
HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates

 ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of HML7 Loci and Solitary LTRs in Human Genome Assembly GRCh38/hg38
2.2. Genomic Distribution and Context of Integration
2.3. Structural Characterization
2.4. Phylogenetic Analyses
2.5. Estimation of HML7 Loci Time of Integration
3. Results
3.1. Identification of HML7 Loci in Human Genome Assembly GRCh38/hg38
3.2. HML7 Loci Are Not Randomly Distributed among Human Chromosomes
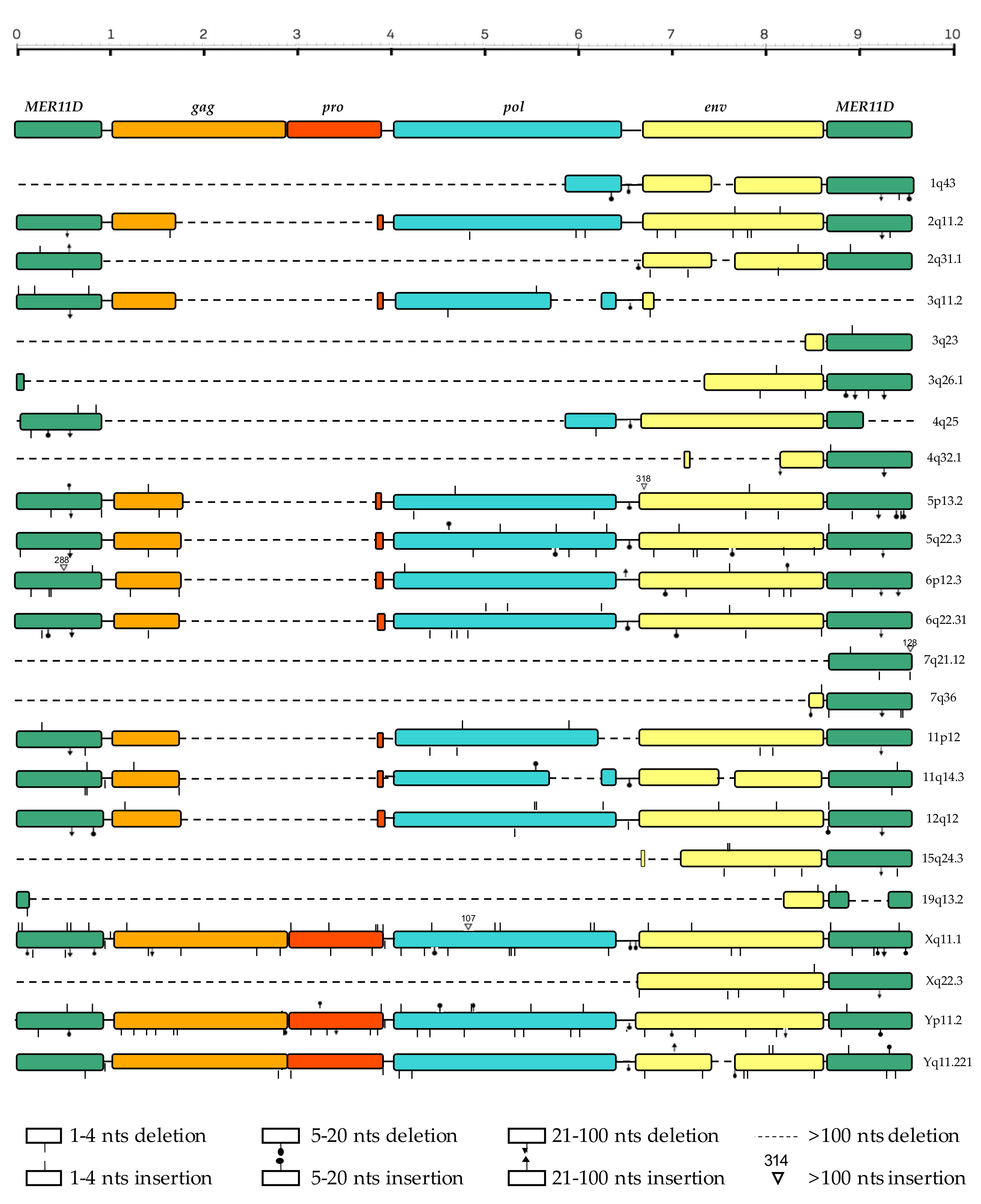
3.3. Structural Characterization
3.4. Phylogenetic Analyses
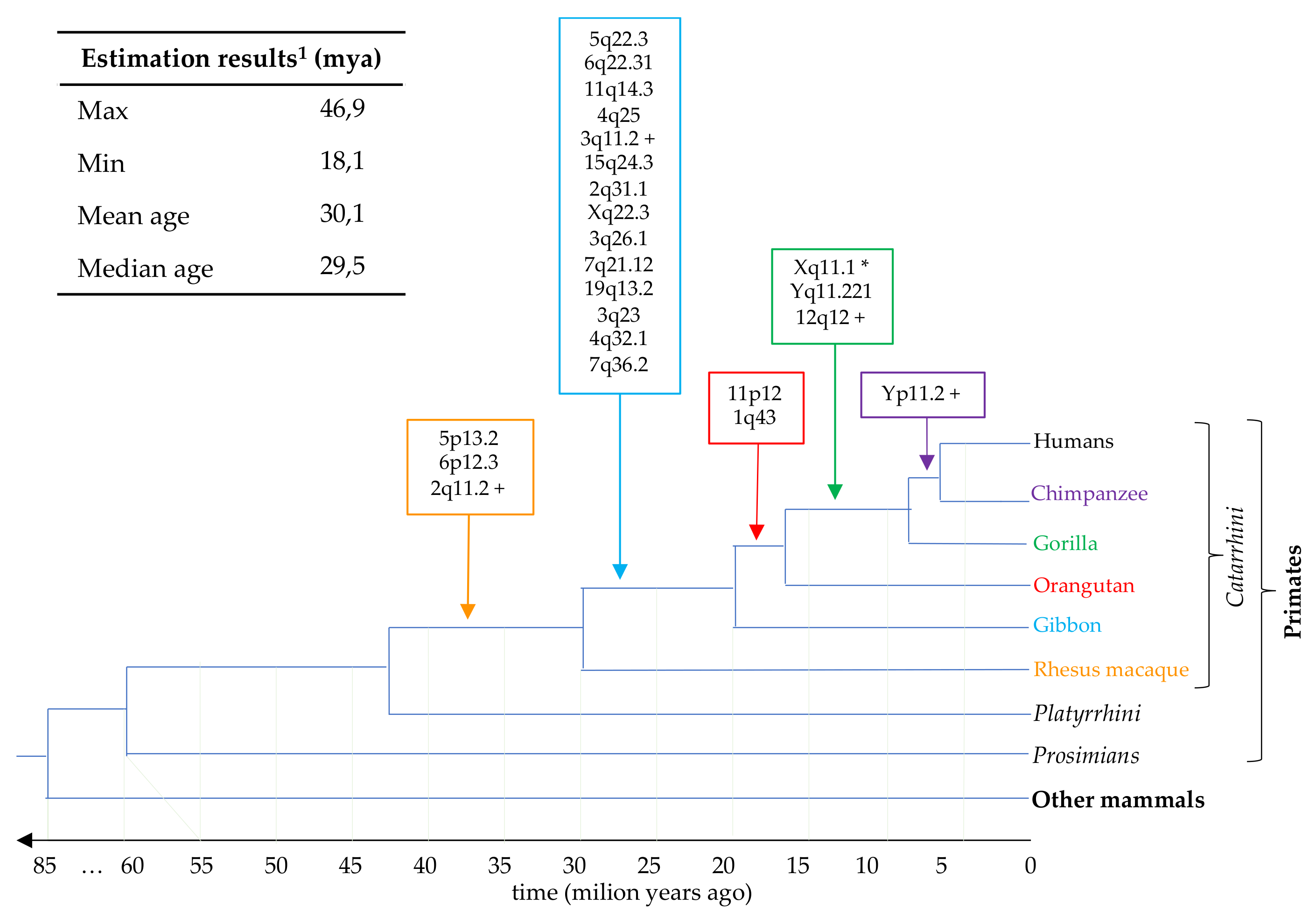
3.5. Estimation of HML7 Loci Time of Integration and Comparative Genomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blond, J.-L.; Lavillette, D.; Cheynet, V.; Bouton, O.; Oriol, G.; Chapel-Fernandes, S.; Mandrand, B.; Mallet, F.; Cosset, F.-L. An Envelope Glycoprotein of the Human Endogenous Retrovirus HERV-W Is Expressed in the Human Placenta and Fuses Cells Expressing the Type D Mammalian Retrovirus Receptor. J. Virol. 2000, 74, 3321–3329. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.-P.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.-Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nat. Cell Biol. 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Mangeney, M.; Renard, M.; Schlecht-Louf, G.; Bouallaga, I.; Heidmann, O.; Letzelter, C.; Richaud, A.; Ducos, B.; Heidmann, T. Placental syncytins: Genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20534–20539. [Google Scholar] [CrossRef] [PubMed]
- Lavialle, C.; Cornelis, G.; Dupressoir, A.; Esnault, C.; Heidmann, O.; Vernochet, C.; Heidmann, T. Paleovirology of ‘ syncytins ’, retroviral env genes exapted for a role in placentation. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120507. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. HERV Envelope Proteins: Physiological Role and Pathogenic Potential in Cancer and Autoimmunity. Front. Microbiol. 2018, 9, 462. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Grandi, N.; Tramontano, E.; Dieci, G. Retrotransposons as Drivers of Mammalian Brain Evolution. Life 2021, 11, 376. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Type W Human Endogenous Retrovirus (HERV-W) Integrations and Their Mobilization by L1 Machinery: Contribution to the Human Transcriptome and Impact on the Host Physiopathology. Viruses 2017, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Grandi, N.; Palanivelu, L.; Tramontano, E.; Lin, L.-T. Contribution of Human Retroviruses to Disease Development—A Focus on the HIV– and HERV–Cancer Relationships and Treatment Strategies. Viruses 2020, 12, 852. [Google Scholar] [CrossRef]
- Mayer, J.; Blomberg, J.; Seal, R.L. A revised nomenclature for transcribed human endogenous retroviral loci. Mob. DNA 2011, 2, 7. [Google Scholar] [CrossRef]
- Sperber, G.; Airola, T.; Jern, P.; Blomberg, J. Automated recognition of retroviral sequences in genomic data–RetroTector. Nucleic Acids Res. 2007, 35, 4964–4976. [Google Scholar] [CrossRef]
- Vargiu, L.; Rodriguez-Tomé, P.; Sperber, G.O.; Cadeddu, M.; Grandi, N.; Blikstad, V.; Tramontano, E.; Blomberg, J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 2016, 13, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Cadeddu, M.; Blomberg, J.; Tramontano, E. Contribution of type W human endogenous retroviruses to the human genome: Characterization of HERV-W proviral insertions and processed pseudogenes. Retrovirology 2016, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Cadeddu, M.; Blomberg, J.; Mayer, J.; Tramontano, E. HERV-W group evolutionary history in non-human primates: characterization of ERV-W orthologs in Catarrhini and related ERV groups in Platyrrhini. BMC Evol. Biol. 2018, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Sperber, G.O.; Ahlsén, G.; Blomberg, J. Sequence Variability, Gene Structure, and Expression of Full-Length Human Endogenous Retrovirus H. J. Virol. 2005, 79, 6325–6337. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef]
- Marchi, E.; Kanapin, A.; Magiorkinis, G.; Belshaw, R. Unfixed Endogenous Retroviral Insertions in the Human Population. J. Virol. 2014, 88, 9529–9537. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Flockerzi, A.; Burkhardt, S.; Schempp, W.; Meese, E.; Mayer, J. Human Endogenous Retrovirus HERV-K14 Families: Status, Variants, Evolution, and Mobilization of Other Cellular Sequences. J. Virol. 2005, 79, 2941–2949. [Google Scholar] [CrossRef]
- Mayer, J.; Meese, E.U. The Human Endogenous Retrovirus Family HERV-K(HML-3). Genomics 2002, 80, 331–343. [Google Scholar] [CrossRef]
- Seifarth, W.; Baust, C.; Murr, A.; Skladny, H.; Krieg-Schneider, F.; Blusch, J.; Werner, T.; Hehlmann, R.; Leib-Mösch, C. Proviral Structure, Chromosomal Location, and Expression of HERV-K-T47D, a Novel Human Endogenous Retrovirus Derived from T47D Particles. J. Virol. 1998, 72, 8384–8391. [Google Scholar] [CrossRef] [PubMed]
- Lavie, L.; Medstrand, P.; Schempp, W.; Meese, E.; Mayer, J. Human Endogenous Retrovirus Family HERV-K(HML-5): Status, Evolution, and Reconstruction of an Ancient Betaretrovirus in the Human Genome. J. Virol. 2004, 78, 8788–8798. [Google Scholar] [CrossRef]
- Pisano, M.P.; Grandi, N.; Cadeddu, M.; Blomberg, J.; Tramontano, E. Comprehensive Characterization of the Human Endogenous Retrovirus HERV-K(HML-6) Group: Overview of Structure, Phylogeny, and Contribution to the Human Genome. J. Virol. 2019, 93, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Cadeddu, M.; Pisano, M.P.; Esposito, F.; Blomberg, J.; Tramontano, E. Identification of a novel HERV-K(HML10): Comprehensive characterization and comparative analysis in non-human primates provide insights about HML10 proviruses structure and diffusion. Mob. DNA 2017, 8, 15. [Google Scholar] [CrossRef]
- Broecker, F.; Horton, R.; Heinrich, J.; Franz, A.; Schweiger, M.-R.; Lehrach, H.; Moelling, K. The intron-enriched HERV-K(HML-10) family suppresses apoptosis, an indicator of malignant transformation. Mob. DNA 2016, 7, 1–17. [Google Scholar] [CrossRef]
- Arroyo, M.; Bautista, R.; Larrosa, R.; Cobo, M.Á.; Claros, M.G. Biomarker potential of repetitive-element transcriptome in lung cancer. PeerJ 2019, 7, e8277. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N.; et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [PubMed]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef]
- Jern, P.; O Sperber, G.; Blomberg, J. Use of Endogenous Retroviral Sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology 2005, 2, 50. [Google Scholar] [CrossRef]
- Thomas, J.; Perron, H.; Feschotte, C. Variation in proviral content among human genomes mediated by LTR recombination. Mob. DNA 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Contreras-Galindo, R.; Kaplan, M.H.; He, S.; Contreras-Galindo, A.C.; Gonzalez-Hernandez, M.J.; Kappes, F.; Dube, D.; Chan, S.M.; Robinson, D.; Meng, F.; et al. HIV infection reveals widespread expansion of novel centromeric human endogenous retroviruses. Genome Res. 2013, 23, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Zahn, J.; Kaplan, M.H.; Fischer, S.; Dai, M.; Meng, F.; Saha, A.K.; Cervantes, P.; Chan, S.M.; Dube, D.; Omenn, G.S.; et al. Expansion of a novel endogenous retrovirus throughout the pericentromeres of modern humans. Genome Biol. 2015, 16, 1–24. [Google Scholar] [CrossRef]
- Saha, A.K.; Mourad, M.; Kaplan, M.H.; Chefetz, I.; Malek, S.N.; Buckanovich, R.; Markovitz, D.M.; Contreras-Galindo, R. The Genomic Landscape of Centromeres in Cancers. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D. Evolution of recombination rates between sex chromosomes. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160456. [Google Scholar] [CrossRef] [PubMed]
- Pisano, M.P.; Grandi, N.; Tramontano, E. High-Throughput Sequencing Is a Crucial Tool to Investigate the Contribution of Human Endogenous Retroviruses (HERVs) to Human Biology and Development. Viruses 2020, 12, 633. [Google Scholar] [CrossRef] [PubMed]
- Zsíros, J.; Jebbink, M.; Lukashov, V.; Voûte, P.; Berkhout, B. Biased nucleotide composition of the genome of HERV-K related endogenous retroviruses and its evolutionary implications. J. Mol. Evol. 1999, 48, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalianAPOBEC3genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef]
- Arunkumar, G.; Melters, D.P. Centromeric Transcription: A Conserved Swiss-Army Knife. Genes 2020, 11, 911. [Google Scholar] [CrossRef]
- Pisano, M.P.; Tabone, O.; Bodinier, M.; Grandi, N.; Textoris, J.; Mallet, F.; Tramontano, E. RNA-seq transcriptome analysis reveals LTR-retrotransposons modulation in Human Peripheral Blood Mononuclear Cells (PBMCs) after in vivo Lipopolysaccharides (LPS) injection. J. Virol. 2020, 94, e00587-20. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Pisano, M.P.; Scognamiglio, S.; Pessiu, E.; Tramontano, E. Comprehensive Analysis of HERV Transcriptome in HIV+ Cells: Absence of HML2 Activation and General Downregulation of Individual HERV Loci. Viruses 2020, 12, 481. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus 1 | Strand | Coordinates | Age (Milion Years) | o.c.a 2 | Reference |
|---|---|---|---|---|---|
| 1q43 | + | 242457056–242460436 | 22.9 | orangutan | Vargiu et al. |
| 2q11.2 P | + | 101237831–101245098 | 32.3 | rhesus | Vargiu et al. |
| 2q31.1 | − | 170615659–170619450 | 24.3 | gibbon | this study |
| 3q11.2 P | + | 94647178–94651193 | 46.9 | gibbon | Vargiu et al. |
| 3q23 | + | 142206726–142207843 | 29.9 | gibbon | this study |
| 3q26.1 | + | 165546697–165548895 | 37.0 | gibbon | this study |
| 4q25 | − | 109855914–109859920 | 27.9 | gibbon | Vargiu et al. |
| 4q32.1 | − | 160255828–160257162 | 22.0 | gibbon | this study |
| 5p13.2 | − | 34460957–34468537 | 29.8 | rhesus * | Vargiu et al. |
| 5q22.3 | − | 114142954–114150230 | 29.3 | gibbon | Vargiu et al. |
| 6p12.3 | − | 49501277–49508842 | 29.3 | rhesus * | Vargiu et al. |
| 6q22.31 | + | 121042084–121049347 | 26.6 | gibbon | Vargiu et al. |
| 7q21.12 | − | 87732402–87733417 | 40.0 | gibbon | this study |
| 7q36.2 | − | 153843398–153844485 | 31.0 | gibbon | this study |
| 11p12 | − | 43161738–43168627 | 18.1 | orangutan (gorilla *) | Vargiu et al. |
| 11q14.3 | − | 92943568–92950131 | 26.4 | gibbon | Vargiu et al. |
| 12q12 P | + | 38122838–38130125 | 36.4 | gorilla | Vargiu et al. |
| 15q24.3 | + | 76639167–76641589 | 24.3 | gibbon | this study |
| 19q13.2 | − | 42712688–42713786 | 30.5 | gibbon | this study |
| Xq11.1 C | − | 62707600–62717099 | - | gorilla ° | Vargiu et al. |
| Xq22.3 | − | 105666188–105669044 | 22.0 | gibbon | this study |
| Yq11.221 | − | 15973344–15982688 | 38.2 | gorilla * | Vargiu et al. |
| Yp11.2 | − | 7952560–7961873 | 38.2 | chimp | Vargiu et al. |
| HML7 Locus | Colocalized Gene Info | ||||
|---|---|---|---|---|---|
| Name | Portion | Description | Function | Associated Diseases | |
| 1q43 (+) | PLD5 (−) | intronic, antisense | Phospholipase D family member 5 | Hydrolyzes phosphatidylcholine | Type 7 nephrotic syndrome; hemopneumothorax |
| 2q31.1 (−) | MYO3B (+) | intronic, antisense | Myosin IIIB | Probable actin-based ATPase with protein kinase activity. Required for normal cochlear development and hearing | Autosomal recessive deafness 30; entropion |
| 3q23 (+) | GK5 (−) | intronic, antisense | Glycerol kinase 5 | Glycerol degradation, triacylglycerol biosynthesis | Type 1 Diabetes Mellitus 3 and 7 |
| 3q26.1 (+) | LINC01322 (+) | intronic, sense | long intergenic non-coding RNA 1322 | - | - |
| 4q25 (−) | LRIT3 (+) | intronic, antisense | Leucine rich repeat Ig-Like transmembrane domains 3 | May regulate fibroblast growth factor receptors and affect their post-translational modification | Congenital stationary night blindness |
| 5q22.3 (−) | KCNN2 (+) | intronic, antisense | Potassium calcium-activated channel subfamily N member 2 | Forms a voltage-independent potassium channel activated by intracellular calcium following membrane hyperpolarization | Lingual-facial-buccal dyskinesia and aceruloplasminemia |
| 6p12.3 (−) | GLYATL3 (+) | intronic, antisense | Glycine-N-acyltransferase like 3 | Catalyzes the conjugation of long-chain fatty acyl-CoA thioester and glycine, an intermediate in primary fatty acid biosynthesis | - |
| 7q21.12 (−) | RUNDC3B (+) | intronic, antisense | RUN domain-containing protein 3B | Encodes a predicted RAP2-interacting protein. May play a role in RAS-like GTPase signaling pathways | - |
| 7q36.2 (−) | DPP6 (+) | intronic, antisense | dipeptidyl peptidase like 6, transcript variant 6 | Member of S9B family of serine proteases (without detectable activity). Promotes cell surface expression of KCND2 potassium channel and modulates its gating activity | Autosomal dominant mental retardation; paroxysmal familial ventricular fibrillation |
| 15q24.3 (+) | SCAPER (−) | intronic, antisense | S-phase cyclin A associated protein in the endoplasmic reticulum | Cyclin A/Cdk2 regulatory protein that transiently maintains cyclin A in the cytoplasm | Intellectual developmental disorder and retinitis pigmentosa; brachydactyly |
| Xq22.3 (−) | IL1RAPL2 (+) | intronic, antisense | Interleukin 1 receptor accessory protein like 2 | Orphan receptor in the IL1R superfamily | Cinca syndrome; Muckle-Wells syndrome |
| gag | pro | pol | env | Translation | GC% | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HML7 Locus | Shift | Stop | MA | CA | NC | NC ZnF | Shift | Stop | PR | dUTPase | Shift | Stop | RT | RH | IN ZnB | IN core | Shift | Stop | SU gp | TM hr | gag/pro Shift | pro/pol Shift | |
| 1q43 | 6 | 7 | x | x | 1 | 1 | x | x | 39.6 | ||||||||||||||
| 2q11.2 | x | 3 | 1 | x | x | x | x | 5 | 5 | x | x | 40.6 | |||||||||||
| 2q31.1 | x | x | x | x | 38.8 | ||||||||||||||||||
| 3q11.2 | x | 6 | 2 | x | x | 41.1 | |||||||||||||||||
| 3q23 | x | x | 41.9 | ||||||||||||||||||||
| 3q26.1 | 39.0 | ||||||||||||||||||||||
| 4q25 | 6 | 8 | 1 | 5 | x | x | 39.2 | ||||||||||||||||
| 4q32.1 | x | x | x | 40.6 | |||||||||||||||||||
| 5p13.2 | x | 4 | 5 | x | x | x | 1 | 1 | x | x | 40.3 | ||||||||||||
| 5q22.3 | x | 8 | 4 | x | x | x | x | 9 | 1 | x | 40.2 | ||||||||||||
| 6p12.3 | x | 0 | 6 | x | x | x | 8 | 1 | 40.0 | ||||||||||||||
| 6q22.31 | x | 4 | 8 | x | x | x | x | 4 | 4 | x | x | 40.1 | |||||||||||
| 7q21.12 | 43.0 | ||||||||||||||||||||||
| 7q36.2 | 43.4 | ||||||||||||||||||||||
| 11p12 | x | x | x | x | x | 40.1 | |||||||||||||||||
| 11q14.3 | x | 1 | 1 | x | x | x | 1 | 5 | x | 40.3 | |||||||||||||
| 12q12 | x | 3 | 7 | x | x | x | x | 2 | 2 | x | x | 39.3 | |||||||||||
| 15q24.3 | x | x | 38.8 | ||||||||||||||||||||
| 19q13.2 | x | 40.3 | |||||||||||||||||||||
| Xq11.1 | 1 | 11 | x | x | x | xx | 2 | 4 | x | x | 8 | 4 | x | x | x | x | 4 | 5 | x | x | 0 | −1 | 40.0 |
| Xq22.3 | 39.7 | ||||||||||||||||||||||
| Yq11.221 | 3 | 7 | x | x | x | xx | 0 | 3 | x | x | 3 | 7 | x | x | x | x | 6 | 5 | x | x | 0 | −1 | 39.6 |
| Yp11.2 | 0 | 3 | x | x | x | xx | 2 | 2 | x | x | 11 | 8 | x | x | x | 7 | 7 | x | x | 0 | 0 | 40.0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grandi, N.; Pisano, M.P.; Pessiu, E.; Scognamiglio, S.; Tramontano, E. HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates. Biology 2021, 10, 439. https://doi.org/10.3390/biology10050439
Grandi N, Pisano MP, Pessiu E, Scognamiglio S, Tramontano E. HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates. Biology. 2021; 10(5):439. https://doi.org/10.3390/biology10050439
Chicago/Turabian StyleGrandi, Nicole, Maria Paola Pisano, Eleonora Pessiu, Sante Scognamiglio, and Enzo Tramontano. 2021. "HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates" Biology 10, no. 5: 439. https://doi.org/10.3390/biology10050439
APA StyleGrandi, N., Pisano, M. P., Pessiu, E., Scognamiglio, S., & Tramontano, E. (2021). HERV-K(HML7) Integrations in the Human Genome: Comprehensive Characterization and Comparative Analysis in Non-Human Primates. Biology, 10(5), 439. https://doi.org/10.3390/biology10050439