
Identifying an lncRNA-Related ceRNA Network to Reveal Novel Targets for a Cutaneous Squamous Cell Carcinoma

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Collection from the GEO Database
2.2. Data Preprocessing and Screening for DElncRNAs and DEmRNAs
2.3. Construction of the cSCC-Associated lncRNA-miRNA-mRNA Network
2.4. Functional Enrichment Analysis
2.5. Establishment of the Protein-Protein Interaction (PPI) Network and the Core ceRNA Subnetwork
2.6. Expression Validation in an External Dataset
3. Results
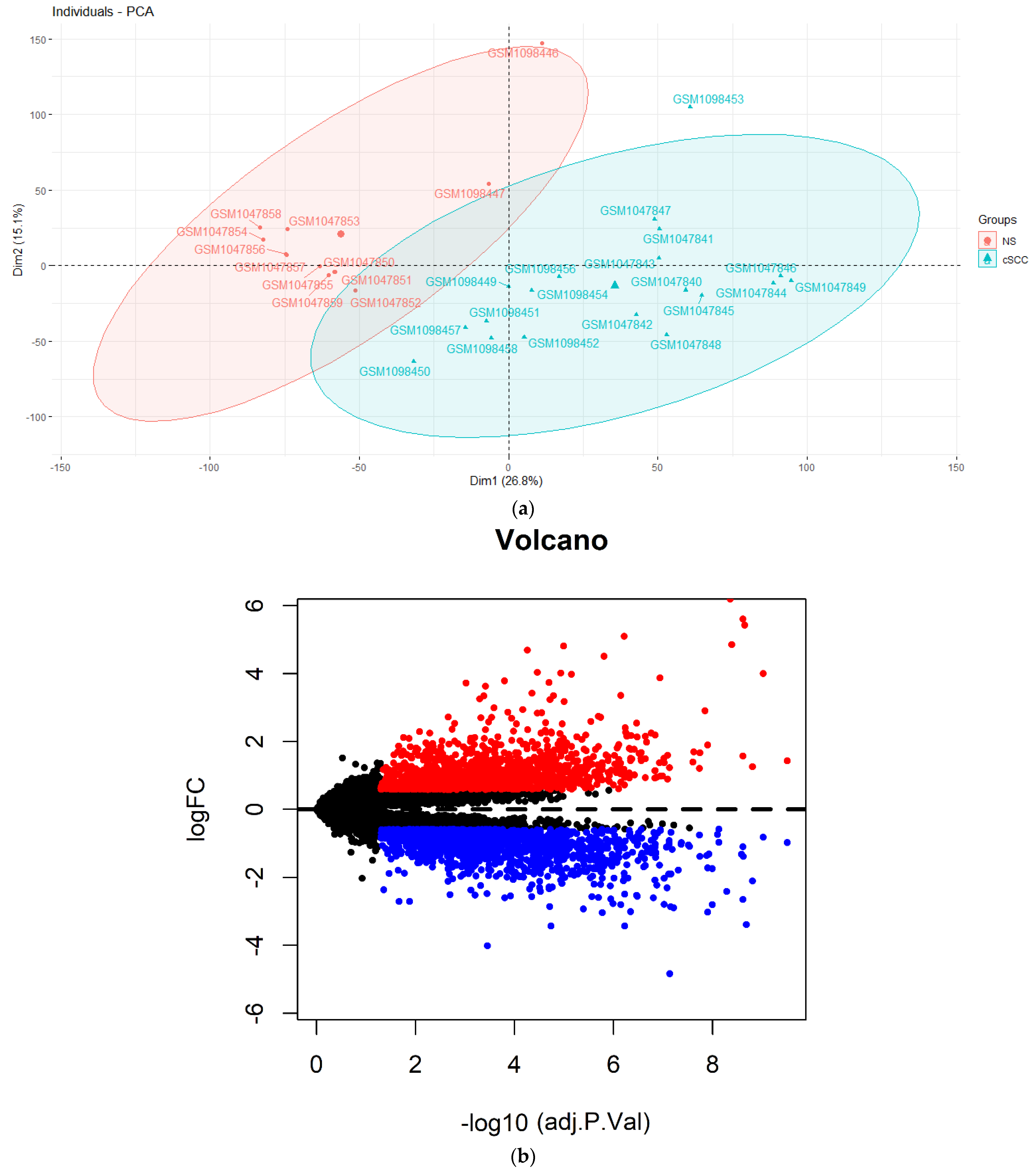
3.1. A Total Of 24 DElncRNAs and 3221 DEmRNAs Were Identified between cSCC and NS
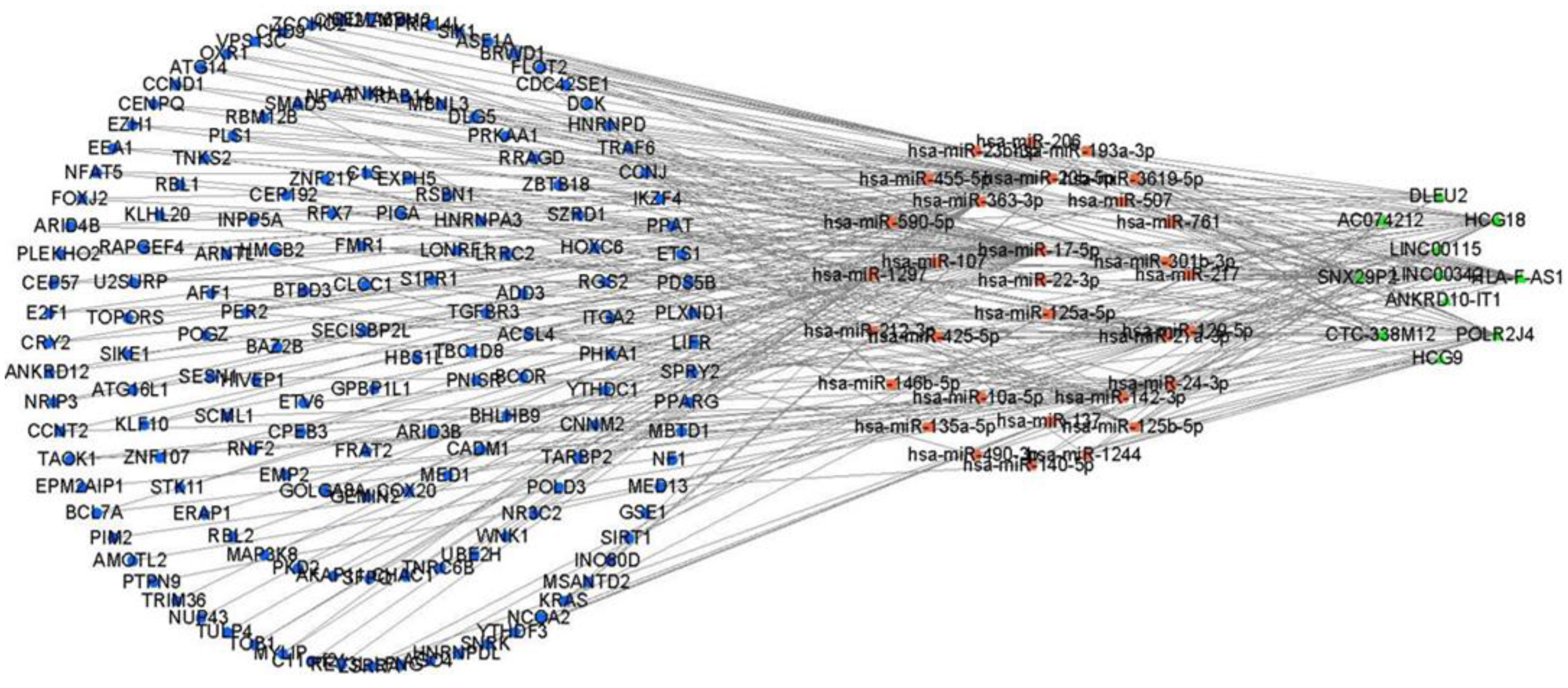
3.2. The cSCC-Associated ceRNA Network Establishment
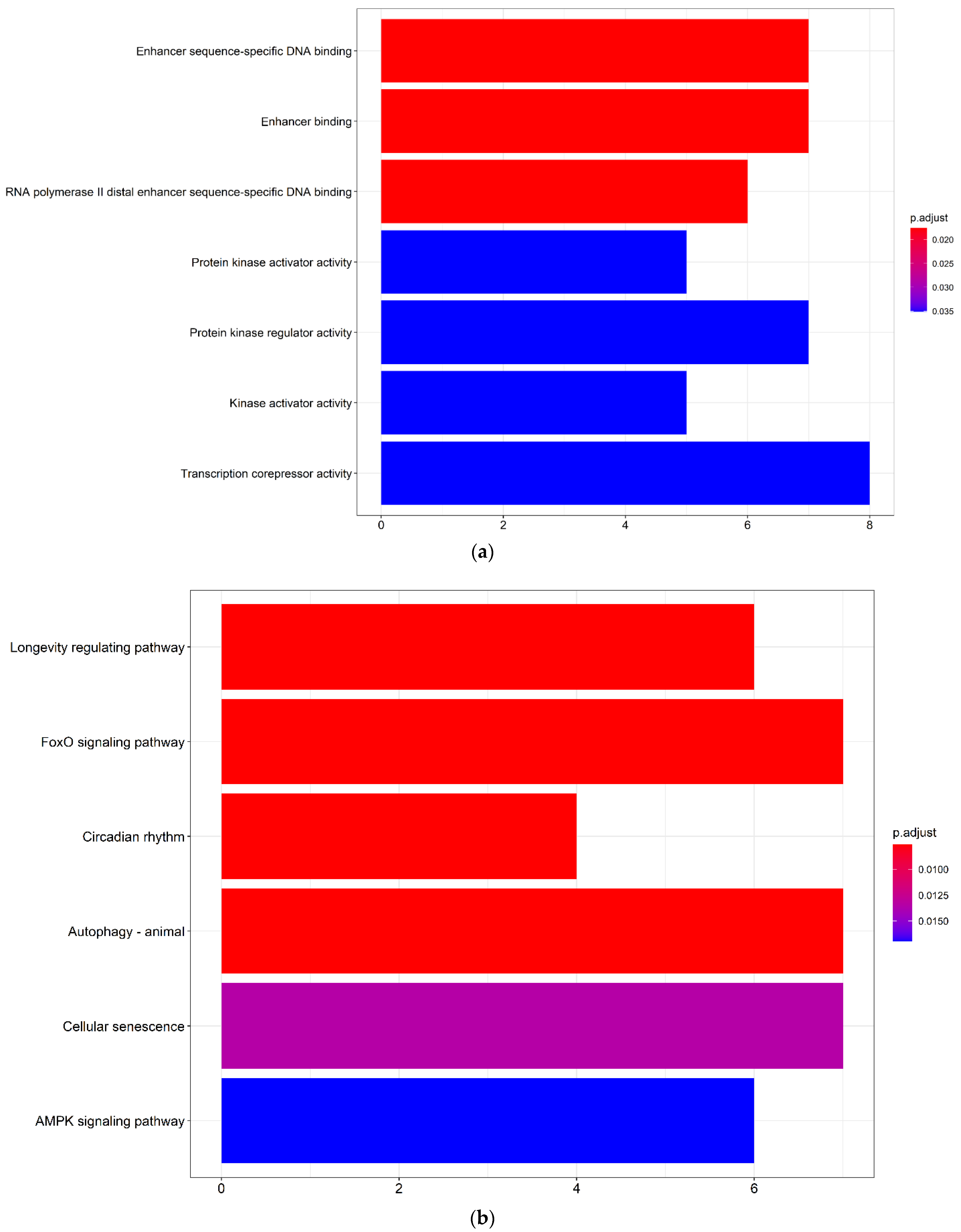
3.3. DEmRNAs of the ceRNA Network Were Associated with Enhancer-Binding, Protein Kinase Regulator Activity and Autophagy
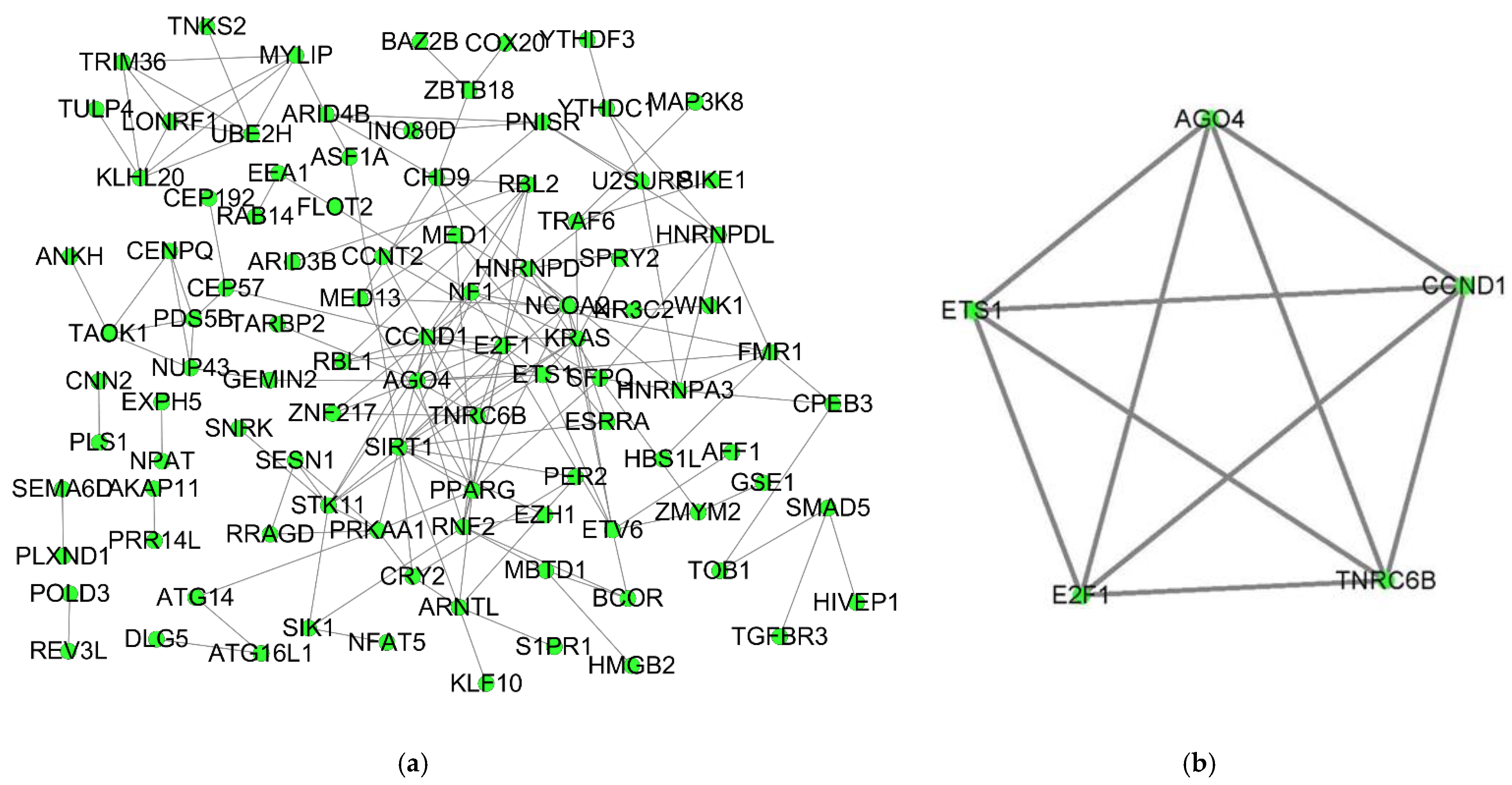
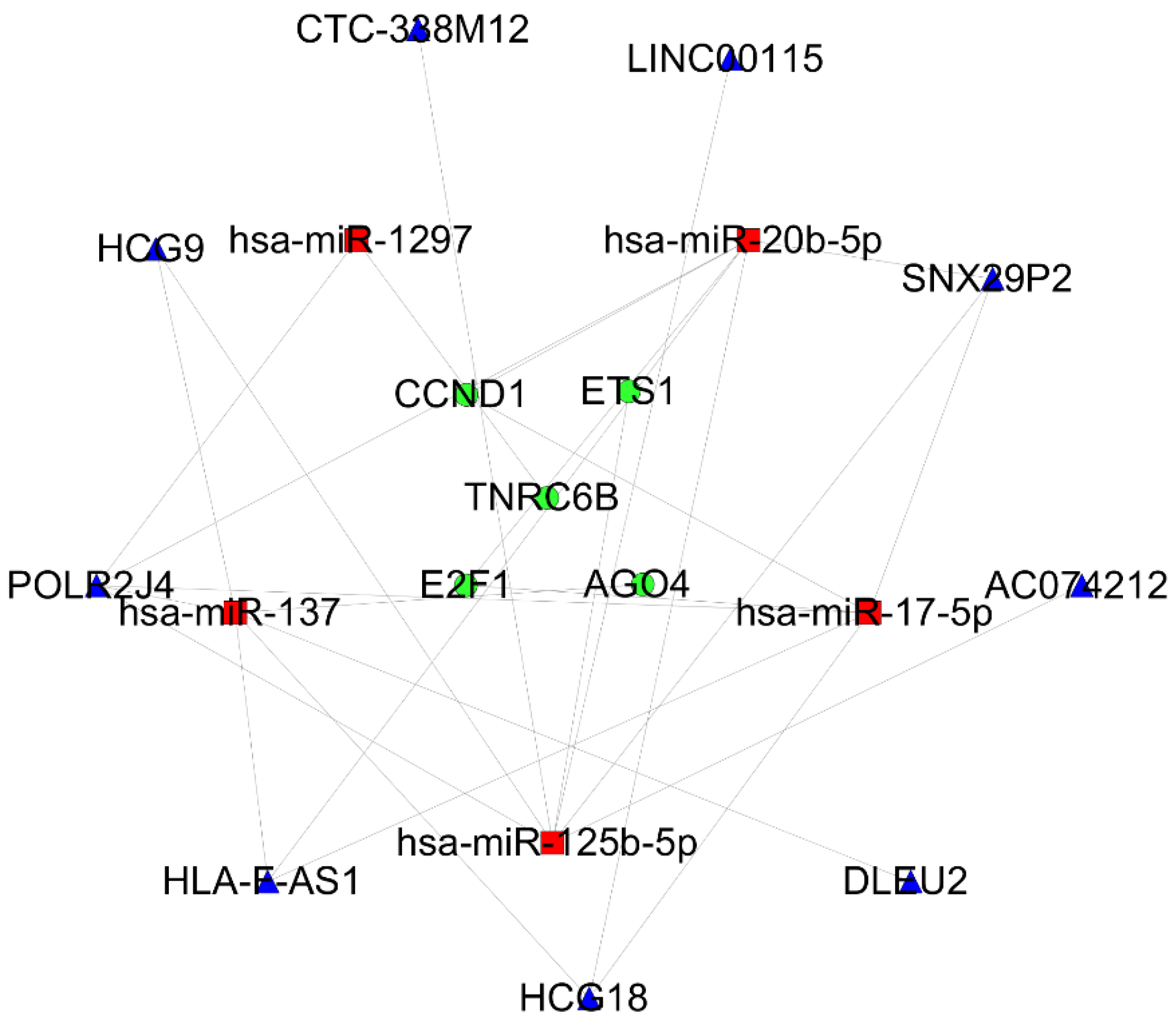
3.4. PPI Network and the Core ceRNA Subnetwork Construction
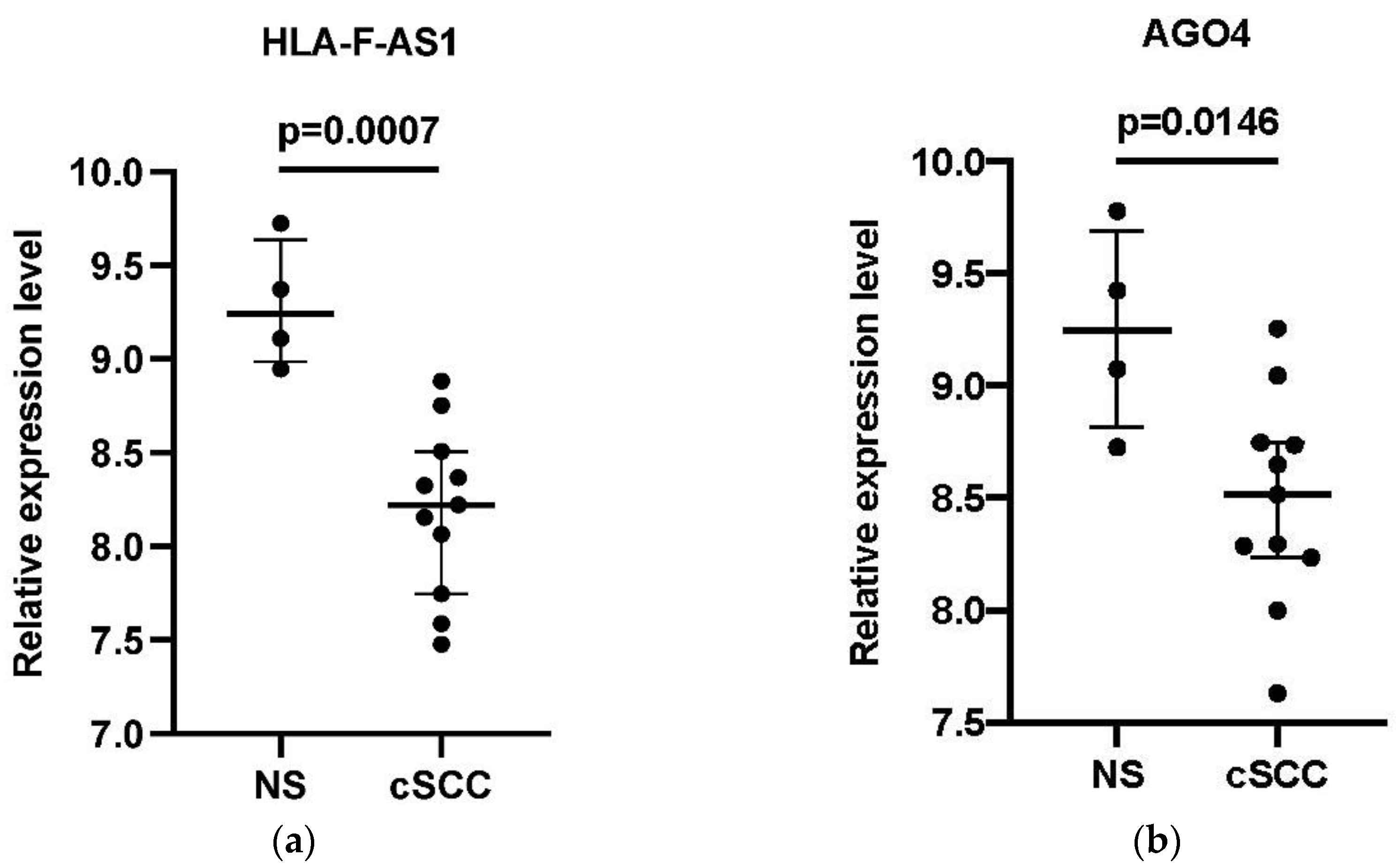
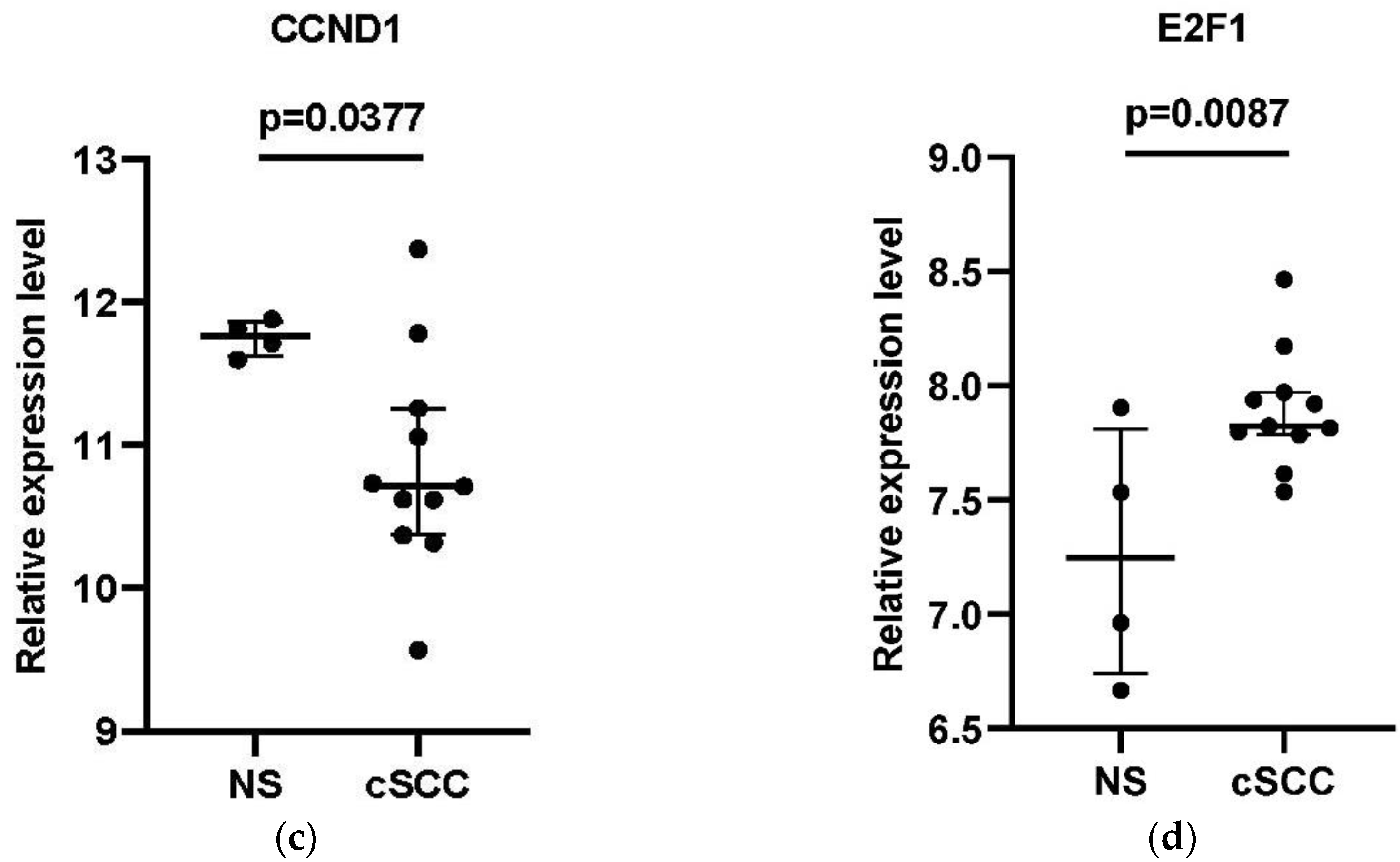
3.5. Verification of the DEGs in the Core ceRNA Subnetwork
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SCC | Squamous Cell Carcinoma |
| cSCC | Cutaneous Squamous Cell Carcinoma |
| SCCIS | Squamous Cell Carcinoma (SCC) In Situ |
| NcRNAs | Non-Coding RNAs |
| LncRNAs | Long Non-Coding RNAs |
| MiRNAs | MicroRNAs |
| CeRNAs | Competing Endogenous RNAs |
| NS | Normal Skin |
| DEGs | Differentially Expressed Genes |
| DElncRNAs | Differentially Expressed LncRNAs |
| DEmRNAs | Differentially Expressed MRNAs |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| BP | Biological Processes |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| STRING | Search Tool for the Retrieval of Interacting Genes |
| MCODE | Molecular Complex Detection |
| FDR | False Discovery Rate |
| FC | Fold Change |
| PCA | Principal Component Analysis |
| PCC | Pearson’s Correlation Coefficient |
| CDC20 | Cell Division Cycle 20 |
| EGFR | Epidermal Growth Factor Receptor |
| PRAF2 | Prenylated Rab Acceptor 1 Domain Family Member 2 |
| FoxO | Forkhead box O |
| HLA-F-AS1 | HLA-F Antisense RNA 1 |
| CCND1 | Cyclin D1 |
| E2F1 | E2F Transcription Factor 1 |
| AGO4 | Argonaute-4 |
| LAD | Lung Adenocarcinoma |
| CRC | Colorectal Cancer |
| PFN1 | Profilin 1 |
| ESCC | Esophageal Squamous Cell Carcinoma |
| TSCC | Tongue Squamous Cell Carcinoma Progression |
| LSCC | Laryngeal Squamous Cell Carcinoma |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| OSCC | Oral Squamous Cell Carcinoma |
| RdDM | RNA-Dependent DNA Methylation |
| DNMT3A | DNA Methyltransferase 3A |
| GBM | Glioblastoma Multiforme |
| AK | Actinic Keratosis |
| NEAT1 | Nuclear Paraspeckle Assembly Transcript 1 |
| RT-PCR | Real-time Polymerase Chain Reaction |
| 3′ UTRs | 3′ Untranslated Regions |
| TRABD | TraB Domain Containing |
References
- Motaparthi, K.; Kapil, J.P.; Velazquez, E.F. Cutaneous Squamous Cell Carcinoma: Review of the Eighth Edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv. Anat. Pathol. 2017, 24, 171–194. [Google Scholar] [CrossRef]
- Waldman, A.; Schmults, C. Cutaneous Squamous Cell Carcinoma. Hematol. Oncol. Clin. N. Am. 2019, 33, 1–12. [Google Scholar] [CrossRef]
- Fernandez Figueras, M.T. From actinic keratosis to squamous cell carcinoma: Pathophysiology revisited. J. Eur. Acad. Derm. Venereol. 2017, 31, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Soura, E.; Gagari, E.; Stratigos, A. Advanced cutaneous squamous cell carcinoma: How is it defined and what new therapeutic approaches are available. Curr. Opin. Oncol. 2019, 31, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Bai, X.; Zhou, Y.; Chen, P.; Yang, M.; Xu, J. MicroRNA-142-5p induces cancer stem cell-like properties of cutaneous squamous cell carcinoma via inhibiting PTEN. J. Cell. Biochem. 2018, 119, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Dai, Y.; Niu, X.; Zhou, M. miR-27a Downregulation Promotes Cutaneous Squamous Cell Carcinoma Progression via Targeting EGFR. Front. Oncol. 2020, 9, 1565. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Selvatici, R.; Di Domenico, M.; Marci, R.; Vesce, F.; Tognon, M.; Martini, F. Methylation loss at H19 imprinted gene correlates with methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples from infertile males. Epigenetics 2013, 8, 990–997. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Riillo, C.; Scionti, F.; Grillone, K.; Polerà, N.; Caracciolo, D.; Arbitrio, M.; Tagliaferri, P.; Tassone, P. miRNAs and lncRNAs as Novel Therapeutic Targets to Improve Cancer Immunotherapy. Cancers (Basel) 2021, 13, 1587. [Google Scholar] [CrossRef]
- Mei, X.L.; Zhong, S. Long noncoding RNA LINC00520 prevents the progression of cutaneous squamous cell carcinoma through the inactivation of the PI3K/Akt signaling pathway by downregulating EGFR. Chin. Med. J. (Engl.) 2019, 132, 454–465. [Google Scholar] [CrossRef]
- Piipponen, M.; Heino, J.; Kähäri, V.M.; Nissinen, L. Long non-coding RNA PICSAR decreases adhesion and promotes migration of squamous carcinoma cells by downregulating α2β1 and α5β1 integrin expression. Biol. Open 2018, 7, bio037044. [Google Scholar] [CrossRef]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef]
- López-Urrutia, E.; Bustamante Montes, L.P.; Ladrón de Guevara Cervantes, D.; Pérez-Plasencia, C.; Campos-Parra, A.D. Crosstalk Between Long Non-coding RNAs, Micro-RNAs and mRNAs: Deciphering Molecular Mechanisms of Master Regulators in Cancer. Front. Oncol. 2019, 9, 669. [Google Scholar] [CrossRef]
- Li, Y.; Chen, D.; Gao, X.; Li, X.; Shi, G. LncRNA NEAT1 Regulates Cell Viability and Invasion in Esophageal Squamous Cell Carcinoma through the miR-129/CTBP2 Axis. Dis. Markers 2017, 9, 5314649. [Google Scholar] [CrossRef]
- Ding, J.; Yang, C.; Yang, S. LINC00511 interacts with miR-765 and modulates tongue squamous cell carcinoma progression by targeting LAMC2. J. Oral. Pathol. Med. 2018, 47, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, X.; Feng, C.; Shang, X.; Xi, Y. Long Non-Coding RNA HCP5 Facilitates Cell Invasion And Epithelial-Mesenchymal Transition In Oral Squamous Cell Carcinoma By miR-140-5p/SOX4 Axis. Cancer Manag. Res. 2019, 11, 10455–10462. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.J.; Sun, Y.; Zhang, D.W.; Zhang, P. Long non-coding RNA HOTAIR functions as a competitive endogenous RNA to regulate PRAF2 expression by sponging miR-326 in cutaneous squamous cell carcinoma. Cancer Cell Int. 2019, 19, 270. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Konishi, T. Principal component analysis for designed experiments. BMC Bioinform. 2015, 16, S7. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Management of advanced and high-stage tumors. J. Am. Acad. Derm. 2018, 78, 249–261. [Google Scholar] [CrossRef]
- Brougham, N.D.; Dennett, E.R.; Cameron, R.; Tan, S.T. The incidence of metastasis from cutaneous squamous cell carcinoma and the impact of its risk factors. J. Surg. Oncol. 2012, 106, 811–815. [Google Scholar] [CrossRef]
- Piipponen, M.; Nissinen, L.; Farshchian, M.; Riihilä, P.; Kivisaari, A.; Kallajoki, M.; Peltonen, J.; Peltonen, S.; Kähäri, V.M. Long Noncoding RNA PICSAR Promotes Growth of Cutaneous Squamous Cell Carcinoma by Regulating ERK1/2 Activity. J. Investig. Derm. 2016, 136, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, S.; Li, J.; Li, Z.; Wang, Y.; Li, X. lncRNA TINCR participates in ALA-PDT-induced apoptosis and autophagy in cutaneous squamous cell carcinoma. J. Cell. Biochem. 2019, 120, 13893–13902. [Google Scholar] [CrossRef]
- Endo-Munoz, L.; Dahler, A.; Teakle, N.; Rickwood, D.; Hazar-Rethinam, M.; Abdul-Jabbar, I.; Sommerville, S.; Dickinson, I.; Kaur, P.; Paquet-Fifield, S.; et al. E2F7 can regulate proliferation, differentiation, and apoptotic responses in human keratinocytes: Implications for cutaneous squamous cell carcinoma formation. Cancer Res. 2009, 69, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Das Mahapatra, K.; Pasquali, L.; Søndergaard, J.N.; Lapins, J.; Nemeth, I.B.; Baltás, E.; Kemény, L.; Homey, B.; Moldovan, L.-I.; Kjems, J.; et al. A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci. Rep. 2020, 10, 3637. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Chin, S.S.; Wang, D.; Liu, S.; Sinha, S.; Garrett-Sinha, L.A. Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J. Cell Sci. 2010, 123, 3566–3575. [Google Scholar] [CrossRef]
- Chun, J.N.; Cho, M.; Park, S.; So, I.; Jeon, J.-H. The conflicting role of E2F1 in prostate cancer: A matter of cell context or interpretational flexibility? Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188336. [Google Scholar] [CrossRef]
- Fang, Z.; Lin, M.; Li, C.; Liu, H.; Gong, C. A comprehensive review of the roles of E2F1 in colon cancer. Am. J. Cancer Res. 2020, 10, 757–768. [Google Scholar]
- Hanken, H.; Gröbe, A.; Cachovan, G.; Smeets, R.; Simon, R.; Sauter, G.; Heiland, M.; Blessmann, M. CCND1 amplification and cyclin D1 immunohistochemical expression in head and neck squamous cell carcinomas. Clin. Oral Investig. 2014, 18, 269–276. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Yan, G.; Kang, Y.; Sun, G.; Wang, S.; Zou, R.; Sun, H.; Zeng, K.; Song, H.; et al. BAP18 is involved in upregulation of CCND1/2 transcription to promote cell growth in oral squamous cell carcinoma. EBioMedicine 2020, 53, 102685. [Google Scholar] [CrossRef]
- Zang, Y.; Li, J.; Wan, B.; Tai, Y. circRNA circ-CCND1 promotes the proliferation of laryngeal squamous cell carcinoma through elevating CCND1 expression via interacting with HuR and miR-646. J. Cell. Mol. Med. 2020, 24, 2423–2433. [Google Scholar] [CrossRef]
- Chalertpet, K.; Pin-On, P.; Aporntewan, C.; Patchsung, M.; Ingrungruanglert, P.; Israsena, N.; Mutirangura, A. Argonaute 4 as an Effector Protein in RNA-Directed DNA Methylation in Human Cells. Front. Genet. 2019, 10, 645. [Google Scholar] [CrossRef]
- Cheray, M.; Etcheverry, A.; Jacques, C.; Pacaud, R.; Bougras-Cartron, G.; Aubry, M.; Denoual, F.; Peterlongo, P.; Nadaradjane, A.; Briand, J.; et al. Cytosine methylation of mature microRNAs inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol. Cancer 2020, 19, 36. [Google Scholar] [CrossRef]
- Sand, M.; Hessam, S.; Amur, S.; Skrygan, M.; Bromba, M.; Stockfleth, E.; Gambichler, T.; Bechara, F.G. Expression of oncogenic miR-17-92 and tumor suppressive miR-143-145 clusters in basal cell carcinoma and cutaneous squamous cell carcinoma. J. Derm. Sci. 2017, 86, 142–148. [Google Scholar] [CrossRef]
- Ding, H.; Luo, Y.; Hu, K.; Liu, P.; Xiong, M. Linc00467 promotes lung adenocarcinoma proliferation via sponging miR-20b-5p to activate CCND1 expression. Onco Targets Ther. 2019, 12, 6733–6743. [Google Scholar] [CrossRef]
- Luo, W.; Li, G.; Yi, Z.; Nie, Q.; Zhang, X. E2F1-miR-20a-5p/20b-5p auto-regulatory feedback loop involved in myoblast proliferation and differentiation. Sci. Rep. 2016, 6, 27904. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, H.; Ma, X.; Zeng, Y.; Pan, Y.; Yu, D.; Liu, Z.; Xiang, Y. HLA-F-AS1/miR-330-3p/PFN1 axis promotes colorectal cancer progression. Life Sci. 2020, 254, 117180. [Google Scholar] [CrossRef]
- Yang, Z.; Li, H.; Wang, Z.; Yang, Y.; Niu, J.; Liu, Y.; Sun, Z.; Yin, C. Microarray expression profile of long non-coding RNAs in human lung adenocarcinoma. Thorac. Cancer 2018, 9, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Jia, H.; Zhang, Z.; Li, S. STAT3-induced HLA-F-AS1 promotes cell proliferation and stemness characteristics in triple negative breast cancer cells by upregulating TRABD. Bioorg. Chem. 2021, 109, 104722. [Google Scholar] [CrossRef] [PubMed]
- Garofoli, M.; Volpicella, M.; Guida, M.; Porcelli, L.; Azzariti, A. The Role of Non-Coding RNAs as Prognostic Factor, Predictor of Drug Response or Resistance and Pharmacological Targets, in the Cutaneous Squamous Cell Carcinoma. Cancers (Basel) 2020, 12, 2552. [Google Scholar] [CrossRef]
- El Tayebi, H.M.; Omar, K.; Hegy, S.; El Maghrabi, M.; El Brolosy, M.; Hosny, K.A.; Esmat, G.; Abdelaziz, A.I. Repression of miR-17-5p with elevated expression of E2F-1 and c-MYC in non-metastatic hepatocellular carcinoma and enhancement of cell growth upon reversing this expression pattern. Biochem. Biophys. Res. Commun. 2013, 434, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.; Huang, C.J.; Choo, K.B.; Cheong, S.K.; Kamarul, T. Oxidative Stress Down-Regulates MiR-20b-5p, MiR-106a-5p and E2F1 Expression to Suppress the G1/S Transition of the Cell Cycle in Multipotent Stromal Cells. Int. J. Med. Sci. 2020, 17, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Li, F.; Qiu, J.; Feng, Z.; Xu, Z.; Chen, Z.; Sun, J. Oncogenic miR-20b-5p contributes to malignant behaviors of breast cancer stem cells by bidirectionally regulating CCND1 and E2F1. BMC Cancer 2020, 20, 949. [Google Scholar] [CrossRef]
- Yang, H.; Lin, J.; Jiang, J.; Ji, J.; Wang, C.; Zhang, J. miR-20b-5p functions as tumor suppressor microRNA by targeting cyclinD1 in colon cancer. Cell Cycle 2020, 19, 2939–2954. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNA/miRNA | Gene Targets | Mechanisms in Cancers |
|---|---|---|
| HLA-F-AS1 | MiR-541-3p | Promotion of cell proliferation and stemness in vitro and tumor growth in vivo by the miR-541-3p/TRABD axis in triple-negative breast cancer [50]. Promotion of cell proliferation, migration and invasion and repression of apoptosis in vitro via the miR-330-3p/ PFN1 axis in colorectal cancer [48]. |
| MiR-330-3p | ||
| MiR-17-5p | 3′ UTR of CCND1 | Repression of the CCND1 protein expression via sponging with the CCND1 3′-UTR region in a lung cancer cell [46]. Promotion of cell growth, proliferation, migration and colony formation in vitro in a non-metastatic hepatocellular carcinoma by regulating E2F1 expression [52]. |
| E2F1 | ||
| MiR-20b-5p | 3′ UTR of CCND1 | Promotion of myoblast differentiation and repression of myoblast proliferation by regulating E2F1 expression [47]. Promotion of cell proliferation, G1/S transition and DNA synthesis by downregulating CCND1 and E2F1 expression in multipotent stromal cells [53]. Promotion of cell migration and proliferation and repression of apoptosis of breast cancer stem cells and promoting tumor growth in vivo by bidirectionally regulating the protein levels of CCND1 and E2F1 in breast cancer [54]. Inhibition of the cell cycle, migration and invasion in vitro and the tumorigenesis in vivo by negatively regulating CCND1 in colon cancer [55]. |
| 3′ UTR of E2F1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Dong, Y.; Deng, Y.; Qi, Q.; Wu, M.; Liang, H.; She, Q.; Guo, Q. Identifying an lncRNA-Related ceRNA Network to Reveal Novel Targets for a Cutaneous Squamous Cell Carcinoma. Biology 2021, 10, 432. https://doi.org/10.3390/biology10050432
Xu Y, Dong Y, Deng Y, Qi Q, Wu M, Liang H, She Q, Guo Q. Identifying an lncRNA-Related ceRNA Network to Reveal Novel Targets for a Cutaneous Squamous Cell Carcinoma. Biology. 2021; 10(5):432. https://doi.org/10.3390/biology10050432
Chicago/Turabian StyleXu, Yaqin, Yingying Dong, Yunhua Deng, Qianrong Qi, Mi Wu, Hongmei Liang, Qiuyun She, and Qing Guo. 2021. "Identifying an lncRNA-Related ceRNA Network to Reveal Novel Targets for a Cutaneous Squamous Cell Carcinoma" Biology 10, no. 5: 432. https://doi.org/10.3390/biology10050432
APA StyleXu, Y., Dong, Y., Deng, Y., Qi, Q., Wu, M., Liang, H., She, Q., & Guo, Q. (2021). Identifying an lncRNA-Related ceRNA Network to Reveal Novel Targets for a Cutaneous Squamous Cell Carcinoma. Biology, 10(5), 432. https://doi.org/10.3390/biology10050432