
Cell Death via Lipid Peroxidation and Protein Aggregation Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
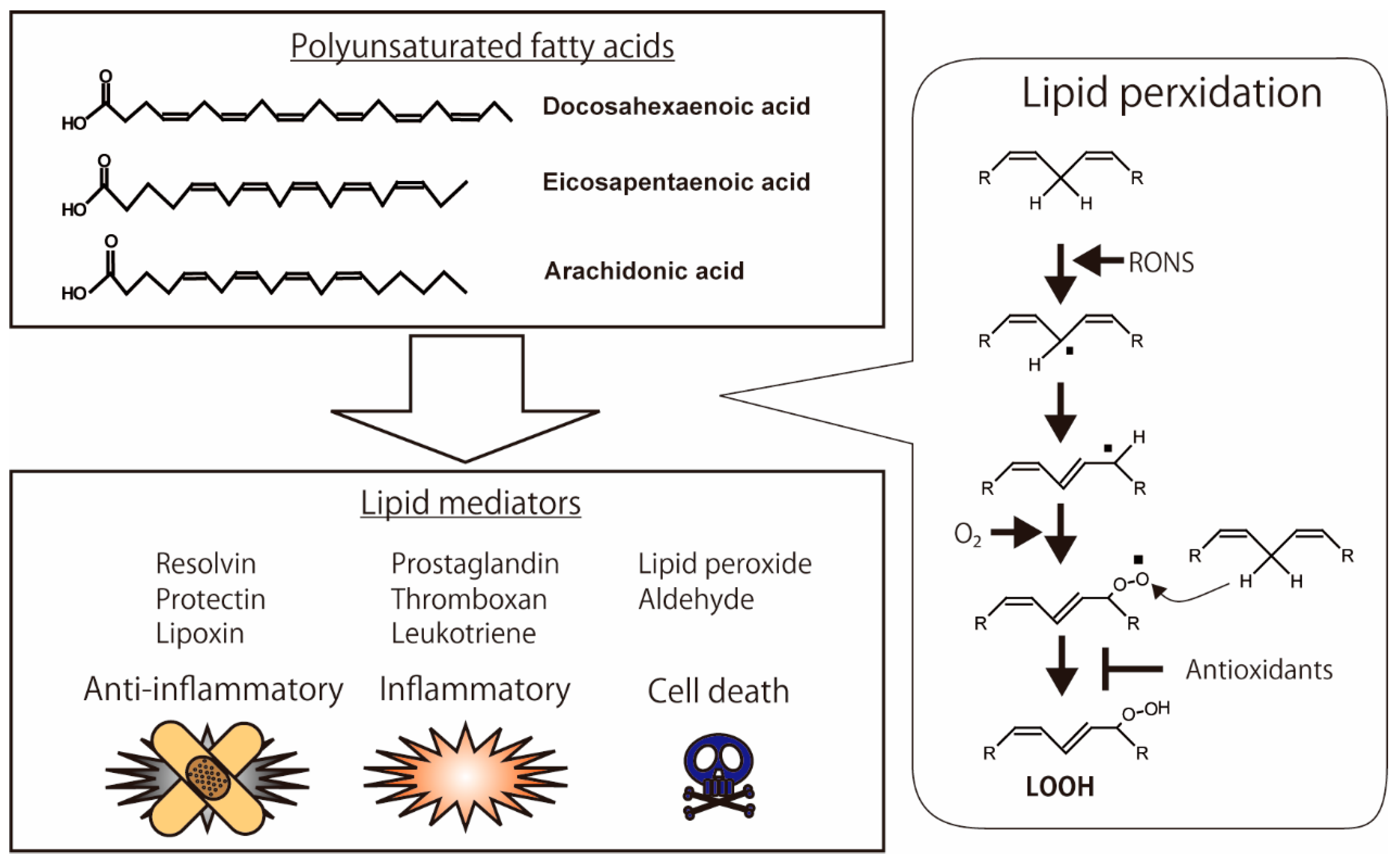
2. Lipid Peroxidation
2.1. Lipid Peroxidation of Polyunsaturated Fatty Acids
2.2. PUFAs and the Peroxidation Products in Neurodegenerative Diseases
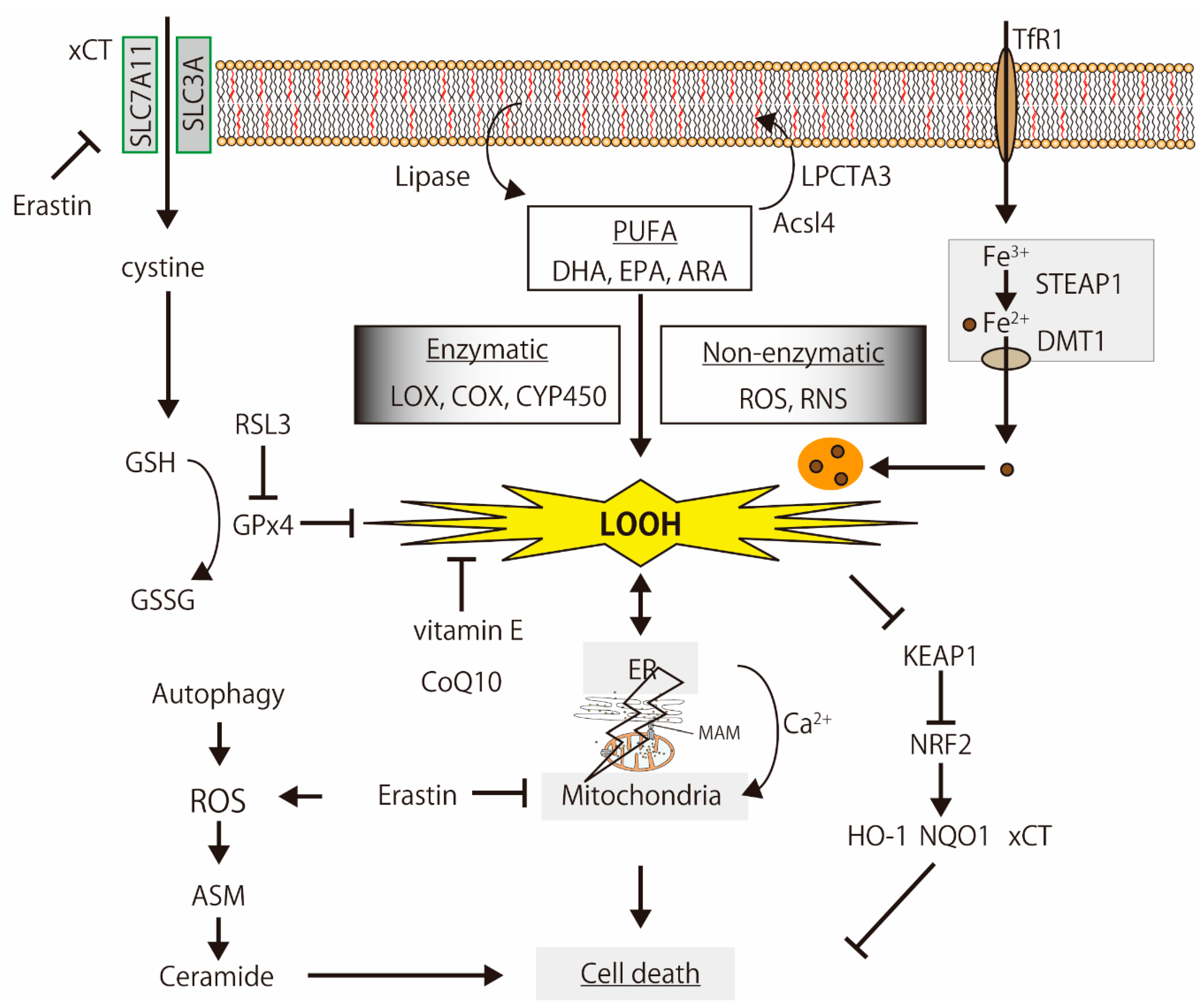
3. Cell Death Induced by Lipid Peroxidation
3.1. Ferroptosis
3.2. Relationship between Lipid Peroxidation and Calcium Signaling Pathway
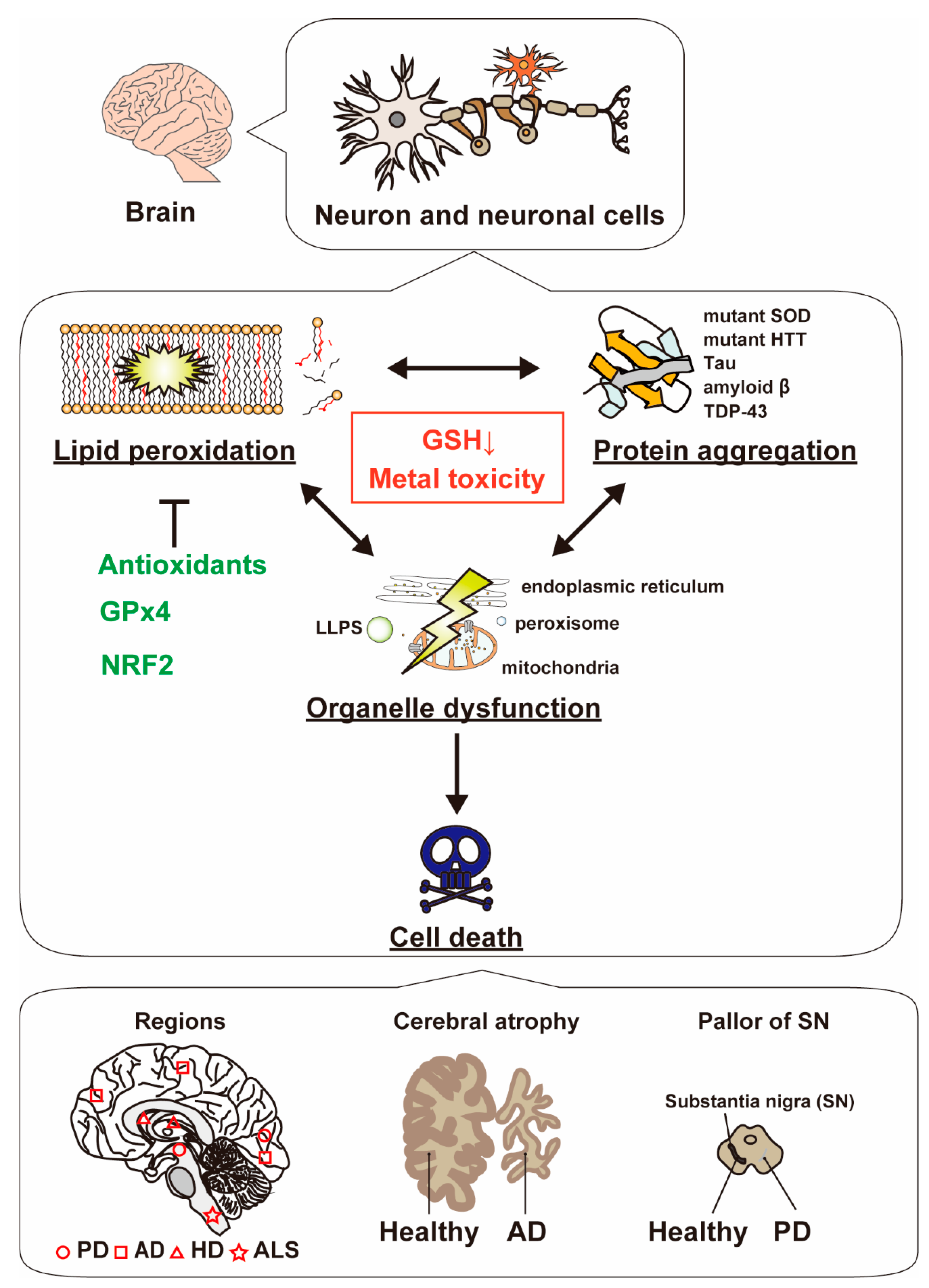
4. Alternation of Organelle Function Regarding Lipid Peroxidation and Protein Aggregation
4.1. Mitochondrial Dysfunction and Misfolded Proteins
4.2. Endoplasmic Reticulum Stress and Lipid Peroxidation

4.3. Other Organelles
4.4. Liquid–Liquid Phase Separation

5. Manipulation of Neurodegeneration by Anti-Oxidative Chemicals and Enzymes
5.1. Anti-Oxidants and Electron Transfer Components
5.2. Metal Ions
5.3. Nuclear Factor (Esrythroid-Derived 2)-Like 2 (NRF2)
5.4. Glutathione Peroxidase 4 (GPx4)
5.5. Other Related Proteins
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Casares, D.; Escriba, P.V.; Rossello, C.A. Membrane lipid composition: Effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef]
- Zeng, M.; Heine, N.; Wilson, K.R. Evidence that Criegee intermediates drive autoxidation in unsaturated lipids. Proc. Natl. Acad. Sci. USA 2020, 117, 4486–4490. [Google Scholar] [CrossRef]
- Ermakov, A.V.; Konkova, M.S.; Kostyuk, S.V.; Izevskaya, V.L.; Baranova, A.; Veiko, N.N. Oxidized extracellular DNA as a stress signal in human cells. Oxid. Med. Cell. Longev. 2013, 2013, 649747. [Google Scholar] [CrossRef]
- Surguchev, A.; Surguchov, A. Conformational diseases: Looking into the eyes. Brain Res. Bull. 2010, 81, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Muthuraman, A.; Rishitha, N.; Paramakrishnan, N.; Mahendran, B.; Ramesh, M. Role of lipid peroxidation process in neurodegenerative disorders. In Lipid Peroxidation Research; Ahmed Mansour, M., Ed.; IntechOpen: London, UK, 2020; ISBN 978-1-83968-547-7. [Google Scholar]
- Ashraf, A.; So, P.W. Spotlight on ferroptosis: Iron-dependent cell death in alzheimer’s disease. Front. Aging Neurosci. 2020, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Kreiser, R.P.; Wright, A.K.; Block, N.R.; Hollows, J.E.; Nguyen, L.T.; LeForte, K.; Mannini, B.; Vendruscolo, M.; Limbocker, R. Therapeutic strategies to reduce the toxicity of misfolded protein oligomers. Int. J. Mol. Sci 2020, 21, 8651. [Google Scholar] [CrossRef]
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 2012, 125, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Iuchi, K. Manipulation of cell fate by fatty acids and oxidized fatty acids. Agric. Biotechnol. 2021, 5, 38–42. [Google Scholar]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef]
- Whittington, R.A.; Planel, E.; Terrando, N. Impaired resolution of inflammation in alzheimer’s disease: A review. Front. Immunol. 2017, 8, 1464. [Google Scholar] [CrossRef]
- Hunyadi, A. The mechanism(s) of action of antioxidants: From scavenging reactive oxygen/nitrogen species to redox signaling and the generation of bioactive secondary metabolites. Med. Res. Rev. 2019, 39, 2505–2533. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Baroni, M.; Mangialasche, F.; Mecocci, P. Vitamin E family: Role in the pathogenesis and treatment of Alzheimer’s disease. Alzheimers Dement. Dement. Transl. Res. Clin. Interv. 2016, 2, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hossain, S.; Al Mamun, A.; Matsuzaki, K.; Arai, H. Docosahexaenoic acid: One molecule diverse functions. Crit. Rev. Biotechnol. 2017, 37, 579–597. [Google Scholar] [CrossRef]
- Greenberg, J.A.; Bell, S.J.; Ausdal, W.V. Omega-3 Fatty Acid supplementation during pregnancy. Rev. Obstet. Gynecol. 2008, 1, 162–169. [Google Scholar]
- Park, Y.H.; Shin, S.J.; Kim, H.S.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.J.; Lim, K.; Kim, J.S.; Kim, J.I.; et al. Omega-3 Fatty acid-type docosahexaenoic acid protects against abeta-mediated mitochondrial deficits and pathomechanisms in alzheimer’s disease-related animal model. Int. J. Mol. Sci. 2020, 21, 3879. [Google Scholar] [CrossRef]
- Massey, K.A.; Nicolaou, A. Lipidomics of oxidized polyunsaturated fatty acids. Free Radic. Biol. Med. 2013, 59, 45–55. [Google Scholar] [CrossRef]
- Iuchi, K.; Ema, M.; Suzuki, M.; Yokoyama, C.; Hisatomi, H. Oxidized unsaturated fatty acids induce apoptotic cell death in cultured cells. Mol. Med. Rep. 2019, 19, 2767–2773. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Maitin, V.; Parathath, S.; Andreo, U.; Lin, S.X.; St Germain, C.; Yao, Z.; Maxfield, F.R.; Williams, K.J.; Fisher, E.A. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: A pathway for late-stage quality control. Proc. Natl. Acad. Sci. USA 2008, 105, 5862–5867. [Google Scholar] [CrossRef]
- Shin, S.K.; Kim, J.H.; Lee, J.H.; Son, Y.H.; Lee, M.W.; Kim, H.J.; Noh, S.A.; Kim, K.P.; Kim, I.G.; Lee, M.J. Docosahexaenoic acid-mediated protein aggregates may reduce proteasome activity and delay myotube degradation during muscle atrophy in vitro. Exp. Mol. Med. 2017, 49, e287. [Google Scholar] [CrossRef] [PubMed]
- Tully, A.M.; Roche, H.M.; Doyle, R.; Fallon, C.; Bruce, I.; Lawlor, B.; Coakley, D.; Gibney, M.J. Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: A case-control study. Br. J. Nutr. 2003, 89, 483–489. [Google Scholar] [CrossRef]
- Song, C.; Shieh, C.H.; Wu, Y.S.; Kalueff, A.; Gaikwad, S.; Su, K.P. The role of omega-3 polyunsaturated fatty acids eicosapentaenoic and docosahexaenoic acids in the treatment of major depression and Alzheimer’s disease: Acting separately or synergistically? Prog. Lipid Res. 2016, 62, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 fatty acids and neurodegenerative diseases: New evidence in clinical trials. Int. J. Mol. Sci. 2019, 20, 4256. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Cui, J.G.; Marcheselli, V.L.; Bodker, M.; Botkjaer, A.; Gotlinger, K.; Serhan, C.N.; Bazan, N.G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shibata, T.; Hisaka, S.; Kawai, Y.; Osawa, T. DHA Hydroperoxides as a potential inducer of neuronal cell death: A mitochondrial dysfunction-mediated pathway. J. Clin. Biochem. Nutr. 2008, 43, 26–33. [Google Scholar] [CrossRef][Green Version]
- Fecchio, C.; Palazzi, L.; de Laureto, P.P. Alpha-Synuclein and polyunsaturated fatty acids: Molecular Basis of the interaction and implication in neurodegeneration. Molecules 2018, 23, 1531. [Google Scholar] [CrossRef]
- De Franceschi, G.; Frare, E.; Pivato, M.; Relini, A.; Penco, A.; Greggio, E.; Bubacco, L.; Fontana, A.; de Laureto, P.P. Structural and morphological characterization of aggregated species of alpha-synuclein induced by docosahexaenoic acid. J. Biol. Chem. 2011, 286, 22262–22274. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-Joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef] [PubMed]
- Hirahashi, J. Omega-3 polyunsaturated fatty acids for the treatment of IgA nephropathy. J. Clin. Med. 2017, 6, 70. [Google Scholar] [CrossRef]
- Ishikado, A.; Morino, K.; Nishio, Y.; Nakagawa, F.; Mukose, A.; Sono, Y.; Yoshioka, N.; Kondo, K.; Sekine, O.; Yoshizaki, T.; et al. 4-Hydroxy hexenal derived from docosahexaenoic acid protects endothelial cells via Nrf2 activation. PLoS ONE 2013, 8, e69415. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Cui, W.; Guo, W.; Liu, H.; Luo, J.; Zhao, L.; Guo, H.; Zheng, L.; Bai, H.; Feng, D.; et al. Acrolein aggravates secondary brain injury after intracerebral hemorrhage through drp1-mediated mitochondrial oxidative damage in mice. Neurosci. Bull. 2020, 36, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, A.; Pierce, G.N.; Ravandi, A. Phospholipid oxidation products in ferroptotic myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H156–H163. [Google Scholar] [CrossRef]
- Matveychuk, D.; Dursun, S.M.; Wood, P.L.; Baker, G.B. Reactive aldehydes and neurodegenerative disorders. Klin. Psikofarmakol. Bülteni Bull. Clin. Psychopharmacol. 2016, 21, 277–288. [Google Scholar] [CrossRef]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3, 2. [Google Scholar] [CrossRef]
- Lizak, B.; Birk, J.; Zana, M.; Kosztyi, G.; Kratschmar, D.V.; Odermatt, A.; Zimmermann, R.; Geiszt, M.; Appenzeller-Herzog, C.; Banhegyi, G. Ca(2+) mobilization-dependent reduction of the endoplasmic reticulum lumen is due to influx of cytosolic glutathione. BMC Biol. 2020, 18, 19. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Alza, N.P.; Iglesias Gonzalez, P.A.; Conde, M.A.; Uranga, R.M.; Salvador, G.A. Lipids at the crossroad of alpha-synuclein function and dysfunction: Biological and pathological implications. Front. Cell. Neurosci. 2019, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef]
- Sheng, J.; Olrichs, N.K.; Gadella, B.M.; Kaloyanova, D.V.; Helms, J.B. Regulation of functional protein aggregation by multiple factors: Implications for the amyloidogenic behavior of the CAP Superfamily proteins. Int. J. Mol. Sci. 2020, 21, 6530. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Allsop, D. Alpha-synuclein interacts with lipoproteins in plasma. J. Mol. Neurosci. 2017, 63, 165–172. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving life through death: Redox biology of lipid peroxidation in ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and its potential role in human diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef]
- Kuch, E.M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brugger, B.; Sreemmel, W.; Ehehalt, R.; Poppelreuther, M.; Fullekrug, J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 2014, 1841, 227–239. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its role in diverse brain diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Corti, O.; Blomgren, K.; Poletti, A.; Beart, P.M. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J. Neurochem. 2020, 154, 354–371. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Gao, X.; Xu, H.; Cui, Y.; Zhang, Y.; Gou, X. The emerging roles of ferroptosis in huntington’s disease. Neuromol. Med. 2019, 21, 110–119. [Google Scholar] [CrossRef]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef]
- Huang, L.; McClatchy, D.B.; Maher, P.; Liang, Z.; Diedrich, J.K.; Soriano-Castell, D.; Goldberg, J.; Shokhirev, M.; Yates, J.R., 3rd; Schubert, D.; et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis. 2020, 11, 828. [Google Scholar] [CrossRef]
- Thayyullathil, F.; Cheratta, A.R.; Alakkal, A.; Subburayan, K.; Pallichankandy, S.; Hannun, Y.A.; Galadari, S. Acid sphingomyelinase-dependent autophagic degradation of GPX4 is critical for the execution of ferroptosis. Cell Death Dis. 2021, 12, 26. [Google Scholar] [CrossRef]
- Pujol-Lereis, L.M. Alteration of sphingolipids in biofluids: Implications for neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 3564. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Hwang, S.W. Modulation of the activities of neuronal ion channels by fatty acid-derived pro-resolvents. Front. Physiol. 2016, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chan, S.L.; Fu, W.; Mattson, M.P. The lipid peroxidation product 4-hydroxynonenal facilitates opening of voltage-dependent Ca2+ channels in neurons by increasing protein tyrosine phosphorylation. J. Biol. Chem. 2002, 277, 24368–24375. [Google Scholar] [CrossRef]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Weber, J.; Schmitt, A.; Stroh, J.; Hailfinger, S.; von Karstedt, S.; Garcia-Saez, A.J. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2020, 1–14. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef] [PubMed]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenerg. 2017, 1858, 615–627. [Google Scholar] [CrossRef]
- Yu, W.; Jin, H.; Huang, Y. Mitochondria-associated membranes (MAMs): A potential therapeutic target for treating Alzheimer’s disease. Clin. Sci. 2021, 135, 109–126. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The interplay between Ca(2+) signaling pathways and neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef]
- Van Laar, V.S.; Berman, S.B. Mitochondrial dynamics in Parkinson’s disease. Exp. Neurol. 2009, 218, 247–256. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective effect of antioxidants in the brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Takahashi, M.; Takahashi, K. Water-soluble CoQ10 as a promising anti-aging agent for neurological dysfunction in brain mitochondria. Antioxidants 2019, 8, 61. [Google Scholar] [CrossRef]
- Osuch, B.; Kucharska, T.; Chmielewska, N.; Maciejak, P.; Szyndler, J.; Płaźnik, A. The role of mitophagy in selected neurodegenerative diseases. Postępy Psychiatr. I Neurol. 2019, 28, 154–161. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bakovic, M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014, 2014, 709828. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 122–126. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Shalaeva, D.N.; Lyamzaev, K.G.; Chernyak, B.V. Does Oxidation of mitochondrial cardiolipin trigger a chain of antiapoptotic reactions? Biochem. Mosc. 2018, 83, 1263–1278. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Kaufman, D.M.; Wu, X.; Scott, B.A.; Itani, O.A.; Van Gilst, M.R.; Bruce, J.E.; Crowder, C.M. Ageing and hypoxia cause protein aggregation in mitochondria. Cell Death Differ. 2017, 24, 1730–1738. [Google Scholar] [CrossRef]
- Biel, T.G.; Aryal, B.; Gerber, M.H.; Trevino, J.G.; Mizuno, N.; Rao, V.A. Mitochondrial dysfunction generates aggregates that resist lysosomal degradation in human breast cancer cells. Cell Death Dis. 2020, 11, 460. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates alpha-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Kobayashi, S.; Seo, H.G. Mutual interaction between oxidative stress and endoplasmic reticulum stress in the pathogenesis of diseases specifically focusing on non-alcoholic fatty liver disease. World J. Biol. Chem. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Kaneko, M.; Imaizumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER stress and disease: Toward prevention and treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef]
- Costa, C.A.D.; Manaa, W.E.; Duplan, E.; Checler, F. The endoplasmic reticulum stress/unfolded protein response and their contributions to parkinson’s disease physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J., 3rd; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Gagliardi, M.; Cotella, D.; Santoro, C.; Cora, D.; Barlev, N.A.; Piacentini, M.; Corazzari, M. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019, 10, 902. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Doval, M.; Majerova, Z.; Lehotsky, J.; Racay, P. Iron-induced lipid peroxidation and protein modification in endoplasmic reticulum membranes. Protection by stobadine. Int. J. Biochem. Cell Biol. 2000, 32, 539–547. [Google Scholar] [CrossRef]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, A.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell. 2010, 40, 787–797. [Google Scholar] [CrossRef]
- Kanemura, S.; Sofia, E.F.; Hirai, N.; Okumura, M.; Kadokura, H.; Inaba, K. Characterization of the endoplasmic reticulum-resident peroxidases GPx7 and GPx8 shows the higher oxidative activity of GPx7 and its linkage to oxidative protein folding. J. Biol. Chem. 2020, 295, 12772–12785. [Google Scholar] [CrossRef]
- Sato, Y.; Kojima, R.; Okumura, M.; Hagiwara, M.; Masui, S.; Maegawa, K.; Saiki, M.; Horibe, T.; Suzuki, M.; Inaba, K. Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 2013, 3, 2456. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Oses-Prieto, J.A.; Pope, L.E.; Burlingame, A.L.; Dixon, S.J.; Renslo, A.R. Reactivity-based probe of the iron(II)-dependent interactome identifies new cellular modulators of ferroptosis. J. Am. Chem. Soc. 2020, 142, 19085–19093. [Google Scholar] [CrossRef]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell. Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Ashabi, G.; Alamdary, S.Z.; Ramin, M.; Khodagholi, F. Reduction of hippocampal apoptosis by intracerebroventricular administration of extracellular signal-regulated protein kinase and/or p38 inhibitors in amyloid beta rat model of Alzheimer’s disease: Involvement of nuclear-related factor-2 and nuclear factor-kappaB. Basic Clin. Pharmacol. Toxicol. 2013, 112, 145–155. [Google Scholar] [CrossRef]
- Mota, S.I.; Costa, R.O.; Ferreira, I.L.; Santana, I.; Caldeira, G.L.; Padovano, C.; Fonseca, A.C.; Baldeiras, I.; Cunha, C.; Letra, L.; et al. Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1852, 1428–1441. [Google Scholar] [CrossRef]
- Mukaigasa, K.; Tsujita, T.; Nguyen, V.T.; Li, L.; Yagi, H.; Fuse, Y.; Nakajima-Takagi, Y.; Kato, K.; Yamamoto, M.; Kobayashi, M. Nrf2 activation attenuates genetic endoplasmic reticulum stress induced by a mutation in the phosphomannomutase 2 gene in zebrafish. Proc. Natl. Acad. Sci. USA 2018, 115, 2758–2763. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, N.Y.; Cho, D.H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef]
- Uzor, N.E.; McCullough, L.D.; Tsvetkov, A.S. Peroxisomal dysfunction in neurological diseases and brain aging. Front. Cell. Neurosci. 2020, 14, 44. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G. Peroxisome: The new player in ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 273. [Google Scholar] [CrossRef]
- Cui, W.; Liu, D.; Gu, W.; Chu, B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar alpha-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020, 295, 17497–17513. [Google Scholar] [CrossRef] [PubMed]
- Kaemmerer, E.; Schutt, F.; Krohne, T.U.; Holz, F.G.; Kopitz, J. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1342–1347. [Google Scholar] [CrossRef]
- Alborzinia, H.; Ignashkova, T.I.; Dejure, F.R.; Gendarme, M.; Theobald, J.; Wolfl, S.; Lindemann, R.K.; Reiling, J.H. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun. Biol. 2018, 1, 210. [Google Scholar] [CrossRef]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. Alpha-synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef] [PubMed]
- Elbaum-Garfinkle, S. Matter over mind: Liquid phase separation and neurodegeneration. J. Biol. Chem. 2019, 294, 7160–7168. [Google Scholar] [CrossRef]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative diseases: Regenerative mechanisms and novel therapeutic approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef]
- Cragnolini, A.B.; Lampitella, G.; Virtuoso, A.; Viscovo, I.; Panetsos, F.; Papa, M.; Cirillo, G. Regional brain susceptibility to neurodegeneration: What is the role of glial cells? Neural Regen. Res. 2020, 15, 838–842. [Google Scholar] [CrossRef]
- Joselow, M.M.; Troiano, R.A.; Bogden, J.D. Copper, zinc, magnesium, and calcium in plasma and cerebrospinal fluid of patients with neurological diseases. Clin. Chem. 1977, 23, 485–489. [Google Scholar] [CrossRef]
- Parnetti, L.; Castrioto, A.; Chiasserini, D.; Persichetti, E.; Tambasco, N.; El-Agnaf, O.; Calabresi, P. Cerebrospinal fluid biomarkers in Parkinson disease. Nat. Rev. Neurol. 2013, 9, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.Y.; Xu, X.; Li, X.C. Cardiovascular diseases: Oxidative damage and antioxidant protection. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3091–3096. [Google Scholar]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Littarru, G.P.; Tiano, L. Clinical aspects of coenzyme Q10: An update. Nutrition 2010, 26, 250–254. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, A. Protective effect of edaravone on cyclophosphamide induced oxidative stress and neurotoxicity in rats. Curr. Drug Saf. 2019, 14, 209–216. [Google Scholar] [CrossRef]
- Catalgol, B.; Ozer, N.K. Protective effects of vitamin E against hypercholesterolemia-induced age-related diseases. Genes Nutr. 2012, 7, 91–98. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Geir Moller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.S. Molecular mechanisms behind free radical scavengers function against oxidative stress. Antioxidants 2017, 6, 51. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.F.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’Autreaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic reticulum transport of glutathione by Sec61 is regulated by Ero1 and Bip. Mol. Cell. 2017, 67, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Camacho, J.D.; Bernier, M.; Lopez-Lluch, G.; Navas, P. Coenzyme Q10 supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Arenas-de Larriva, A.P.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; Lopez-Miranda, J. Coenzyme Q10 supplementation for the reduction of oxidative stress: Clinical implications in the treatment of chronic diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.M.; Manor, D. Vitamin E and neurodegeneration. Neurobiol. Dis. 2015, 84, 78–83. [Google Scholar] [CrossRef]
- Ogawa, S.; Shinkawa, M.; Hirase, R.; Tsubomura, T.; Iuchi, K.; Hara, S. Development of water-insoluble vehicle comprising natural cyclodextrin—vitamin E complex. Antioxidants 2021, 10, 490. [Google Scholar] [CrossRef]
- Agmon, E.; Stockwell, B.R. Lipid homeostasis and regulated cell death. Curr. Opin. Chem. Biol. 2017, 39, 83–89. [Google Scholar] [CrossRef]
- Maor, I.; Hayek, T.; Coleman, R.; Aviram, M. Plasma LDL oxidation leads to its aggregation in the atherosclerotic apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Hasanbasic, S.; Jahic, A.; Berbic, S.; Znidaric, M.T.; Zerovnik, E. Inhibition of protein aggregation by several antioxidants. Oxid. Med. Cell. Longev. 2018, 2018, 8613209. [Google Scholar] [CrossRef] [PubMed]
- Budimir, A. Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm. 2011, 61, 1–14. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in neurodegeneration - cause or consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in parkinson’s disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Bharathi; Indi, S.S.; Rao, K.S. Copper- and iron-induced differential fibril formation in alpha-synuclein: TEM study. Neurosci. Lett. 2007, 424, 78–82. [Google Scholar] [CrossRef]
- Mayes, J.; Tinker-Mill, C.; Kolosov, O.; Zhang, H.; Tabner, B.J.; Allsop, D. beta-amyloid fibrils in Alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J. Biol. Chem. 2014, 289, 12052–12062. [Google Scholar] [CrossRef]
- Han, J.; Lee, H.J.; Kim, K.Y.; Lee, S.J.C.; Suh, J.M.; Cho, J.; Chae, J.; Lim, M.H. Tuning Structures and Properties for Developing Novel Chemical Tools toward Distinct Pathogenic Elements in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 800–808. [Google Scholar] [CrossRef]
- Yi, Y.; Lin, Y.; Han, J.; Lee, H.J.; Park, N.; Nam, G.; Park, Y.S.; Lee, Y.-H.; Lim, M.H. Impact of sphingosine and acetylsphingosines on the aggregation and toxicity of metal-free and metal-treated amyloid-β. Chem. Sci. 2021, 12, 2456–2466. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc signal in brain diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Bowers, K.; Srai, S.K.S. The trafficking of metal ion transporters of the Zrt- and Irt-like protein family. Traffic 2018, 19, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Xu, K.; Yoo, J.; Chen, T.T.; Andrews, G.; Noebels, J.L. Knockout of Zn transporters Zip-1 and Zip-3 attenuates seizure-induced CA1 neurodegeneration. J. Neurosci. 2011, 31, 97–104. [Google Scholar] [CrossRef]
- Chen, P.H.; Wu, J.; Xu, Y.; Ding, C.C.; Mestre, A.A.; Lin, C.C.; Yang, W.H.; Chi, J.T. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis. 2021, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tian, H.; Li, X.; Mao, L.; Zhao, X.; Lin, J.; Lin, S.; Xu, C.; Liu, Y.; Guo, Y.; et al. Zinc promotes functional recovery after spinal cord injury by activating Nrf2/HO-1 defense pathway and inhibiting inflammation of NLRP3 in nerve cells. Life Sci. 2020, 245, 117351. [Google Scholar] [CrossRef]
- Kiechle, M.; Grozdanov, V.; Danzer, K.M. The role of lipids in the initiation of alpha-Synuclein misfolding. Front. Cell Dev. Biol. 2020, 8, 562241. [Google Scholar] [CrossRef] [PubMed]
- Warmlander, S.; Osterlund, N.; Wallin, C.; Wu, J.; Luo, J.; Tiiman, A.; Jarvet, J.; Graslund, A. Metal binding to the amyloid-beta peptides in the presence of biomembranes: Potential mechanisms of cell toxicity. J. Biol. Inorg. Chem. 2019, 24, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Bellezza, I. Oxidative stress in age-related macular degeneration: Nrf2 as Therapeutic target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular mechanism of cellular oxidative stress sensing by keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Roberts, B.R.; Bush, A.I.; Hare, D.J. Selenium, selenoproteins and neurodegenerative diseases. Metallomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Pitts, M.W.; Byrns, C.N.; Ogawa-Wong, A.N.; Kremer, P.; Berry, M.J. Selenoproteins in nervous system development and function. Biol. Trace Elem. Res. 2014, 161, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iuchi, K.; Takai, T.; Hisatomi, H. Cell Death via Lipid Peroxidation and Protein Aggregation Diseases. Biology 2021, 10, 399. https://doi.org/10.3390/biology10050399
Iuchi K, Takai T, Hisatomi H. Cell Death via Lipid Peroxidation and Protein Aggregation Diseases. Biology. 2021; 10(5):399. https://doi.org/10.3390/biology10050399
Chicago/Turabian StyleIuchi, Katsuya, Tomoka Takai, and Hisashi Hisatomi. 2021. "Cell Death via Lipid Peroxidation and Protein Aggregation Diseases" Biology 10, no. 5: 399. https://doi.org/10.3390/biology10050399
APA StyleIuchi, K., Takai, T., & Hisatomi, H. (2021). Cell Death via Lipid Peroxidation and Protein Aggregation Diseases. Biology, 10(5), 399. https://doi.org/10.3390/biology10050399