Metabolic Reprogramming: Strategy for Ischemic Stroke Treatment by Ischemic Preconditioning


Simple Summary
Abstract
1. Introduction
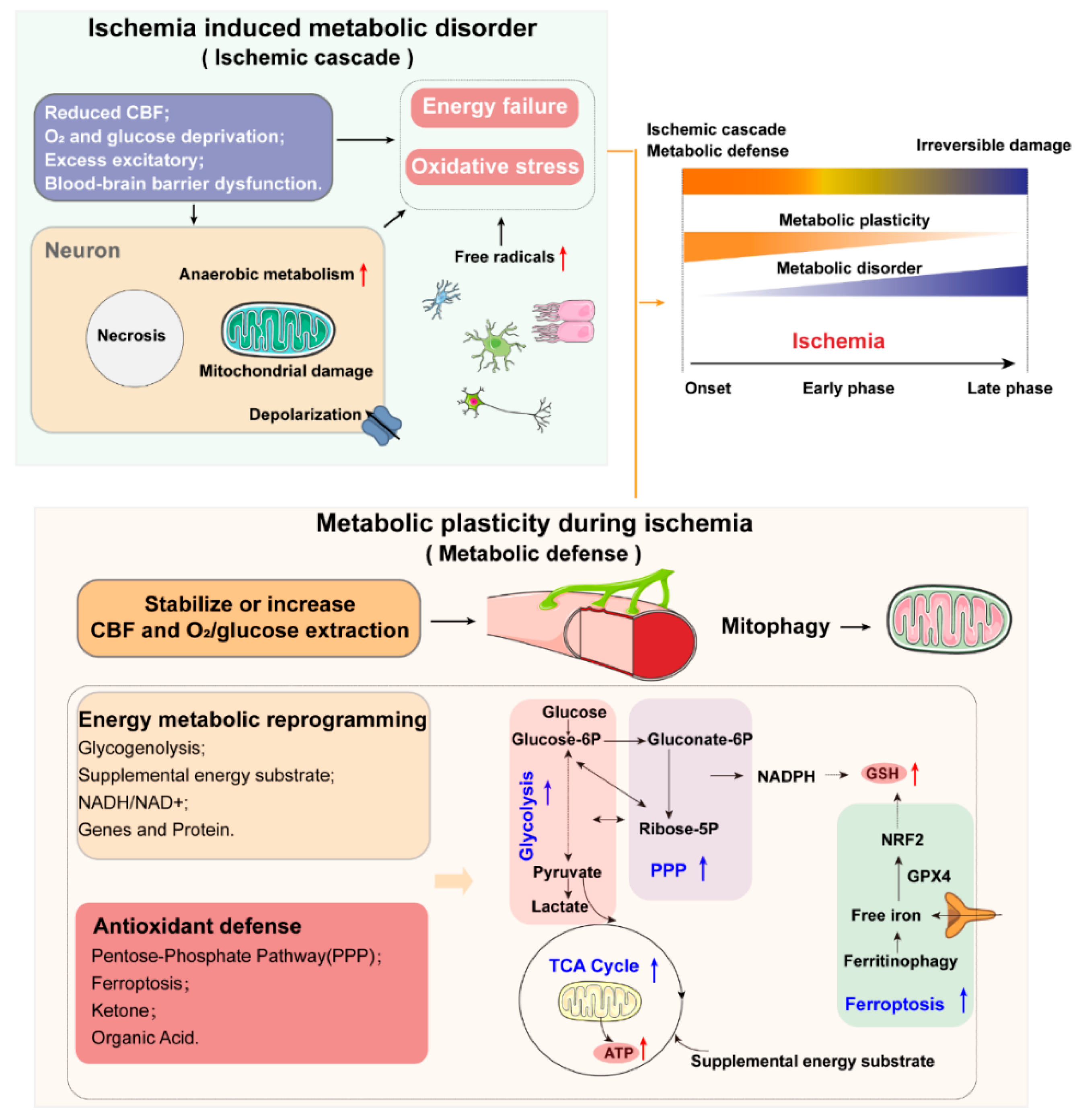
1.1. Metabolic Disorder and Metabolic Plasticity in Ischemic Stroke: Key Considerations and Major Features
1.2. Cerebral Blood Flow
1.3. Mitochondria and Energy Metabolic Reprogramming
1.3.1. Mitochondria and Energy Substrate and Supply
1.3.2. Genes and Protein Related with Energy Metabolism
1.4. Oxidative Stress and Antioxidant Defense
1.4.1. Oxidative Stress
1.4.2. Antioxidant Defense
1.5. Mitochondria and Mitophagy
1.6. Metabolic Syndrome and Ischemic Stroke
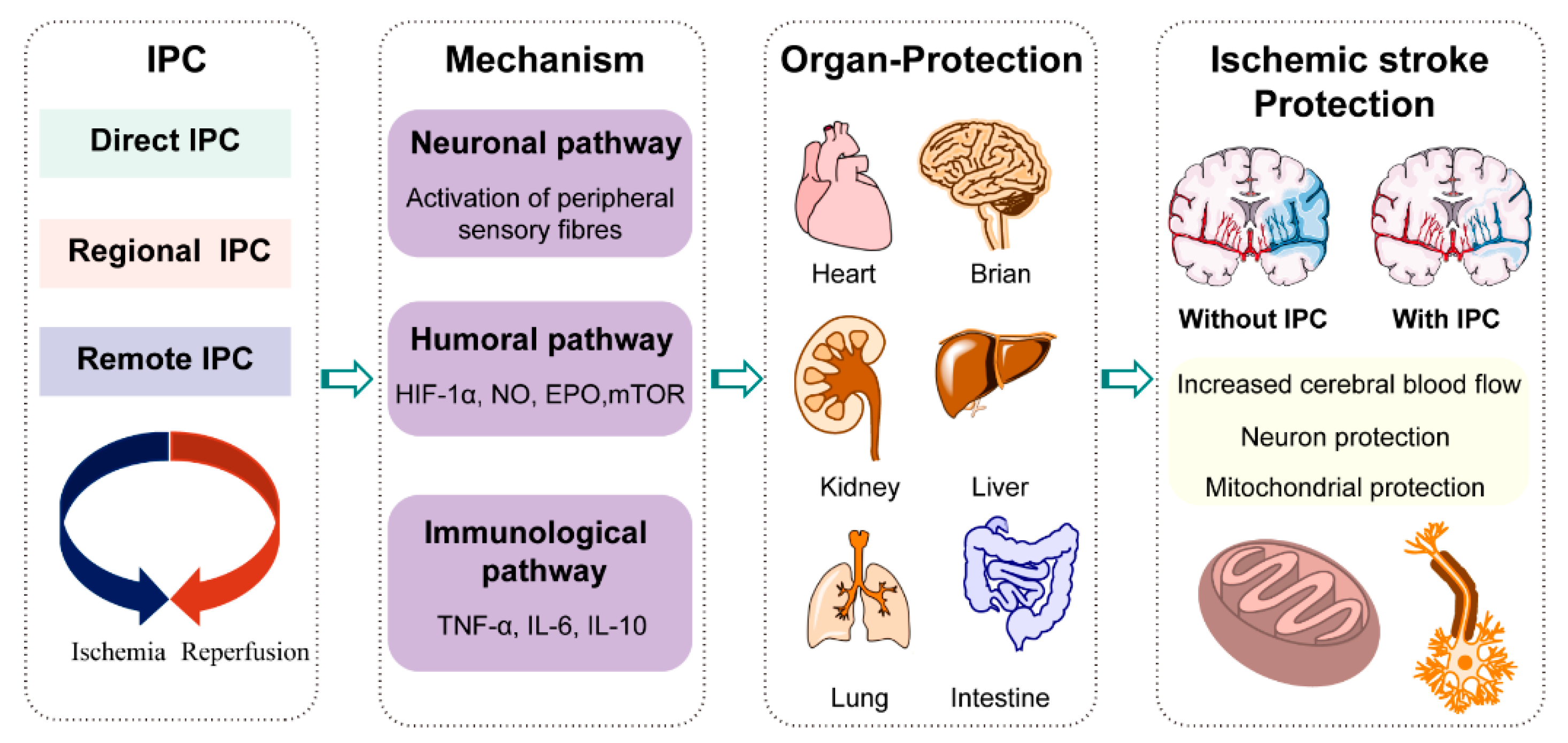
2. Metabolic Reprogramming in Ischemic Stroke Treatment by Ischemic Preconditioning
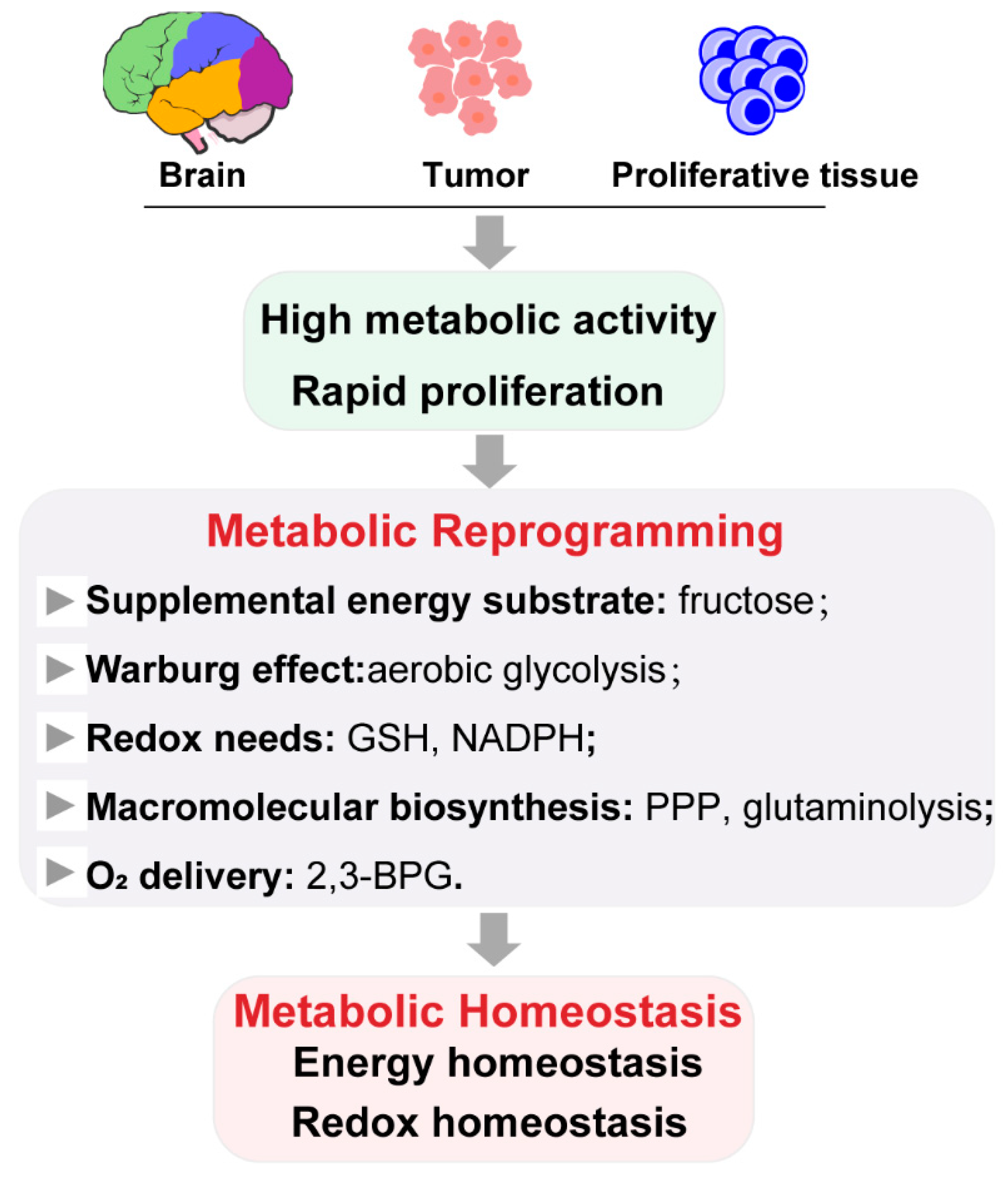
2.1. Role of Metabolic Reprogramming in Metabolic Homeostasis
2.2. Metabolic Reprogramming by Ischemic Preconditioning
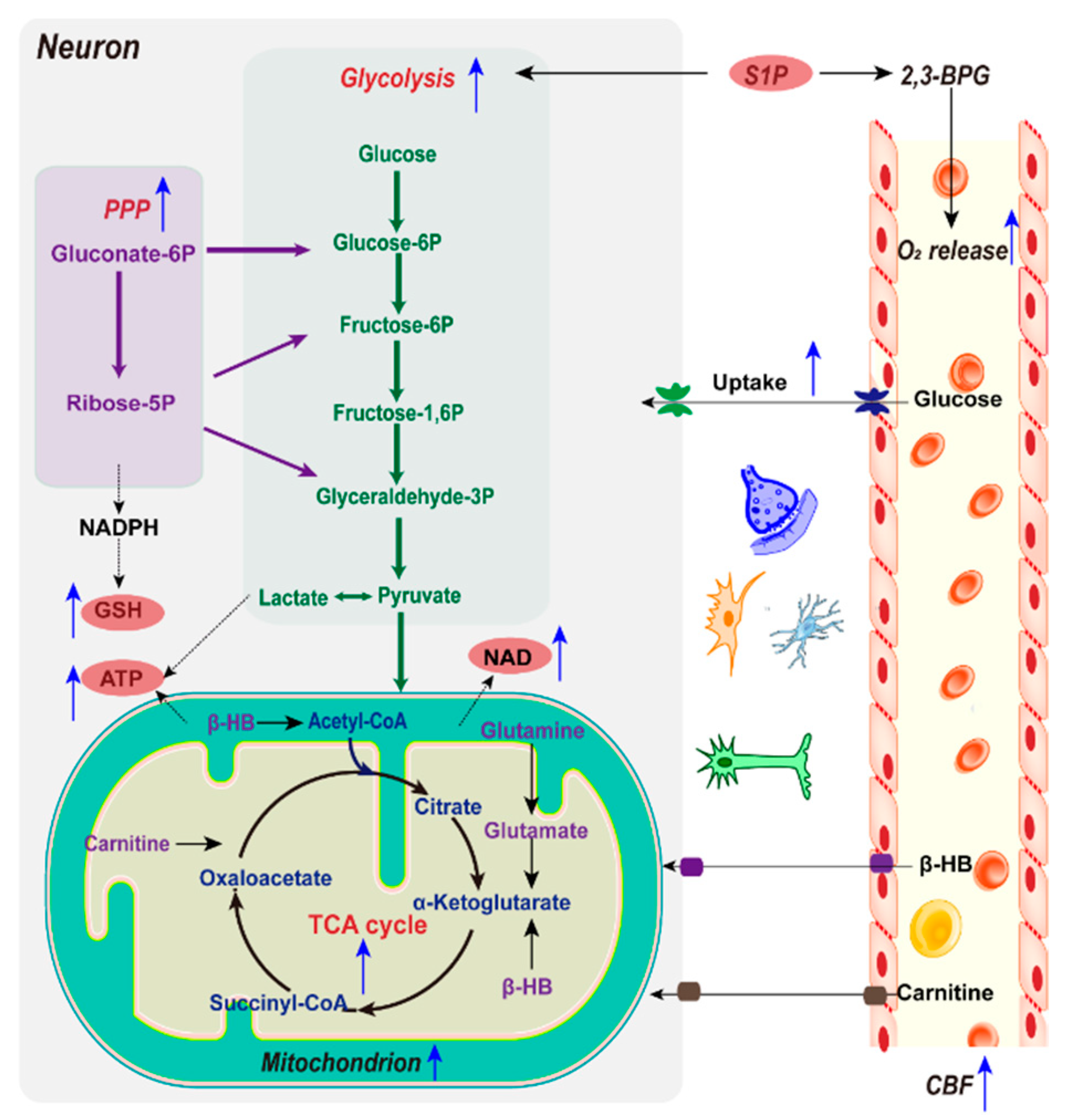
2.2.1. Glucose and Mitochondria
2.2.2. Glycolysis
2.2.3. NAD+/NADH
2.2.4. NADPH and GSH
2.2.5. Alternative Energy Substrates
2.2.6. Sphingosine 1-Phosphate and O2 Delivery
2.3. Spatiotemporal Variation in IPC Metabolomic Reprogramming
2.4. Influence of Aging on IPC Metabolic Reprogramming
2.5. Astrocyte-Neuron Interactions in IPC Metabolic Reprogramming
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, M.; Wang, H.; Zeng, X.; Yin, P.; Zhu, J.; Chen, W.; Li, X.; Wang, L.; Wang, L.; Liu, Y.; et al. Mortality, morbidity, and risk factors in China and its provinces, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 394, 1145–1158. [Google Scholar] [CrossRef]
- Brainin, M.; Feigin, V.L.; Norrving, B.; Martins, S.C.O.; Hankey, G.J.; Hachinski, V. Global prevention of stroke and dementia: The WSO Declaration. Lancet Neurol. 2020, 19, 487–488. [Google Scholar] [CrossRef]
- Chen, S.Y.; Liu, J.W.; Wang, Y.H.; Huang, J.Y.; Chen, S.C.; Yang, S.F.; Wang, P.H. The Conditions Under Which Piracetam Is Used and the Factors That Can Improve National Institute of Health Stroke Scale Score in Ischemic Stroke Patients and the Importance of Previously Unnoticed Factors from a Hospital-Based Observational Study in Taiwan. J. Clin. Med. 2019, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Endres, M. Found in translation preclinical stroke research predicts human pathophysiology clinical phenotypes, and therapeutic outcomes. Stroke 2014, 45, 1510–1518. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Menon, B.K.; van Zwam, W.H.; Dippel, D.W.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.; van der Lug, A.; de Miquel, M.A.; et al. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Blauenfeldt, R.A.; Andersen, G.; Hougaard, K.D.; Hoda, M.N.; Ding, Y.; Ji, X. Remote ischaemic conditioning—A new paradigm of self-protection in the brain. Nat. Rev. Neurol. 2015, 11, 698–710. [Google Scholar] [CrossRef]
- Meng, R.; Asmaro, K.; Meng, L.; Liu, Y.; Ma, C.; Xi, C.; Li, G.; Ren, C.; Luo, Y.; Ling, F.; et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 2012, 79, 1853–1861. [Google Scholar] [CrossRef]
- Morawetz, R.B.; Crowell, R.H.; De Girolami, U. Regional cerebral blood flow thresholds during cerebral ischemia. Fed. Proc. 1979, 38, 2493–2494. [Google Scholar]
- Bang, O.Y.; Saver, J.L.; Kim, S.J.; Kim, G.M.; Chung, C.S.; Ovbiagele, B. Collateral flow predicts response to endovascular therapy for acute ischemic stroke. Stroke 2011, 42, 693–699. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, V.J.; Lasley, R.D. Adenosine receptor-mediated cardioprotection: Are all 4 subtypes required or redundant? J. Cardiovasc. Pharmacol. Ther. 2012, 17, 21–33. [Google Scholar] [CrossRef]
- Xi, Q.; Cheranov, S.Y.; Jaggar, J.H. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ. Res. 2005, 97, 354–362. [Google Scholar] [CrossRef]
- Busija, D.W.; Rutkai, I.; Dutta, S.; Katakam, P.V. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr. Physiol. 2016, 6, 1529–1548. [Google Scholar]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, W.; Huang, J.; Liu, X.; Zhang, H.; Zhang, N. Metabolomic investigation of regional brain tissue dysfunctions induced by global cerebral ischemia. BMC Neurosci. 2016, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Raf, B.; Rishi, S.; Annick, W. Evaluation of lactate as a marker of metabolic stress and cause of secondary damage in acute ischemic stroke or TIA. Clin. Chim. Acta 2008, 397, 27–31. [Google Scholar]
- Geng, J.L.; Zhang, Y.; Li, S.J.; Li, S.N.; Wang, J.K.; Wang, H.; Aa, J.Y.; Wang, G.J. Metabolomic Profiling Reveals That Reprogramming of Cerebral Glucose Metabolism is Involved in Ischemic Preconditioning Induced Neuroprotection in a Rodent Model of Ischemic Stroke. J. Proteome Res. 2019, 18, 57–68. [Google Scholar] [CrossRef]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000, 15, 6804–6810. [Google Scholar] [CrossRef]
- Suh, S.W.; Bergher, J.P.; Anderson, C.M.; Treadway, J.L.; Fosgerau, K.; Swanson, R.A. Astrocyte glycogen sustains neuronal activity during hypoglycemia: Studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*-S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl) propyl]-1H-indole-2-carboxamide). J. Pharmacol. Exp. Ther. 2007, 321, 45–50. [Google Scholar] [CrossRef]
- Berthet, C.; Lei, H.T.; Thevenet, J.; Gruetter, R. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 1780–1789. [Google Scholar] [CrossRef]
- Berthet, C.; Castillo, X.; Magistretti, P.J.; Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: Extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 2012, 34, 329–335. [Google Scholar] [CrossRef]
- Bouzat, P.; Sala, N.; Suys, T.; Zerlauth, J.B. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 2014, 40, 412–421. [Google Scholar] [CrossRef]
- Rothman, D.L.; Behar, K.L.; Hyder, F. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: Implications for brain function. Annu. Rev. Physiol. 2003, 65, 401–427. [Google Scholar] [CrossRef]
- Luo, L.L.; Li, Y.F.; Shan, H.M.; Wang, L.P.; Yuan, F.; Ma, Y.Y.; Li, W.L.; He, T.T.; Wang, Y.Y.; Qu, M.J.; et al. L-glutamine protects mouse brain from ischemic injury via up-regulating heat shock protein 70. CNS Neurosci. Ther. 2019, 25, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guo, M.; Wang, X.; Zhao, Y.X.; Zhao, Q.; Ding, H.Y.; Dong, Q.; Cui, M. Ischemic preconditioning with a ketogenic diet improves brain ischemic tolerance through increased extracellular adenosine levels and hypoxia-inducible factors. Brain Res. 2017, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Baranovicova, E.; Grendar, M.; Kalenska, D.; Tomascova, A.; Cierny, D.; Lehotsky, J. NMR metabolomic study of blood plasma in ischemic and ischemically preconditioned rats: An increased level of ketone bodies and decreased content of glycolytic products 24 h after global cerebral ischemia. J. Physiol. Biochem. 2018, 74, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.L.; Aa, J.Y.; Feng, S.Q.; Wang, S.Y.; Wang, P.; Zhang, Y.; Ouyang, B.C. Exploring the neuroprotective effects of ginkgolides injection in a rodent model of cerebral ischemia-reperfusion injury by GC-MS based metabolomic profiling. J. Pharm. Biomed. Anal. 2017, 142, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Williamson, D.H.; Bates, M.W.; Page, M.A.; Hawkins, R.A. The role of ketone bodies in caloric homeostasis. Adv. Enzyme Regul. 1971, 9, 387–409. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Xu, J.; Khoury, N.; Jackson, C.W.; Escobar, I.; Stegelmann, S.D.; Dave, K.R.; Perez-Pinzon, M.A. Ischemic Neuroprotectant PKCepsilon Restores Mitochondrial Glutamate Oxaloacetate Transaminase in the Neuronal NADH Shuttle after Ischemic Injury. Transl. Stroke Res. 2020, 11, 418–432. [Google Scholar] [CrossRef]
- Wang, S.; Xing, Z.; Vosler, P.S.; Yin, H.; Li, W.; Zhang, F.; Signore, A.P.; Stetler, R.A.; Gao, Y.; Chen, J. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: Role of enhanced DNA repair. Stroke 2008, 39, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.H.; Wei, G.W.; Wang, D.M.; Wang, Q.; Tang, X.N.; Shi, J.; Zhang, P.; Lu, H.F. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front. Biosci. 2007, 12, 2728–2734. [Google Scholar] [CrossRef]
- Wang, P.; Miao, C.Y. NAMPT as a Therapeutic Target against Stroke. Trends Pharmacol. Sci. 2015, 36, 891–905. [Google Scholar] [CrossRef]
- Giusti, B.; Saracini, C.; Bolli, P.; Magi, A.; Martinelli, I. Early-onset Ischaemic Stroke: Analysis of 58 Polymorphisms in 17 Genes Involved in Methionine Metabolism. Thromb. Haemost. 2010, 10, 231–242. [Google Scholar]
- Stankovic, S.; Majkic-Singh, N. Genetic aspects of ischemic stroke coagulation homocysteine, and lipoprotein metabolism as potential risk factors Critical Reviews in Clinical Laboratory. Sciences 2010, 47, 72–123. [Google Scholar]
- Rink, C.; Gnyawali, S.; Peterson, L.; Khanna, S. Oxygen-inducible glutamate oxaloacetate transaminase as protective switch transforming neurotoxic glutamate to metabolic fuel during acute ischemic stroke. Antioxid. Redox Signal 2011, 14, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhao, H.; Zhang, X.; Lou, Z.; Dong, X. UHPLC-Q-TOF-MS based serum metabonomics revealed the metabolic perturbations of ischemic stroke and the protective effect of RKIP in rat models. Mol. Biosyst. 2016, 12, 1831–1841. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Awooda, H.A. Pathophysiology of Cerebral Ischemia: Role of Oxidative/Nitrosative Stress. J. Biosci. Med. 2019, 7, 20–28. [Google Scholar] [CrossRef]
- Li, M.; Zhou, Z.P.; Sun, M.; Cao, L.; Chen, J.; Qin, Y.Y.; Gu, J.H.; Han, F.; Sheng, R.; Wu, J.C.; et al. Reduced Nicotinamide Adenine Dinucleotide Phosphate, a Pentose Phosphate Pathway Product, Might Be a Novel Drug Candidate for Ischemic Stroke. Stroke 2016, 47, 187–195. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, S.; Gong, X.; Tam, S.; Xiao, D.; Liu, S.; Tao, Y. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 2020, 19, 39. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M. Gleason Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Tang, X.; Fang, M.; Cheng, R.; Zhang, Z.; Wang, Y.; Shen, C.; Han, Y.; Lu, Q.; Du, Y.; Liu, Y.; et al. Iron-Deficiency and Estrogen Are Associated with Ischemic Stroke by Up-Regulating Transferrin to Induce Hypercoagulability. Circ. Res. 2020, 127, 651–663. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Li, X.; Yang, X.; Yan, J.; Shi, P.; Ba, L.; Cao, Y.; Wang, P. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 2019, 235, 116795. [Google Scholar] [CrossRef]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.A.; Porter, D.W.; Martin, W.G.; Castranova, V. Ozone-induced lipid-peroxidation and membrane leakage in isolated rat alveolar macrophages- protective effects of taurine. J. Nutr. Biochem. 1991, 2, 308–313. [Google Scholar] [CrossRef]
- Bae, J.E.; Kang, G.M.; Min, S.H.; Jo, D.S.; Jung, Y.K.; Kim, K.; Kim, M.S.; Cho, D.H. Primary cilia mediate mitochondrial stress responses to promote dopamine neuron survival in a Parkinson’s disease model. Cell Death Dis. 2019, 10, 952. [Google Scholar] [CrossRef]
- Ham, P.B.; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309. [Google Scholar] [CrossRef]
- Lemasters, J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade, M.R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Katayama, H.; Hama, H.; Nagasawa, K.; Kurokawa, H.; Sugiyama, M.; Ando, R.; Funata, M.; Yoshida, N.; Homma, M.; Nishimura, T.; et al. Visualizing and Modulating Mitophagy for Therapeutic Studies of Neurodegeneration. Cell 2020, 181, 1176–1187. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Sarrafzadegan, N.; Gharipour, M.; Sadeghi, M.; Nezafati, P.; Talaie, M.; Oveisgharan, S.; Nouri, F.; Khosravi, A. Metabolic Syndrome and the Risk of Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2017, 26, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Tong, D.M.; Chen, X.D.; Yang, T.H.; Zhou, Y.T.; Ma, X.B. Metabolic Syndrome Is a Strong Risk Factor for Minor Ischemic Stroke and Subsequent Vascular Events. PLoS ONE 2016, 11, e0156243. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Quan, W.; Lu, D.; Wang, Y.; Zhang, H.H.; Liu, S.; Jiang, R.C.; Zhou, Y.Y. Association between Metabolic Syndrome and Cognitive Impairment after Acute Ischemic Stroke: A Cross-Sectional Study in a Chinese Population. PLoS ONE 2016, 11, e0167327. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Kim, J.S.; Labreuche, J.; Charles, H.; Abtan, J.; Bejot, Y.; Cabrejo, L.; Cha, J.K.; Ducrocq, G.; Giroud, M.; et al. A Comparison of Two LDL Cholesterol Targets after Ischemic Stroke. N. Engl. J. Med. 2020, 382, 9. [Google Scholar] [CrossRef] [PubMed]
- Lekoubou, A.; Ovbiagele, B.; Markovic, D.; Sanossian, N.; Towfighi, A. Age, sex, and race/ethnic temporal trends in metabolic syndrome prevalence among individuals with myocardial infarction or stroke in the United States. J. Neurol. Sci. 2017, 376, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Marchiq, I.; Pouysségur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H+ symporters. J. Mol. Med. 2015, 94, 155–171. [Google Scholar] [CrossRef]
- Chen, W.L.; Jin, X.; Wang, M.; Liu, D.; Luo, Q.; Tian, H.; Cai, L.; Meng, L.; Bi, R.; Wang, L.; et al. GLUT5-mediated fructose utilization drives lung cancer growth by stimulating fatty acid synthesis and AMPK/mTORC1 signaling. JCI Insight 2020, 5, e131596. [Google Scholar] [CrossRef]
- Ardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Servick, K. Reprogrammed cells could tackle brain damage. Science 2018, 362, 736–737. [Google Scholar] [CrossRef]
- Bahadoran, A.; Bezavada, L.; Smallwood, H.S. Fueling influenza and the immune response: Implications for metabolic reprogramming during influenza infection and immune metabolism. Immunol. Rev. 2020, 295, 140–166. [Google Scholar] [CrossRef]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, J.; Ren, Z.; Sun, R.; Zhou, Y.; Zhang, Q.; Wang, Q.; Cui, G.; Li, J.; Li, A.; et al. Switching from Fatty Acid Oxidation to Glycolysis Improves the Outcome of Acute-On-Chronic Liver Failure. Adv. Sci. 2020, 7, 1902996. [Google Scholar] [CrossRef]
- Sun, K.; Zhang, Y.; D’Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.; Huang, A.; Wen, Y.E.; et al. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat. Commun. 2016, 7, 12086. [Google Scholar] [CrossRef]
- Xie, T.; Chen, C.; Peng, Z.; Brown, B.C.; Reisz, J.A.; Xu, P.; Zhou, Z.; Song, A.; Zhang, Y.; Bogdanov, M.V.; et al. Erythrocyte Metabolic Reprogramming by Sphingosine 1-Phosphate in Chronic Kidney Disease and Therapies. Circ. Res. 2020, 127, 360–375. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Wang, H.; He, Z.; Zhang, Y.; Zhang, J. (1)H NMR metabolic signature of cerebrospinal fluid following repetitive lower-limb remote ischemia preconditioning. Neurochem. Int. 2018, 116, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.W.; Escobar, I.; Xu, J.; Perez-Pinzon, M.A. Effects of ischemic preconditioning on mitochondrial and metabolic neruoprotection: 5′ adenosine monophosphate-activated protein kinase and sirtuins. Brain Circ. 2018, 4, 54–61. [Google Scholar]
- Parsons, M.W.; Barber, P.A.; Desmond, P.M.; Baird, T.A.; Darby, D.G.; Byrnes, G.; Tress, B.M.; Davis, S.M. Acute hyperglycemia adversely affects stroke outcome: A magnetic resonance imaging and spectroscopy study. Ann. Neurol. 2002, 52, 20–28. [Google Scholar] [CrossRef]
- Morris-Blanco, K.C.; Cohan, C.H.; Neumann, J.T.; Sick, T.J.; Perez-Pinzon, M.A. Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J. Cereb. Blood Flow Metab. 2014, 34, 1024–1032. [Google Scholar] [CrossRef]
- Laursen, M.R.; Hansen, J.; Elkjaer, C.; Stavnager, N.; Nielsen, C.B.; Pryds, K.; Johnsen, J.; Nielsen, J.M.; Botker, H.E.; Johannsen, M. Untargeted metabolomics reveals a mild impact of remote ischemic conditioning on the plasma metabolome and alpha-hydroxybutyrate as a possible cardioprotective factor and biomarker of tissue ischemia. Metabolomics 2017, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.P.; Markhard, A.L.; Shah, H.; Sharma, R.; Skinner, O.S.; Clish, C.B.; Deik, A.; Patgiri, A.; Hsu, Y.H.; Masia, R.; et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 2020, 583, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, B.L.; Sutton, R.L.; Fukushima, M.; Harris, N.G.; Hovda, D.A.; Lee, S.M. Upregulation of pentose phosphate pathway and preservation oftricarboxylic acid cycle flux after experimental brain injury. J. Neurotrauma 2005, 22, 1052–1065. [Google Scholar] [CrossRef]
- Sato, H.; Nomura, S.; Maebara, K.; Sato, K.; Tamba, M.; Bannai, S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem. Biophys. Res. Commun. 2004, 325, 109–116. [Google Scholar] [CrossRef]
- Hirayama, Y.; Koizumi, S. Astrocytes and ischemic tolerance. Neurosci. Res. 2018, 126, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.V.; Perez-Pinzon, M.A. Ischemic preconditioning treatment of astrocytes transfers ischemic tolerance to neurons. Cond. Med. 2017, 1, 2–8. [Google Scholar] [PubMed]
- Glenn, T.C.; Martin, N.A.; Horning, M.A. Lactate: Brain fuel in human traumatic brain injury: A comparison with normal healthy control subjects. J. Neurotrauma 2015, 32, 820–832. [Google Scholar] [CrossRef]
- Vessey, D.A.; Li, L.; Honbo, N.; Karliner, J.S. Sphingosine 1-phosphate is an important endogenous cardioprotectant released by ischemic pre- and postconditioning. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1429–H1435. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, J.; Epstein, A.A.; Kurczy, M.E.; Benton, P.H.; Uritboonthai, W.; Fox, H.S.; Boska, M.D.; Gendelman, H.E.; Siuzdak, G. Brain region mapping using global metabolomics. Chem. Biol. 2014, 21, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Shariatgorji, M.; Nilsson, A.; Fridjonsdottir, E.; Vallianatou, T.; Källback, P.; Katan, L.; Sävmarker, J.; Mantas, I.; Zhang, X.; Bezard, E.; et al. Comprehensive mapping of neurotransmitter networks by MALDI–MS imaging. Nat. Methods 2019, 16, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019, 29, 1258–1273. [Google Scholar] [CrossRef]
- Hu, S.; Dong, H.; Zhang, H.; Wang, S.; Hou, L.; Chen, S. Noninvasive limb remote ischemic preconditioning contributes neuroprotective effects via activation of adenosine a1 receptor and redox status after transient focal cerebral ischemia in rats. Brain Res. 2012, 1459, 81–90. [Google Scholar] [CrossRef]
- de Jonge, R.; de Jong, J.W. Ischemic preconditioning and glucose metabolism during low-flow ischemia: Role of the adenosine a receptor. Cardiovasc. Res. 1999, 43, 909–918. [Google Scholar] [CrossRef]
- Paez, D.T.; Garces, M.; Calabro, V.; Bin, E.P.; D’Annunzio, V.; Del Mauro, J. Adenosine a1 receptors and mitochondria: Targets of remote ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H743–H750. [Google Scholar] [CrossRef]
- Durukan, A.; Tatlisumak, T. Preconditioning-induced ischemic tolerance: A window into endogenous gearing for cerebroprotection. Exp. Transl. Stroke Med. 2010, 2, 2. [Google Scholar] [CrossRef]
- Zhou, D.; Ding, J.Y.; Ya, J.Y.; Pan, L.Q.; Bai, C.B.; Guan, J.W.; Wang, Z.G.; Jin, K.X.; Yang, Q.; Ji, X.M.; et al. Efficacy of remote ischemic conditioning on improving WMHs and cognition in very elderly patients with intracranial atherosclerotic stenosis. Aging 2019, 11, 634–648. [Google Scholar] [CrossRef]
- Jennifer, D. Ischemic preconditioning in 18- to 20-month-old gerbils long-term survival with functional outcome measures. Stroke 1999, 30, 1240–1246. [Google Scholar]
- Yarian, C.S.; Toroser, D.; Sohal, R.S. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech. Ageing Dev. 2006, 127, 79–84. [Google Scholar] [CrossRef]
- Hirayama, Y.; Ikeda, M.Y.; Notomi, S.; Enaida, H.; Kinouchi, H.; Koizumi, S. Astrocyte-mediated ischemic tolerance. J. Neurosci. 2015, 35, 3794–3805. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Martin, J.L. Regulation of glycogen metabolism: Physiological, pharmacological and pathological aspects. In Astrocytes: Pharmacology and Function; Murphy, S., Ed.; Academic Press: San Diego, CA, USA, 1993; pp. 243–265. [Google Scholar]
- Hatten, M.E. Neuronal regulation of astroglial morphology and proliferation in vitro. J. Cell Biol. 1985, 100, 384–396. [Google Scholar] [CrossRef]
- Zeiger, S.L.; McKenzie, J.R.; Stankowski, J.N.; Martin, J.A.; Cliffel, D.E.; McLaughlin, B. Neuron specific metabolic adaptations following multi-day exposures to oxygen glucose deprivation. Biochim. Biophys. Acta 2010, 1802, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Arrell, D.K.; Elliott, S.T.; Kane, L.A.; Guo, Y.; Ko, Y.H.; Pedersen, P.L. Proteomic analysis of pharmacological preconditioning. Circ. Res. 2006, 99, 706–714. [Google Scholar] [CrossRef]
- Della Morte, D.; Abete, P.; Gallucci, F.; Scaglione, A.; D’Ambrosio, D.; Gargiulo, G.; De Rosa, G.; Dave, K.R.; Lin, H.W.; Cacciatore, F.; et al. Transient ischemic attack before nonlacunar ischemic stroke in the elderly. J. Stroke Cerebrovasc. Dis. 2008, 17, 257–262. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Main Results | Reference |
|---|---|---|
| Cohort study of 5398 adults aged 35 years or older followed for 10 years | Stroke incidence rates for those with and without MetS were 2.6% and 1.1%, respectively. | [58] |
| Cohort study of 1361 outpatients | 40.2% ischemic stroke individuals were diagnosed with MetS. | [59] |
| Cross-sectional study of 840 patients | MetS patients had a 3.542-fold increased odds ratio (OR) for cognitive impairment. | [60] |
| Trial of 2860 patients and followed them for 3.5 years | When ischemic stroke occurred, patients who had a target LDL cholesterol level of 90–110 mg per deciliter had a higher risk of subsequent cardiovascular events than those who had a target range of less than 70 mg per deciliter. | [61] |
| U.S. National Health and Nutrition Examination Surveys of 12,502 adults during 1999–2010 | MetS prevalence was 61.2% in stroke survivors. | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Han, R.; Zhou, B. Metabolic Reprogramming: Strategy for Ischemic Stroke Treatment by Ischemic Preconditioning. Biology 2021, 10, 424. https://doi.org/10.3390/biology10050424
Liang J, Han R, Zhou B. Metabolic Reprogramming: Strategy for Ischemic Stroke Treatment by Ischemic Preconditioning. Biology. 2021; 10(5):424. https://doi.org/10.3390/biology10050424
Chicago/Turabian StyleLiang, Jing, Rongrong Han, and Bing Zhou. 2021. "Metabolic Reprogramming: Strategy for Ischemic Stroke Treatment by Ischemic Preconditioning" Biology 10, no. 5: 424. https://doi.org/10.3390/biology10050424
APA StyleLiang, J., Han, R., & Zhou, B. (2021). Metabolic Reprogramming: Strategy for Ischemic Stroke Treatment by Ischemic Preconditioning. Biology, 10(5), 424. https://doi.org/10.3390/biology10050424