
Integrated Signals of Jasmonates, Sugars, Cytokinins and Auxin Influence the Initial Growth of the Second Buds of Chrysanthemum after Decapitation




Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, Tissue Preparation, and RNA Extraction
2.2. Measurements of Endogenous Plant Hormones
2.3. PacBio cDNA Library Construction and SMRT Sequencing
2.4. Illumina cDNA Library Construction and Sequencing
2.5. Prediction of Alternative Splicing and lncRNAs and Annotation of Transcription Factors (TFs)
2.6. Functional Annotation and Identification of Differently Expressed Genes (DEGs)
3. Results
3.1. Sequence Samples of Different Time Points via ISO-Seq and RNA-Seq
3.2. Features of the TGS Transcriptome and Functional Annotations
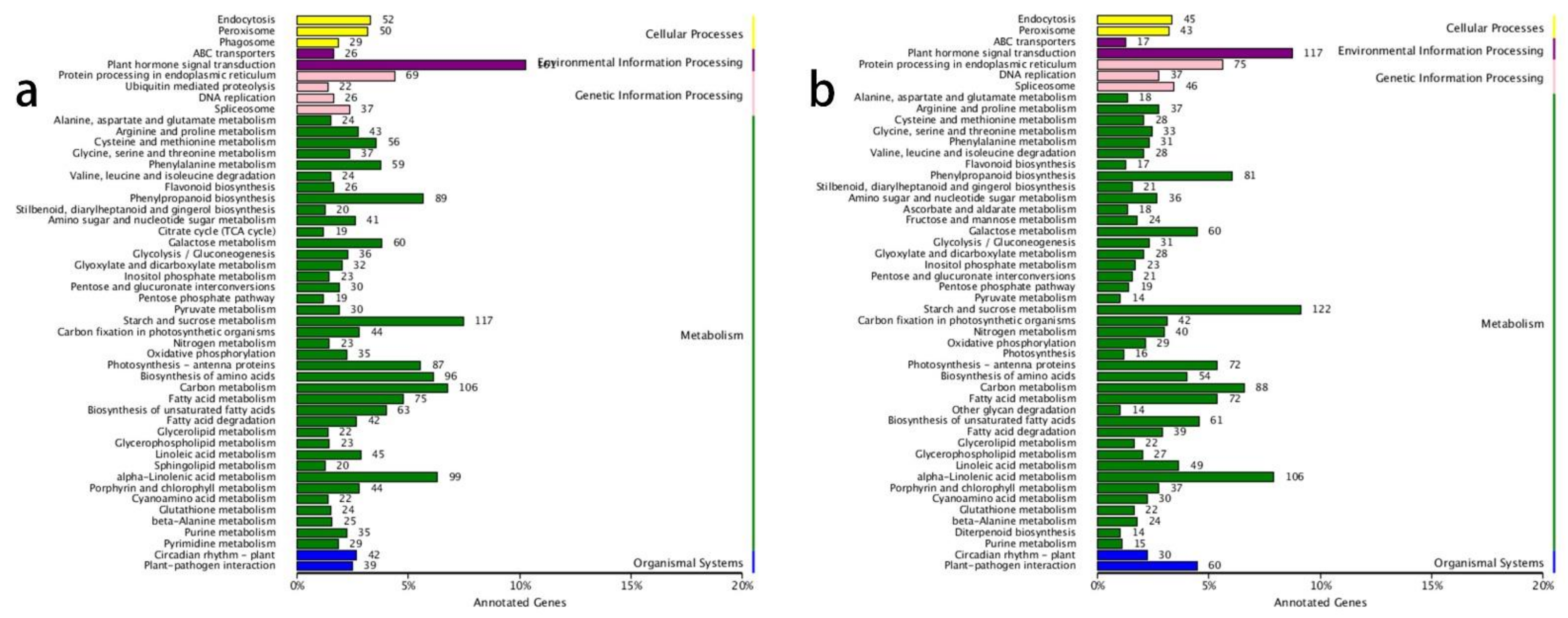
3.3. GO and KEGG Annotation of DEGs
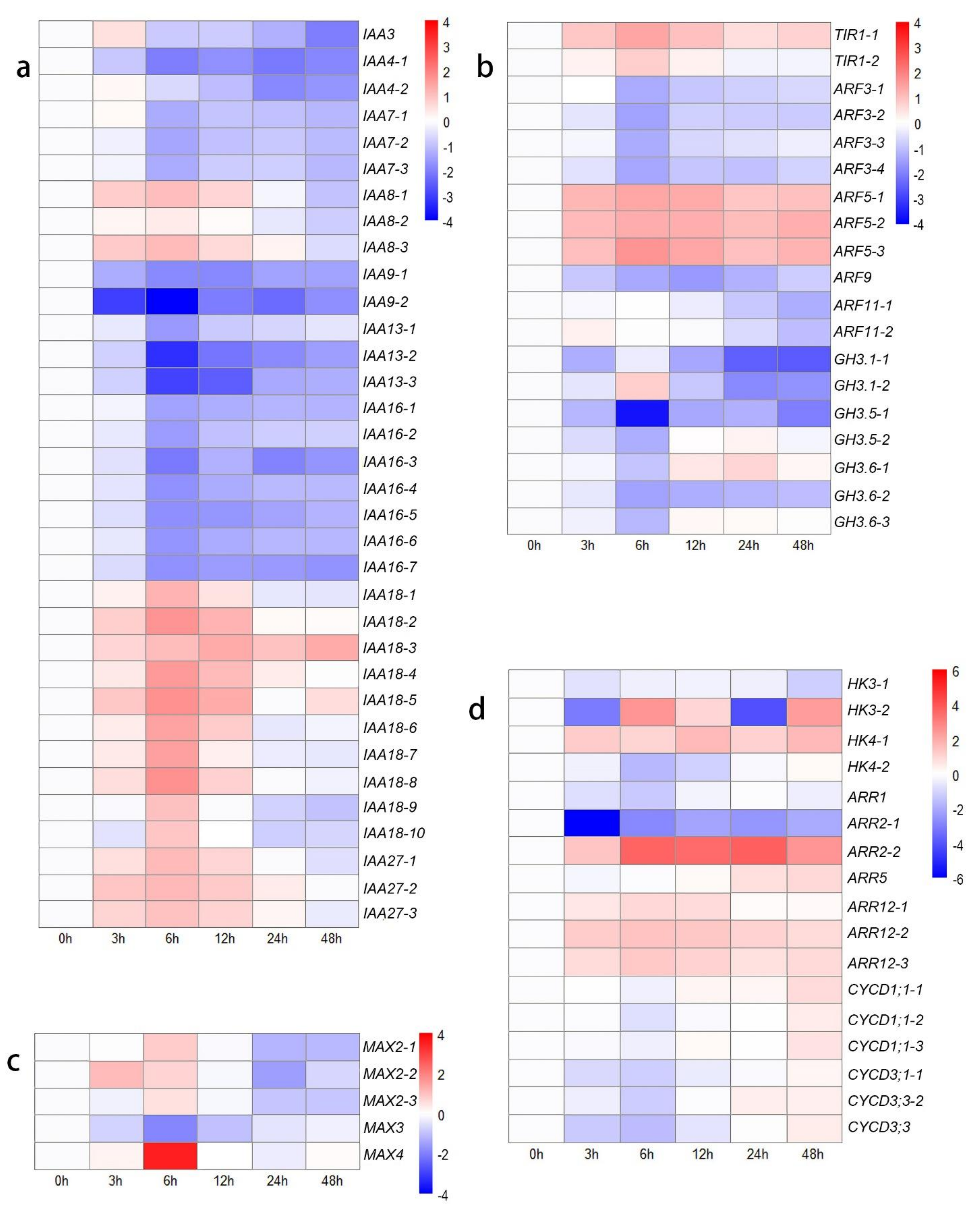
3.4. DEGs Related to Hormones
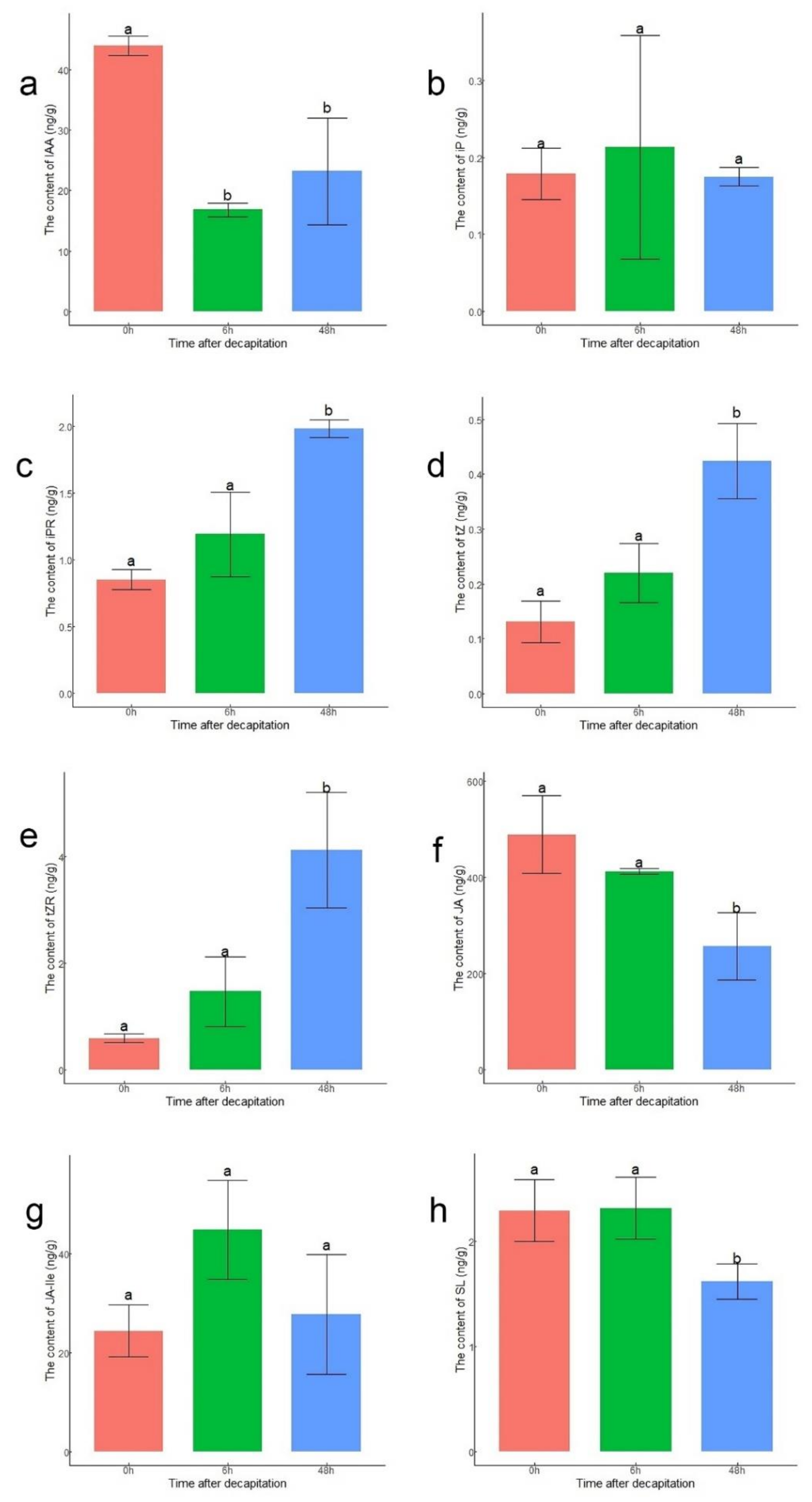
3.5. Analysis of Hormone Contents in Buds after Decapitation
3.6. DEGs Related to Metabolic Activity
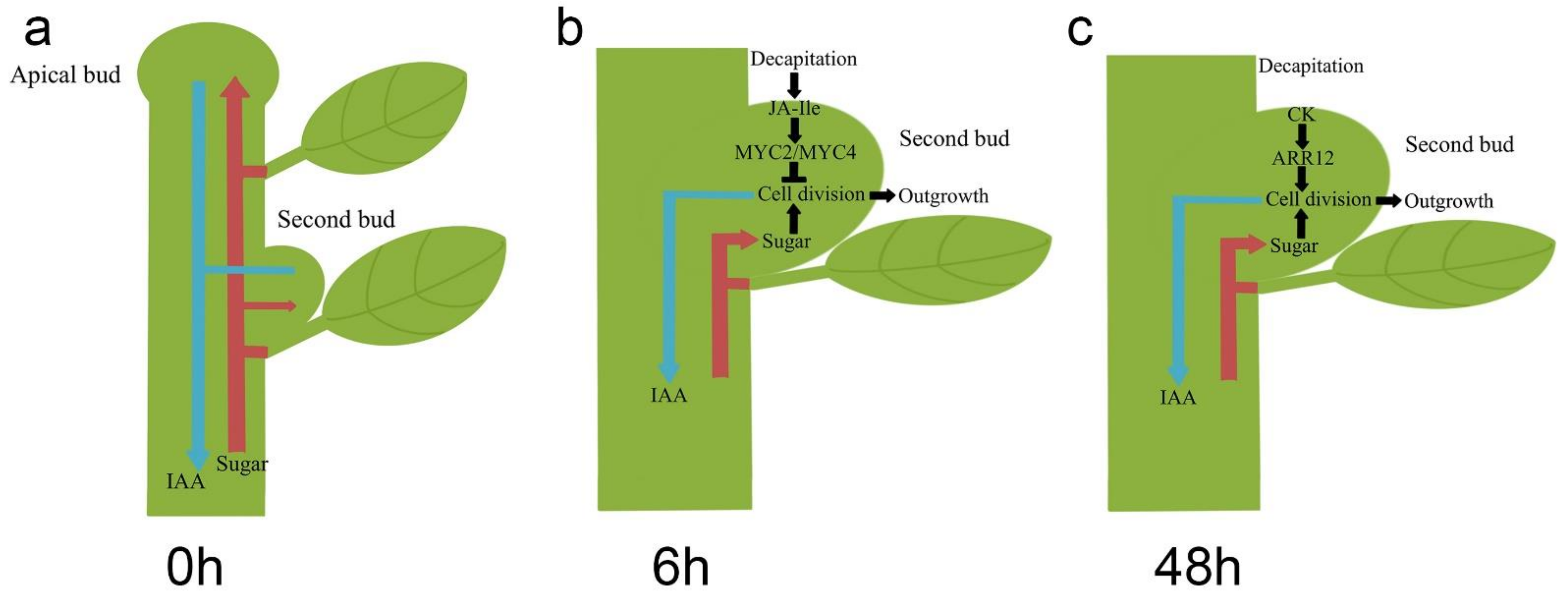
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McSteen, P.; Leyser, O. Shoot branching. Annu. Rev. Plant Biol. 2005, 56, 353. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ‘Green revolution’genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef]
- Shimizu-Sato, S.; Mori, H. Control of outgrowth and dormancy in axillary buds. Plant Physiol. 2001, 127, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Domagalska, M.A.; Leyser, O. Signal integration in the control of shoot branching. Nat. Rev. Mol. Cell Biol. 2011, 12, 211–221. [Google Scholar] [CrossRef]
- Rameau, C.; Bertheloot, J.; Leduc, N.; Andrieu, B.; Foucher, F.; Sakr, S. Multiple pathways regulate shoot branching. Front. Plant Sci. 2015, 5, 741. [Google Scholar] [CrossRef]
- Thimann, K.V.; Skoog, F. Studies on the growth hormone of plants: III. The inhibiting action of the growth substance on bud development. Proc. Natl. Acad. Sci. USA 1933, 19, 714. [Google Scholar] [CrossRef]
- Thimann, K.V.; Skoog, F. On the inhibition of bud development and other functions of growth substance in Vicia faba. Proc. R. Soc. Lond. B 1934, 114, 317–339. [Google Scholar]
- Booker, J.; Chatfield, S.; Leyser, O. Auxin acts in xylem-associated or medullary cells to mediate apical dominance. Plant Cell 2003, 15, 495–507. [Google Scholar] [CrossRef]
- Prasad, T.; Li, X.; Abdel-Rahman, A.; Hosokawa, Z.; Cloud, N.; Lamotte, C.; Cline, M. Does auxin play a role in the release of apical dominance by shoot inversion in Ipomoea nil? Ann. Bot. 1993, 71, 223–229. [Google Scholar] [CrossRef]
- Miyawaki, K.; Matsumoto-Kitano, M.; Kakimoto, T. Expression of cytokinin biosynthetic isopentenyltransferase genes in Arabidopsis: Tissue specificity and regulation by auxin, cytokinin, and nitrate. Plant J. 2004, 37, 128–138. [Google Scholar] [CrossRef]
- Nordström, A.; Tarkowski, P.; Tarkowska, D.; Norbaek, R.; Åstot, C.; Dolezal, K.; Sandberg, G. Auxin regulation of cytokinin biosynthesis in Arabidopsis thaliana: A factor of potential importance for auxin–cytokinin-regulated development. Proc. Natl. Acad. Sci. USA 2004, 101, 8039–8044. [Google Scholar] [CrossRef]
- Tanaka, M.; Takei, K.; Kojima, M.; Sakakibara, H.; Mori, H. Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. 2006, 45, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.; Stirnberg, P.; Beveridge, C.; Leyser, O. Interactions between auxin and strigolactone in shoot branching control. Plant Physiol. 2009, 151, 400–412. [Google Scholar] [CrossRef]
- Alder, A.; Jamil, M.; Marzorati, M.; Bruno, M.; Vermathen, M.; Bigler, P.; Ghisla, S.; Bouwmeester, H.; Beyer, P.; Al-Babili, S. The path from β-carotene to carlactone, a strigolactone-like plant hormone. Science 2012, 335, 1348–1351. [Google Scholar] [CrossRef] [PubMed]
- Sachs, T. The induction of transport channels by auxin. Planta 1975, 127, 201–206. [Google Scholar] [CrossRef]
- Li, C.J.; Bangerth, F. Autoinhibition of indoleacetic acid transport in the shoots of two-branched pea (Pisum sativum) plants and its relationship to correlative dominance. Physiol. Plant. 1999, 106, 415–420. [Google Scholar] [CrossRef]
- Ongaro, V.; Leyser, O. Hormonal control of shoot branching. J. Exp. Bot. 2008, 59, 67–74. [Google Scholar] [CrossRef]
- Chen, C.-M.; Ertl, J.R.; Leisner, S.M.; Chang, C.-C. Localization of cytokinin biosynthetic sites in pea plants and carrot roots. Plant Physiol. 1985, 78, 510–513. [Google Scholar] [CrossRef]
- Dewitte, W.; Scofield, S.; Alcasabas, A.A.; Maughan, S.C.; Menges, M.; Braun, N.; Collins, C.; Nieuwland, J.; Prinsen, E.; Sundaresan, V.; et al. Arabidopsis CYCD3 D-type cyclins link cell proliferation and endocycles and are rate-limiting for cytokinin responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14537–14542. [Google Scholar] [CrossRef]
- Schaller, G.E.; Street, I.H.; Kieber, J.J. Cytokinin and the cell cycle. Curr. Opin. Plant Biol. 2014, 21, 7–15. [Google Scholar] [CrossRef]
- Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pagès, V.; Dun, E.A.; Pillot, J.-P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.-C.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Umehara, M.; Hanada, A.; Yoshida, S.; Akiyama, K.; Arite, T.; Takeda-Kamiya, N.; Magome, H.; Kamiya, Y.; Shirasu, K.; Yoneyama, K.; et al. Inhibition of shoot branching by new terpenoid plant hormones. Nature 2008, 455, 195–200. [Google Scholar] [CrossRef]
- Barbier, F.F.; Dun, E.A.; Kerr, S.C.; Chabikwa, T.G.; Beveridge, C.A. An update on the signals controlling shoot branching. Trends Plant Sci. 2019, 24, 220–236. [Google Scholar] [CrossRef]
- Waters, M.T.; Gutjahr, C.; Bennett, T.; Nelson, D.C. Strigolactone signaling and evolution. Trends Plant Sci. 2017, 68, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lin, Q.; Zhu, L.; Ren, Y.; Zhou, K.; Shabek, N.; Wu, F.; Mao, H.; Dong, W.; Gan, L.; et al. D14-SCFD3-dependent degradation of D53 regulates strigolactone signalling. Nature 2013, 504, 406–410. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Xiong, G.; Liu, H.; Chen, F.; Wang, L.; Meng, X.; Liu, G.; Yu, H.; Yuan, Y.; et al. DWARF 53 acts as a repressor of strigolactone signalling in rice. Nature 2013, 504, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, N.; Taylor, C.; Leyser, O. Strigolactone can promote or inhibit shoot branching by triggering rapid depletion of the auxin efflux protein PIN1 from the plasma membrane. PLoS. Biol. 2013, 11, e1001474. [Google Scholar] [CrossRef]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef]
- Patrick, J.W.; Botha, F.C.; Birch, R.G. Metabolic engineering of sugars and simple sugar derivatives in plants. Plant Biotechnol. J. 2013, 11, 142–156. [Google Scholar] [CrossRef]
- Mason, M.G.; Ross, J.J.; Babst, B.A.; Wienclaw, B.N.; Beveridge, C.A. Sugar demand, not auxin, is the initial regulator of apical dominance. Proc. Natl. Acad. Sci. USA 2014, 111, 6092–6097. [Google Scholar] [CrossRef] [PubMed]
- Barbier, F.F.; Lunn, J.E.; Beveridge, C.A. Ready, steady, go! A sugar hit starts the race to shoot branching. Curr. Opin. Plant Biol. 2015, 25, 39–45. [Google Scholar] [CrossRef]
- Kebrom, T.H. A growing stem inhibits bud outgrowth–the overlooked theory of apical dominance. Front. Plant Sci. 2017, 8, 1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, X.; Xi, L.; Li, J.; Zhao, R.; Ma, N.; Zhao, L. Roles of DgBRC1 in regulation of lateral branching in chrysanthemum (Dendranthema × grandiflora cv. Jinba). PLoS ONE 2013, 8, e61717. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- He, J.; Lin, S.; Yu, Z.; Song, A.; Guan, Z.; Fang, W.; Chen, S.; Zhang, F.; Jiang, J.; Chen, F.; et al. Identification of 5S and 45S rDNA sites in Chrysanthemum species by using oligonucleotide fluorescence in situ hybridization (Oligo-FISH). Mol. Biol. Rep. 2021, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Pan, J.; Wang, H.; Reiter, R.J.; Li, X.; Mou, Z.; Zhang, J.; Yao, Z.; Zhao, D.; Yu, D. Melatonin inhibits seed germination by crosstalk with abscisic acid, gibberellin, and auxin in Arabidopsis. J. Pineal Res. 2021, e12736. [Google Scholar]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Suza, W.P.; Staswick, P.E. The role of JAR1 in jasmonoyl-L-isoleucine production during Arabidopsis wound response. Planta 2008, 227, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K.; Manners, J.M. MYC2: The master in action. Mol. Plant 2013, 6, 686–703. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Calvo, P.; Chini, A.; Fernández-Barbero, G.; Chico, J.-M.; Gimenez-Ibanez, S.; Geerinck, J.; Eeckhout, D.; Schweizer, F.; Godoy, M.; Franco-Zorrilla, J.M.; et al. The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 2011, 23, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Rae, G.M.; David, K.; Wood, M. The dormancy marker DRM1/ARP associated with dormancy but a broader role In Planta. Dev. Biol. J. 2013, 2013. [Google Scholar] [CrossRef]
- Lastdrager, J.; Hanson, J.; Smeekens, S. Sugar signals and the control of plant growth and development. J. Exp. Bot. 2014, 65, 799–807. [Google Scholar] [CrossRef]
- Li, L.; Sheen, J. Dynamic and diverse sugar signaling. Curr. Opin. Plant Biol. 2016, 33, 116–125. [Google Scholar] [CrossRef]
- Abel, S.; Theologis, A. Early genes and auxin action. Plant Physiol. 1996, 111, 9. [Google Scholar] [CrossRef]
- Quint, M.; Gray, W.M. Auxin signaling. Curr. Opin. Plant Biol. 2006, 9, 448–453. [Google Scholar] [CrossRef]
- Vernoux, T.; Brunoud, G.; Farcot, E.; Morin, V.; Van den Daele, H.; Legrand, J.; Oliva, M.; Das, P.; Larrieu, A.; Wells, D.; et al. The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Mol. Syst. Biol. 2011, 7, 508. [Google Scholar] [CrossRef]
- Ckurshumova, W.; Smirnova, T.; Marcos, D.; Zayed, Y.; Berleth, T. Irrepressible MONOPTEROS/ARF5 promotes de novo shoot formation. New Phytol. 2014, 204, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yan, D.-W.; Yuan, T.-T.; Gao, X.; Lu, Y.-T. A gain-of-function mutation in IAA8 alters Arabidopsis floral organ development by change of jasmonic acid level. Plant Mol. Biol. 2013, 82, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Okushima, Y.; Mimura, T.; Tasaka, M.; Fukaki, H. Domain II mutations in CRANE/IAA18 suppress lateral root formation and affect shoot development in Arabidopsis thaliana. Plant Cell Physiol. 2008, 49, 1025–1038. [Google Scholar] [CrossRef]
- Staswick, P.E.; Serban, B.; Rowe, M.; Tiryaki, I.; Maldonado, M.T.; Maldonado, M.C.; Suza, W. Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell 2005, 17, 616–627. [Google Scholar] [CrossRef]
- Ludwig-Müller, J. Auxin conjugates: Their role for plant development and in the evolution of land plants. J. Exp. Bot. 2011, 62, 1757–1773. [Google Scholar] [CrossRef]
- Peer, W.A.; Bandyopadhyay, A.; Blakeslee, J.J.; Makam, S.N.; Chen, R.J.; Masson, P.H.; Murphy, A.S. Variation in expression and protein localization of the PIN family of auxin efflux facilitator proteins in flavonoid mutants with altered auxin transport in Arabidopsis thaliana. Plant Cell 2004, 16, 1898–1911. [Google Scholar] [CrossRef]
- Peer, W.A.; Murphy, A.S. Flavonoids and auxin transport: Modulators or regulators? Trends Plant Sci. 2007, 12, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.P.; Soto-Suárez, M.; Chao, W.S.; Jia, Y.; Anderson, J.V. Transcriptome analysis of paradormancy release in root buds of leafy spurge (Euphorbia esula). Weed Sci. 2005, 53, 795–801. [Google Scholar] [CrossRef]
- Chao, W.S.; Doğramaci, M.; Horvath, D.P.; Anderson, J.V.; Foley, M.E. Phytohormone balance and stress-related cellular responses are involved in the transition from bud to shoot growth in leafy spurge. BMC Plant Biol. 2016, 16, 1–21. [Google Scholar] [CrossRef]
- Min, Z.; Zhao, X.; Li, R.; Yang, B.; Liu, M.; Fang, Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to bud dormancy in grapevine (Vitis vinifera). Sci. Hortic. 2017, 225, 213–220. [Google Scholar] [CrossRef]
- Zha, M.; Imran, M.; Wang, Y.; Xu, J.; Ding, Y.; Wang, S. Transcriptome analysis revealed the interaction among strigolactones, auxin, and cytokinin in controlling the shoot branching of rice. Plant Cell Rep. 2019, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Nuccio, M.L.; Wu, J.; Mowers, R.; Zhou, H.-P.; Meghji, M.; Primavesi, L.F.; Paul, M.J.; Chen, X.; Gao, Y.; Haque, E.; et al. Expression of trehalose-6-phosphate phosphatase in maize ears improves yield in well-watered and drought conditions. Nat. Biotechnol. 2015, 33, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Oszvald, M.; Primavesi, L.F.; Griffiths, C.A.; Cohn, J.; Basu, S.S.; Nuccio, M.L.; Paul, M.J. Trehalose 6-phosphate regulates photosynthesis and assimilate partitioning in reproductive tissue. Plant Physiol. 2018, 176, 2623–2638. [Google Scholar] [CrossRef]
- Liu, W.; Peng, B.; Song, A.; Jiang, J.; Chen, F. Sugar transporter, CmSWEET17, promotes bud outgrowth in Chrysanthemum morifolium. Genes 2020, 11, 26. [Google Scholar] [CrossRef]
- Fichtner, F.; Barbier, F.F.; Feil, R.; Watanabe, M.; Annunziata, M.G.; Chabikwa, T.G.; Höfgen, R.; Stitt, M.; Beveridge, C.A.; Lunn, J.E. Trehalose 6-phosphate is involved in triggering axillary bud outgrowth in garden pea (Pisum sativum L.). Plant J. 2017, 92, 611–623. [Google Scholar] [CrossRef]
- Fichtner, F.; Barbier, F.F.; Annunziata, M.G.; Feil, R.; Olas, J.J.; Mueller-Roeber, B.; Stitt, M.; Beveridge, C.A.; Lunn, J.E. Regulation of shoot branching in Arabidopsis by trehalose 6-phosphate. New Phytol. 2021, 229, 2135–2151. [Google Scholar] [CrossRef]
- Pauwels, L.; Morreel, K.; De Witte, E.; Lammertyn, F.; Van Montagu, M.; Boerjan, W.; Inzé, D.; Goossens, A. Mapping methyl jasmonate-mediated transcriptional reprogramming of metabolism and cell cycle progression in cultured Arabidopsis cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1380–1385. [Google Scholar] [CrossRef]
- Ofer, K.; Gold, D.; Flescher, E. Methyl jasmonate induces cell cycle block and cell death in the amitochondriate parasite Trichomonas vaginalis. Int. J. Parasit. 2008, 38, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Ioio, R.D.; Nakamura, K.; Moubayidin, L.; Perilli, S.; Taniguchi, M.; Morita, M.T.; Aoyama, T.; Costantino, P.; Sabatini, S. A genetic framework for the control of cell division and differentiation in the root meristem. Science 2008, 322, 1380–1384. [Google Scholar] [CrossRef]
- Hill, K.; Mathews, D.E.; Kim, H.J.; Street, I.H.; Wildes, S.L.; Chiang, Y.-H.; Mason, M.G.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J.; et al. Functional characterization of type-B response regulators in the Arabidopsis cytokinin response. Plant Physiol. 2013, 162, 212–224. [Google Scholar] [CrossRef]
- Lara-Núñez, A.; García-Ayala, B.B.; Garza-Aguilar, S.M.; Flores-Sánchez, J.; Sánchez-Camargo, V.A.; Bravo-Alberto, C.E.; Vázquez-Santana, S.; Vázquez-Ramos, J.M. Glucose and sucrose differentially modify cell proliferation in maize during germination. Plant Physiol. Biochem. 2017, 113, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Tarancón, C.; González-Grandío, E.; Oliveros, J.C.; Nicolas, M.; Cubas, P. A conserved carbon starvation response underlies bud dormancy in woody and herbaceous species. Front. Plant Sci. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | BMK-ID | ReadSum | BaseSum | GC(%) | Q30(%) |
|---|---|---|---|---|---|
| 0 h | T01 | 26,767,576 | 8,005,676,720 | 42.83 | 94.51 |
| T02 | 20,950,118 | 6,267,548,322 | 42.61 | 94.65 | |
| T03 | 25,183,097 | 7,514,078,462 | 42.69 | 94.73 | |
| 3 h | T04 | 22,967,608 | 6,873,236,908 | 42.94 | 90.51 |
| T06 | 25,210,897 | 7,548,853,932 | 42.85 | 91.34 | |
| T19 | 28,931,725 | 8,613,977,646 | 44.06 | 91.09 | |
| 6 h | T07 | 24,152,120 | 7,224,006,796 | 43.46 | 94.95 |
| T08 | 20,746,285 | 6,203,429,872 | 42.58 | 94.38 | |
| T09 | 21,172,920 | 6,326,572,354 | 42.60 | 94.41 | |
| 12 h | T10 | 22,846,998 | 6,824,592,758 | 42.62 | 94.66 |
| T11 | 22,734,631 | 6,794,137,036 | 42.45 | 94.47 | |
| T12 | 23,235,704 | 6,939,240,352 | 42.45 | 94.56 | |
| 24 h | T13 | 24,151,007 | 7,220,435,914 | 42.73 | 94.20 |
| T14 | 23,940,581 | 7,158,885,232 | 42.53 | 94.39 | |
| T15 | 22,207,198 | 6,642,610,762 | 42.68 | 94.39 | |
| 48 h | T16 | 23,154,529 | 6,913,758,850 | 43.25 | 94.54 |
| T17 | 22,872,822 | 6,838,511,346 | 42.70 | 94.43 | |
| T18 | 23,455,701 | 7,000,411,940 | 42.81 | 94.59 |
| Library | 1–2K | 2–3K | 3–6K | All |
|---|---|---|---|---|
| ROIs | 113,368 | 105,317 | 78,834 | 297,519 |
| Number of five prime reads | 57,998 | 58,541 | 40,708 | 157,247 |
| Number of three prime reads | 66,054 | 63,609 | 43,561 | 173,224 |
| Number of poly-A reads | 63,848 | 62,527 | 43,423 | 169,798 |
| Number of filtered short reads | 16,314 | 9054 | 2105 | 27,473 |
| Number of non-full-length reads | 49,732 | 47,916 | 42,560 | 140,208 |
| Number of full-length reads | 47,322 | 48,347 | 34,169 | 129,838 |
| Number of full-length non-chimeric reads | 47,124 | 48,196 | 34,068 | 129,388 |
| Average FLNC read Length | 1421 | 2213 | 3619 | 2294 |
| Full-length percentage (FL%) | 41.74% | 45.91% | 43.34% | 43.64% |
| Artificial concatemers(%) | 0.42% | 0.31% | 0.30% | 0.35% |
| All | GO | KEGG | KOG | Pfam | Swissprot | COG | eggNOG | nr | |
|---|---|---|---|---|---|---|---|---|---|
| Annotated_Number | 57,972 | 1789 | 26,357 | 37,374 | 47,880 | 45,307 | 25,052 | 56,606 | 57,629 |
| DEG Set | DEG Number | Up-Regulator | Down-Regulator |
|---|---|---|---|
| 0 h vs. 3 h | 1587 | 869 | 718 |
| 0 h vs. 6 h | 5247 | 2778 | 2469 |
| 0 h vs. 12 h | 2785 | 1469 | 1316 |
| 0 h vs. 24 h | 2773 | 1329 | 1444 |
| 0 h vs. 48 h | 2351 | 1098 | 1253 |
| 3 h vs. 6 h | 381 | 185 | 196 |
| 3 h vs. 12 h | 570 | 294 | 276 |
| 3 h vs. 24 h | 1531 | 680 | 851 |
| 3 h vs. 48 h | 1427 | 611 | 816 |
| 6 h vs. 12 h | 1094 | 451 | 643 |
| 6 h vs. 24 h | 4169 | 1689 | 2480 |
| 6 h vs. 48 h | 4377 | 1783 | 2594 |
| 12 h vs. 24 h | 1176 | 246 | 930 |
| 12 h vs. 48 h | 1633 | 563 | 1070 |
| 24 h vs. 48 h | 600 | 199 | 401 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.; Zhang, L.; Yu, Q.; Zhang, J.; Li, P.; Zhang, Y.; Xing, X.; Ding, L.; Fang, W.; Chen, F.; et al. Integrated Signals of Jasmonates, Sugars, Cytokinins and Auxin Influence the Initial Growth of the Second Buds of Chrysanthemum after Decapitation. Biology 2021, 10, 440. https://doi.org/10.3390/biology10050440
Sun D, Zhang L, Yu Q, Zhang J, Li P, Zhang Y, Xing X, Ding L, Fang W, Chen F, et al. Integrated Signals of Jasmonates, Sugars, Cytokinins and Auxin Influence the Initial Growth of the Second Buds of Chrysanthemum after Decapitation. Biology. 2021; 10(5):440. https://doi.org/10.3390/biology10050440
Chicago/Turabian StyleSun, Daojin, Luyao Zhang, Qi Yu, Jiali Zhang, Peiling Li, Yu Zhang, Xiaojuan Xing, Lian Ding, Weimin Fang, Fadi Chen, and et al. 2021. "Integrated Signals of Jasmonates, Sugars, Cytokinins and Auxin Influence the Initial Growth of the Second Buds of Chrysanthemum after Decapitation" Biology 10, no. 5: 440. https://doi.org/10.3390/biology10050440
APA StyleSun, D., Zhang, L., Yu, Q., Zhang, J., Li, P., Zhang, Y., Xing, X., Ding, L., Fang, W., Chen, F., & Song, A. (2021). Integrated Signals of Jasmonates, Sugars, Cytokinins and Auxin Influence the Initial Growth of the Second Buds of Chrysanthemum after Decapitation. Biology, 10(5), 440. https://doi.org/10.3390/biology10050440