Materials and Marketed Formulations by an HPTLC Method. J. Pharm. Pharmacol. 2000, 52, 445–449. [Google Scholar] [CrossRef]
- Melka Abdo, B. Sennosides Determination of Ethiopian Senna alexandrina Mill Accessions. Nat. Prod. Chem. Res. 2017, 5, 1–4. [Google Scholar] [CrossRef]
- Upadhyay, A.; Chandel, Y.; Nayak, P.S.; Khan, N.A. Sennoside contents in Senna (Cassia angustifolia Vahl.) as influenced by date of leaf picking, packaging material and storage period. J. Stored Prod. Postharvest Res. 2011, 2, 97–103. [Google Scholar]
- Basak, B.B.; Gajbhiye, N.A. Herbage yield and bioactive principle of senna as influenced by micronutrient application in soil. J. Environ. Biol. 2018, 39, 43–49. [Google Scholar] [CrossRef]
- Kuhnert, N.; Molod, H.Y. An efficient total synthesis of chrysophanol and the sennoside C aglycon. Tetrahedron Lett. 2005, 46, 7571–7573. [Google Scholar] [CrossRef]
- Waltenberger, B.; Avula, B.; Ganzera, M.; Khan, I.A.; Stuppner, H.; Khan, S.I. Transport of sennosides and sennidines from Cassia angustifolia and Cassia senna across Caco-2 monolayers—An in vitro model for intestinal absorption. Phytomedicine 2008, 15, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Namba, T.; Akao, T.; Kobashi, K. Metabolism of sennosides by human intestinal bacteria. Pharmacology 1988, 36, 172–179. [Google Scholar] [CrossRef]
- Whitefield, M.; Henrick, K.; Owston, P.G. 1,1′,8,8′-Tetrahydroxy-10,10′-bi-9 (10H)-anthrone. Acta Cryst. 1982, B38, 1248–1251. [Google Scholar]
- Li, P.-C.; Wang, T.-S.; Lee, G.-H.; Liu, Y.-H.; Wang, Y.; Chen, C.-T.; Chao, I. Theoretical Study and X-ray Determination of Bianthrones: Long C-C Bond Length and Preferred Gauche Conformation. J. Org. Chem. 2002, 67, 8002–8009. [Google Scholar] [CrossRef]
- Mandelare, P.E.; Adpressa, D.A.; Kaweesa, E.N.; Zakharov, L.N.; Loesgen, S. Coculture of Two Developmental Stages of a Marine-Derived Aspergillus alliaceus Results in the Production of the Cytotoxic Bianthrone Allianthrone A. J. Nat. Prod. 2018, 81, 1014–1022. [Google Scholar] [CrossRef]
- Ji, N.-Y.; Liang, X.-R.; Sun, R.-R.; Miao, F.-P. A rule to distinguish diastereomeric bianthrones by 1H NMR. RSC Adv. 2014, 4, 7710–7715. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Keith, T.A. AIMALL, version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019.
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Herges, R.; Geuenich, D. Delocalization of electrons in molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Putz, M.V. Chemical Action Concept and Principle. MATCH Commun. Math. Comput. Chem. 2011, 66, 35–63. [Google Scholar]
- Bader, R.F.W. Bond paths are not chemical bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Definition of molecular structure: By choice or by appeal to observation? J. Phys. Chem. A 2010, 114, 7431–7444. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Bader, R.F.W. Effect of twisting a polypeptide on its geometry and electron distribution. J. Phys. Chem. 1994, 98, 4473–4481. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of C–H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. The carbon–carbon bond dissociation energy as a function of the chain length. Chem. Phys. Lett. 2006, 425, 221–224. [Google Scholar] [CrossRef]
- Grimme, S.; Mück-Lichtenfeld, C. Accurate Computation of Structures and Strain Energies of Cyclophanes with Modern DFT Methods. Isr. J. Chem. 2012, 52, 180–192. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Plane/Angle [Deg.] | |
|---|---|---|---|
| A–C | F–H | ||
| Sennidin A | 1 | 17.700 | 17.701 |
| 2 | 22.040 | 20.086 | |
| 3 | 11.944 | 11.913 | |
| 4 | 18.993 | 18.990 | |
| 5 | 20.256 | 22.294 | |
| 6 | 25.014 | 25.017 | |
| Sennidin B | 7 | 16.229 | 17.101 |
| 8 | 14.540 | 16.816 | |
| 9 | 11.855 | 18.733 | |
| 10 | 18.915 | 16.502 | |
| Sennidin C | 11 | 17.479 | 16.784 |
| 12 | 22.503 | 26.483 | |
| 13 | 14.519 | 11.452 | |
| 14 | 13.671 | 14.046 | |
| 15 | 23.246 | 26.664 | |
| 16 | 25.190 | 23.316 | |
| Sennidin D | 17 | 18.724 | 14.341 |
| 18 | 16.381 | 17.255 | |
| 19 | 20.767 | 14.317 | |
| 20 | 20.403 | 15.657 | |
| 21 | 23.629 | 24.659 | |
| A (Figure 5) | - | 17.302 | 17.309 |
| B (Figure 5) | - | 11.947 | 11.947 |
| Compound | Structure | Hydrogen Bond | H⋯O [Å] | O⋯O [Å] | OHO [Deg.] |
|---|---|---|---|---|---|
| Sennidin A | 1 | C(A)-O-H⋯O=C(B) | 1.7142 | 2.5852 | 145.43 |
| C(C)-O-H⋯O=C(B) | 1.706 | 2.5813 | 145.897 | ||
| C(F)-O-H⋯O=C(G) | 1.7142 | 2.5852 | 145.43 | ||
| C(H)-O-H⋯O=C(G) | 1.7061 | 2.5813 | 145.897 | ||
| Sennidin B | 7 | C(A)-O-H⋯O=C(B) | 1.7096 | 2.5841 | 145.868 |
| C(C)-O-H⋯O=C(B) | 1.7142 | 2.5856 | 145.446 | ||
| C(F)-O-H⋯O=C(G) | 1.7135 | 2.585 | 145.494 | ||
| C(H)-O-H⋯O=C(G) | 1.7068 | 2.5818 | 145.919 | ||
| Sennidin C | 11 | C(A)-O-H⋯O=C(B) | 1.715 | 2.5859 | 145.44 |
| C(C)-O-H⋯O=C(B) | 1.7036 | 2.5801 | 146.018 | ||
| C(F)-O-H⋯O=C(G) | 1.7043 | 2.5798 | 145.92 | ||
| C(H)-O-H⋯O=C(G) | 1.705 | 2.5831 | 146.3 | ||
| Sennidin D | 17 | C(A)-O-H⋯O=C(B) | 1.7156 | 2.5887 | 145.805 |
| C(C)-O-H⋯O=C(B) | 1.7478 | 2.6091 | 144.62 | ||
| C(F)-O-H⋯O=C(G) | 1.7001 | 2.578 | 146.235 | ||
| C(H)-O-H⋯O=C(G) | 1.6976 | 2.5748 | 146.066 | ||
| C(H)-C(H2)-O-H⋯O=C(B) | 2.1931 | 3.0922 | 154.333 | ||
| A | - | C(A)-O-H⋯O=C(B) | 1.6862 | 2.5831 | 151.734 |
| C(C)-O-H⋯O=C(B) | 1.7383 | 2.5646 | 146.429 | ||
| C(F)-O-H⋯O=C(G) | 1.7383 | 2.5646 | 146.429 | ||
| C(H)-O-H⋯O=C(G) | 1.6862 | 2.5831 | 151.734 | ||
| B | - | C(A)-O-H⋯O=C(B) | 1.7001 | 2.5783 | 146.268 |
| C(C)-O-H⋯O=C(B) | 1.7077 | 2.5814 | 145.74 | ||
| C(F)-O-H⋯O=C(G) | 1.7077 | 2.5815 | 145.737 | ||
| C(H)-O-H⋯O=C(G) | 1.7002 | 2.5784 | 146.266 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymanski, S.; Majerz, I. In Silico Studies on Sennidines—Natural Dianthrones from Senna. Biology 2021, 10, 468. https://doi.org/10.3390/biology10060468
Szymanski S, Majerz I. In Silico Studies on Sennidines—Natural Dianthrones from Senna. Biology. 2021; 10(6):468. https://doi.org/10.3390/biology10060468
Chicago/Turabian StyleSzymanski, Sebastian, and Irena Majerz. 2021. "In Silico Studies on Sennidines—Natural Dianthrones from Senna" Biology 10, no. 6: 468. https://doi.org/10.3390/biology10060468
APA StyleSzymanski, S., & Majerz, I. (2021). In Silico Studies on Sennidines—Natural Dianthrones from Senna. Biology, 10(6), 468. https://doi.org/10.3390/biology10060468