
Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors

 ,
,  , , , , and
, , , , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Marine Sponge Materials
2.2. Isolation of Scalarane-Based Metabolites 1–15
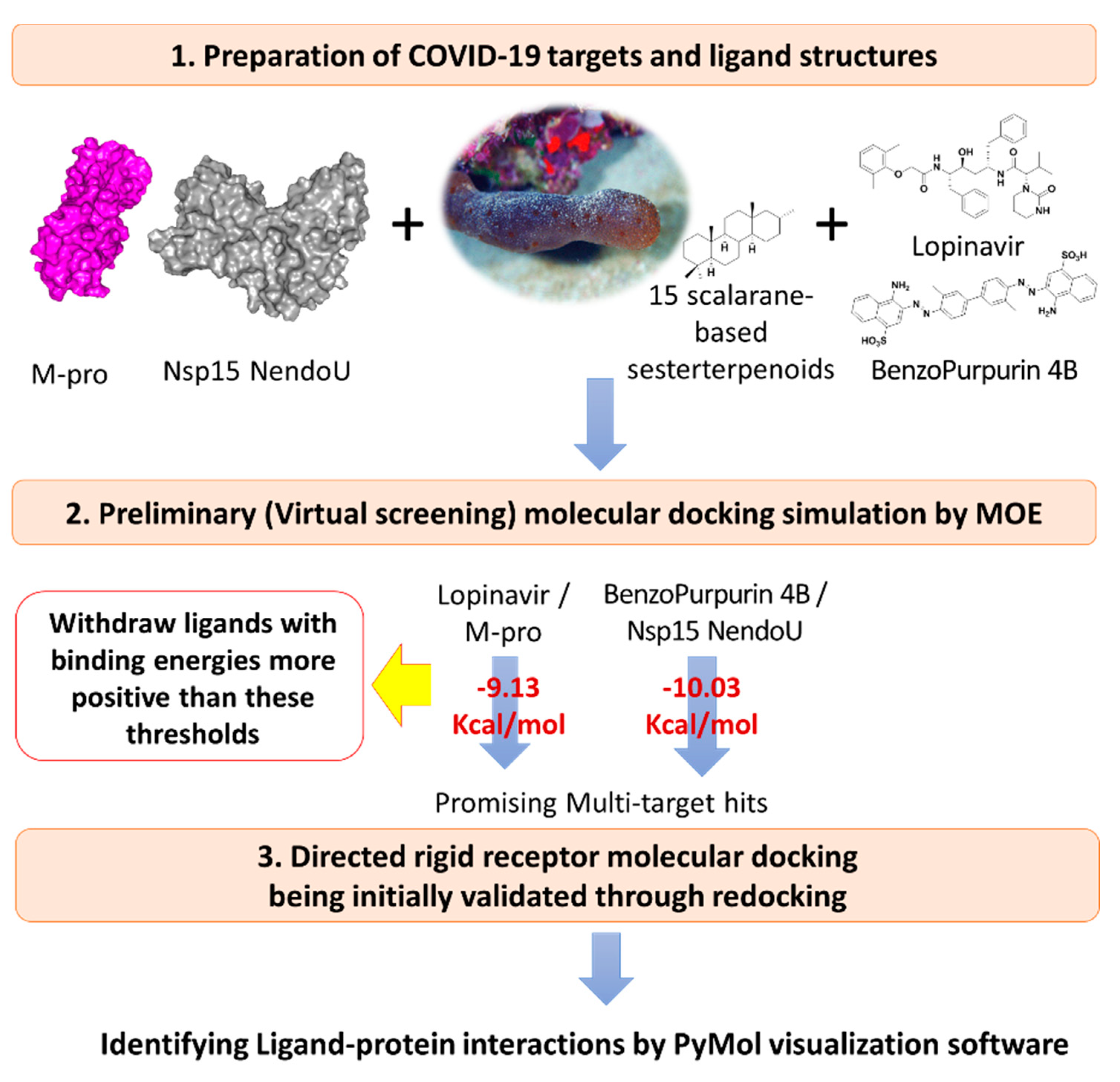
2.3. Multi-Target Docking Analysis of the Investigated Compounds
2.3.1. Ligand Construction and Protein Preparation
2.3.2. Molecular Docking Protocol
2.4. Molecular Dynamics Simulations
2.5. Drug-Likeness Evaluation and In Silico ADME/TOX Prediction
3. Results and Discussion
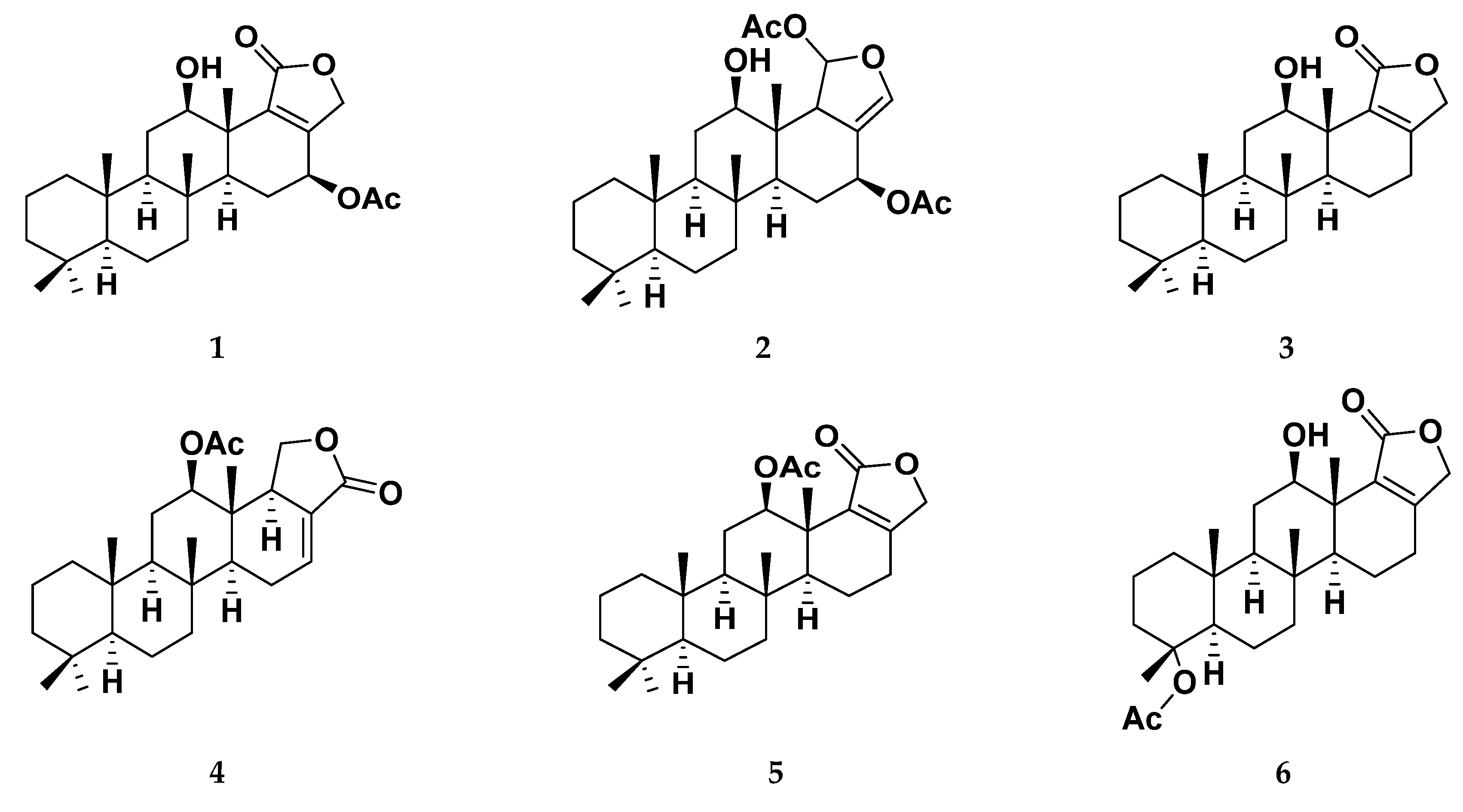
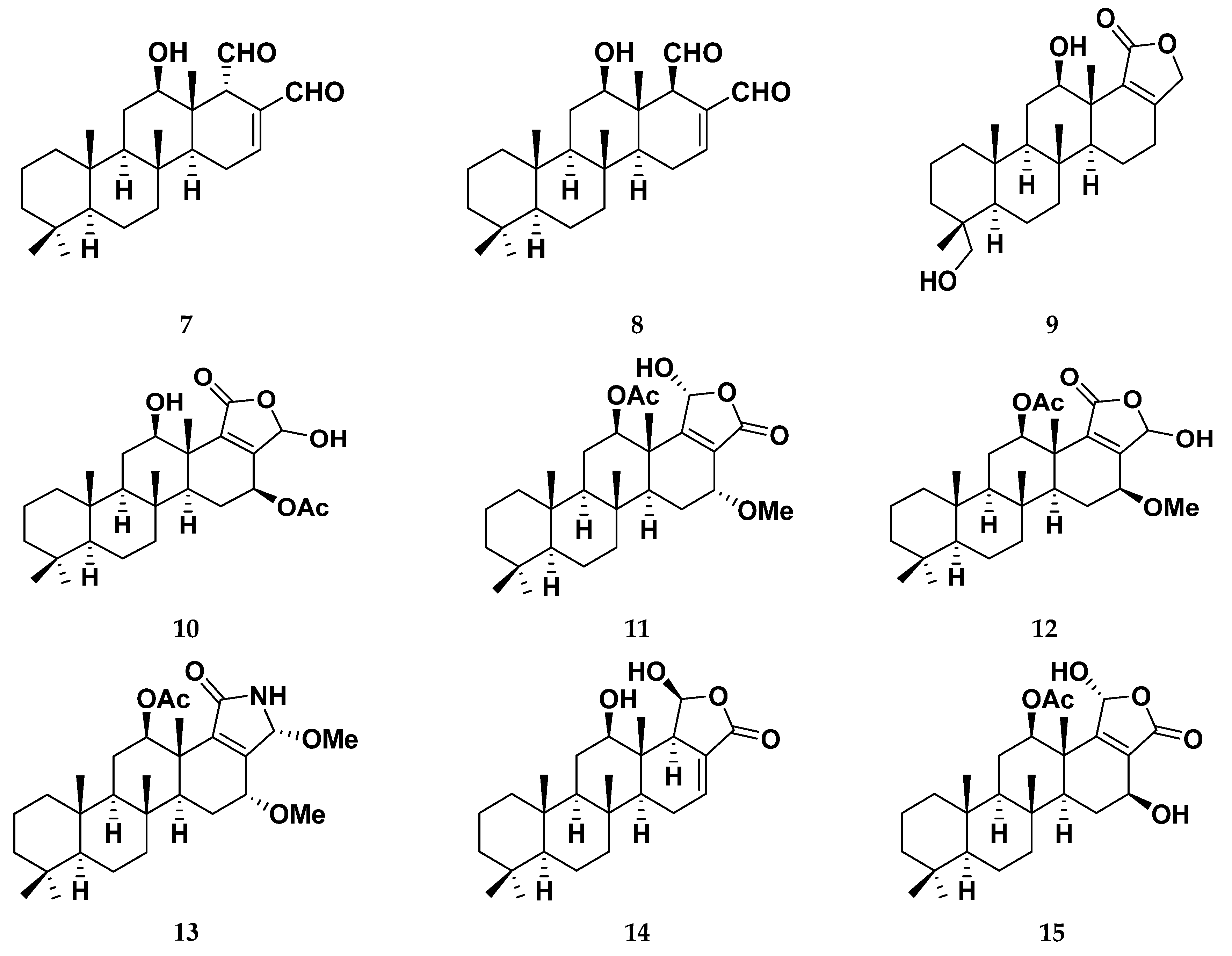
3.1. Isolation and Identification of Scalarane Metabolites 1–15
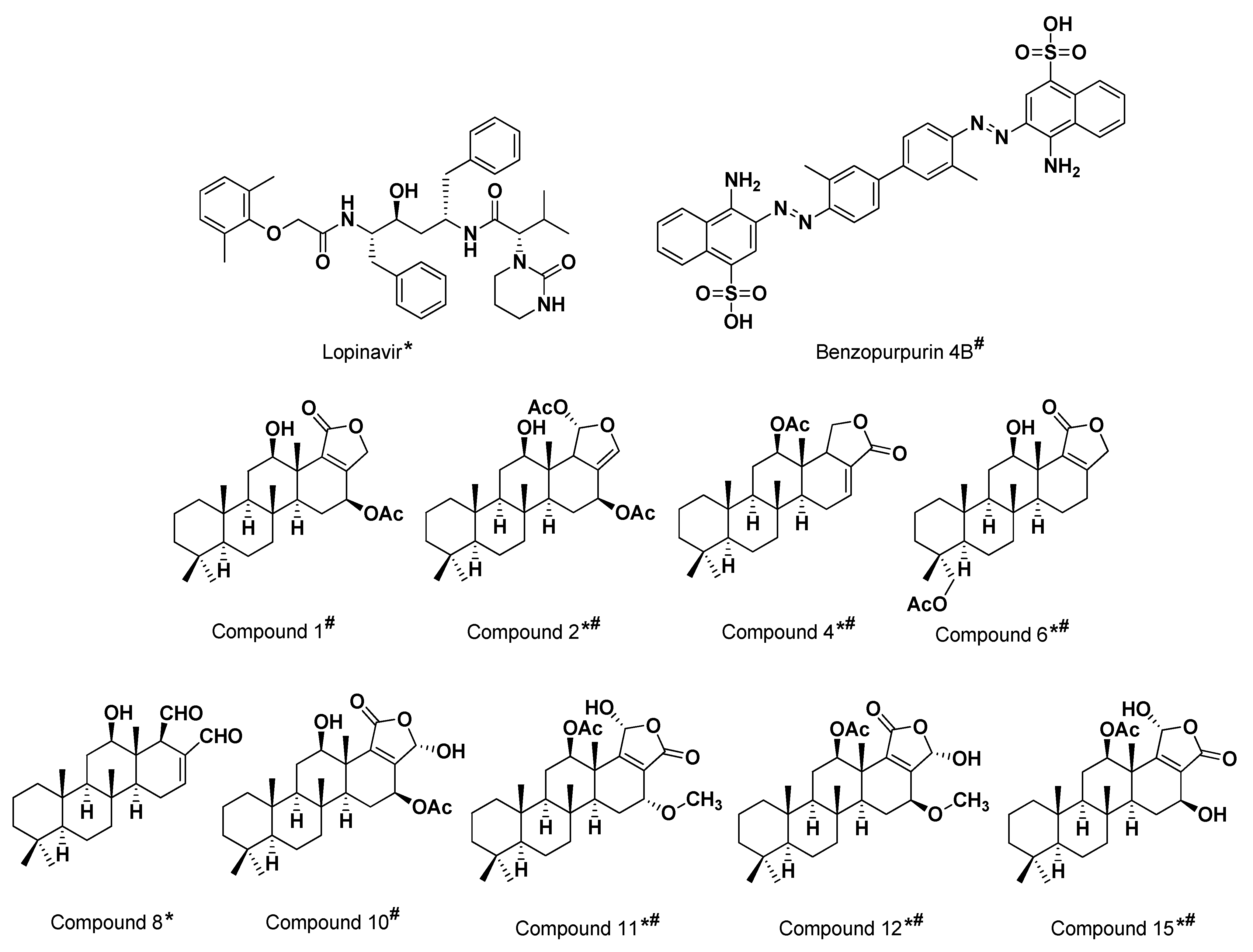
3.2. Multi-Target Molecular Docking and Structural-Based Activity Insights
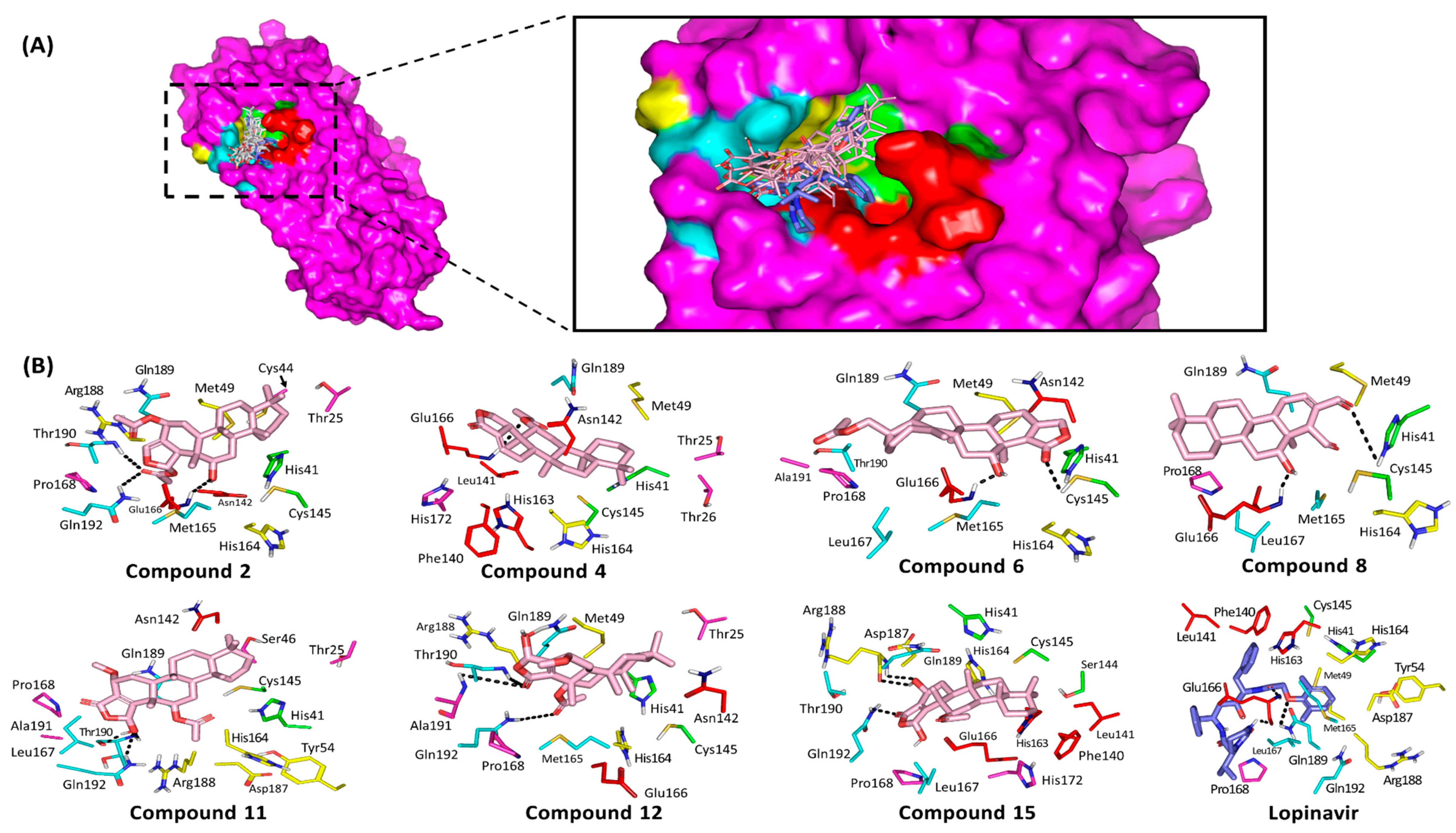
3.3. Ligand/M-Pro Binding Interaction Analysis
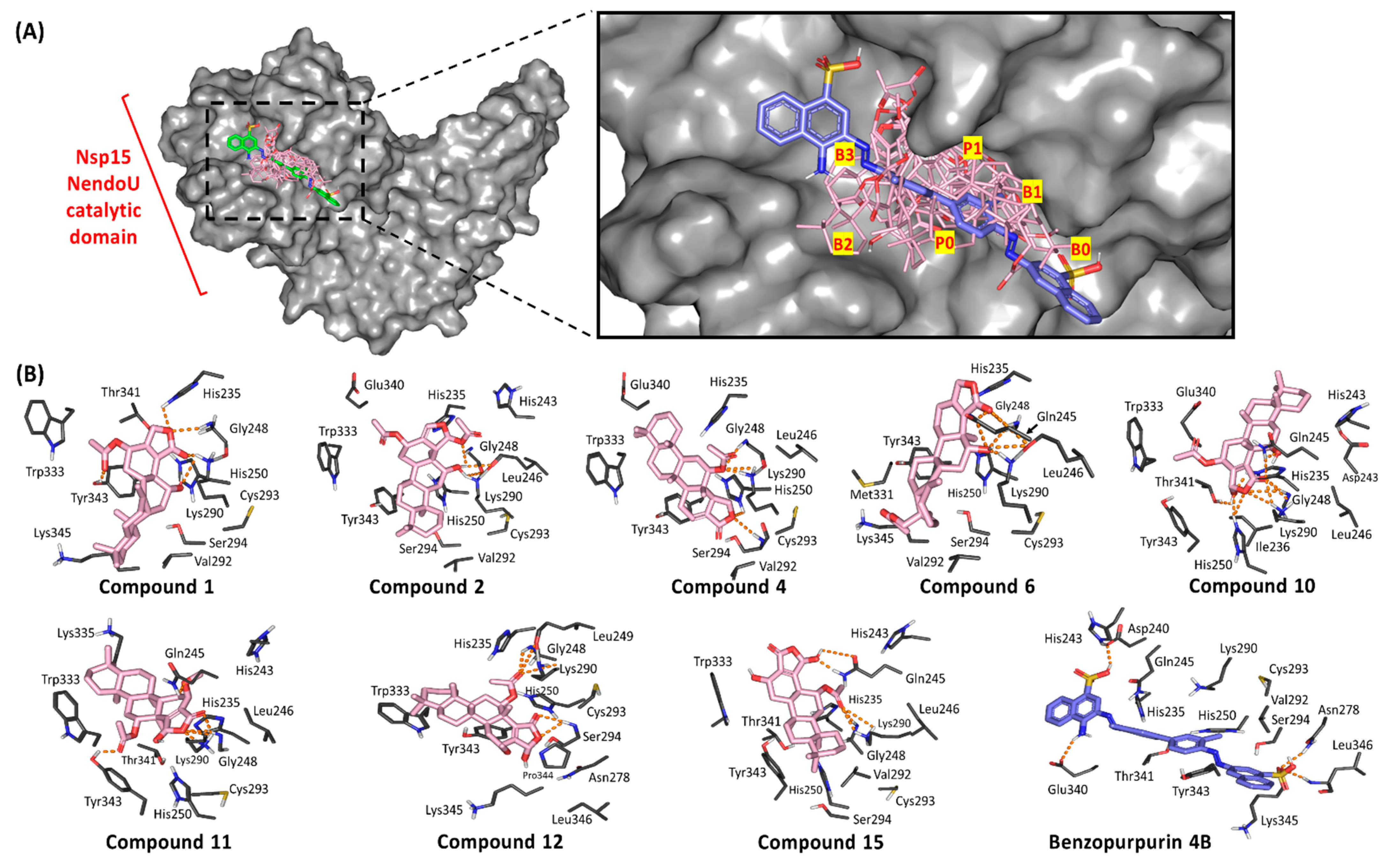
3.4. Ligand/Nsp15 NendoU Binding Interaction Analysis
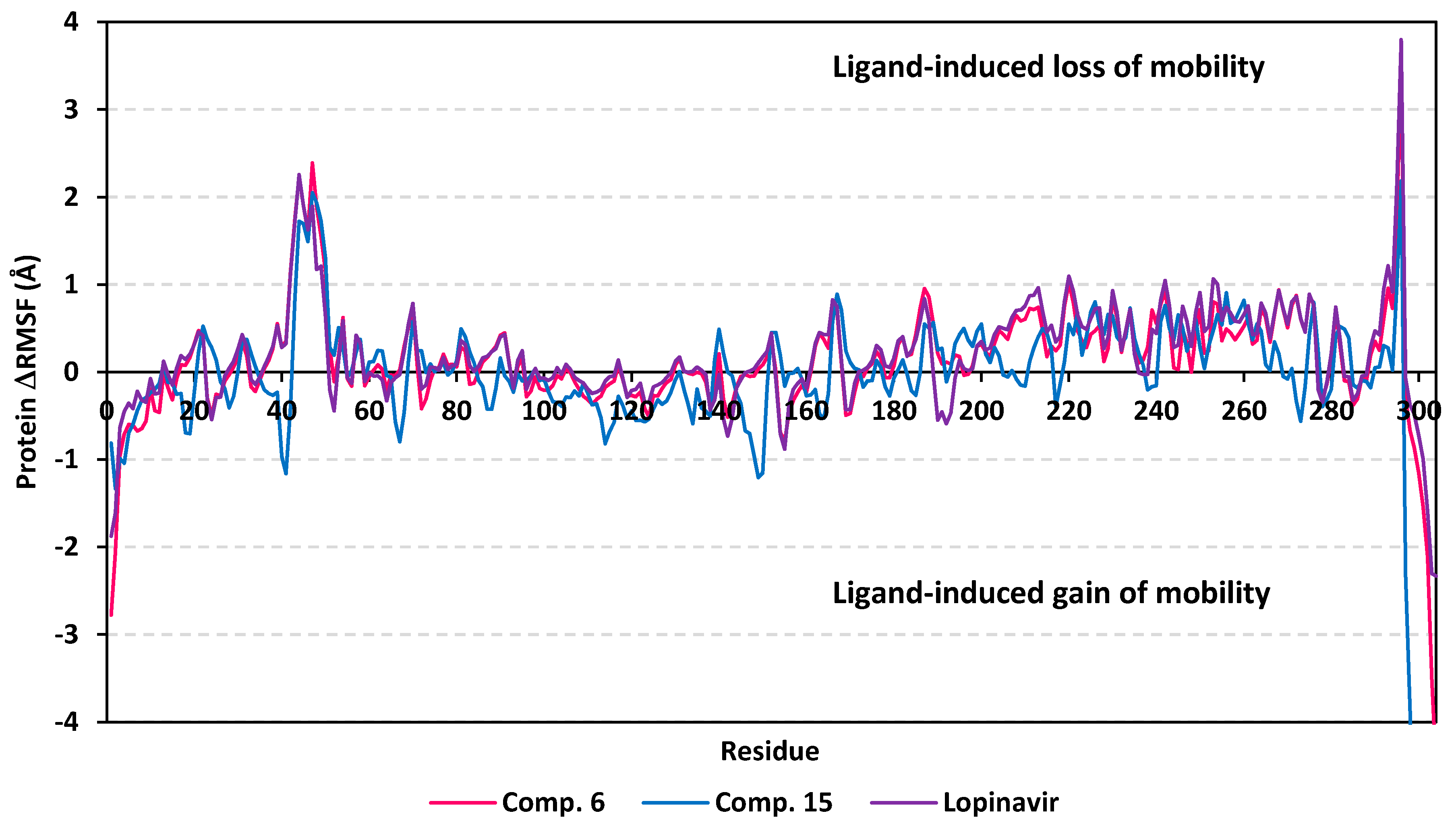
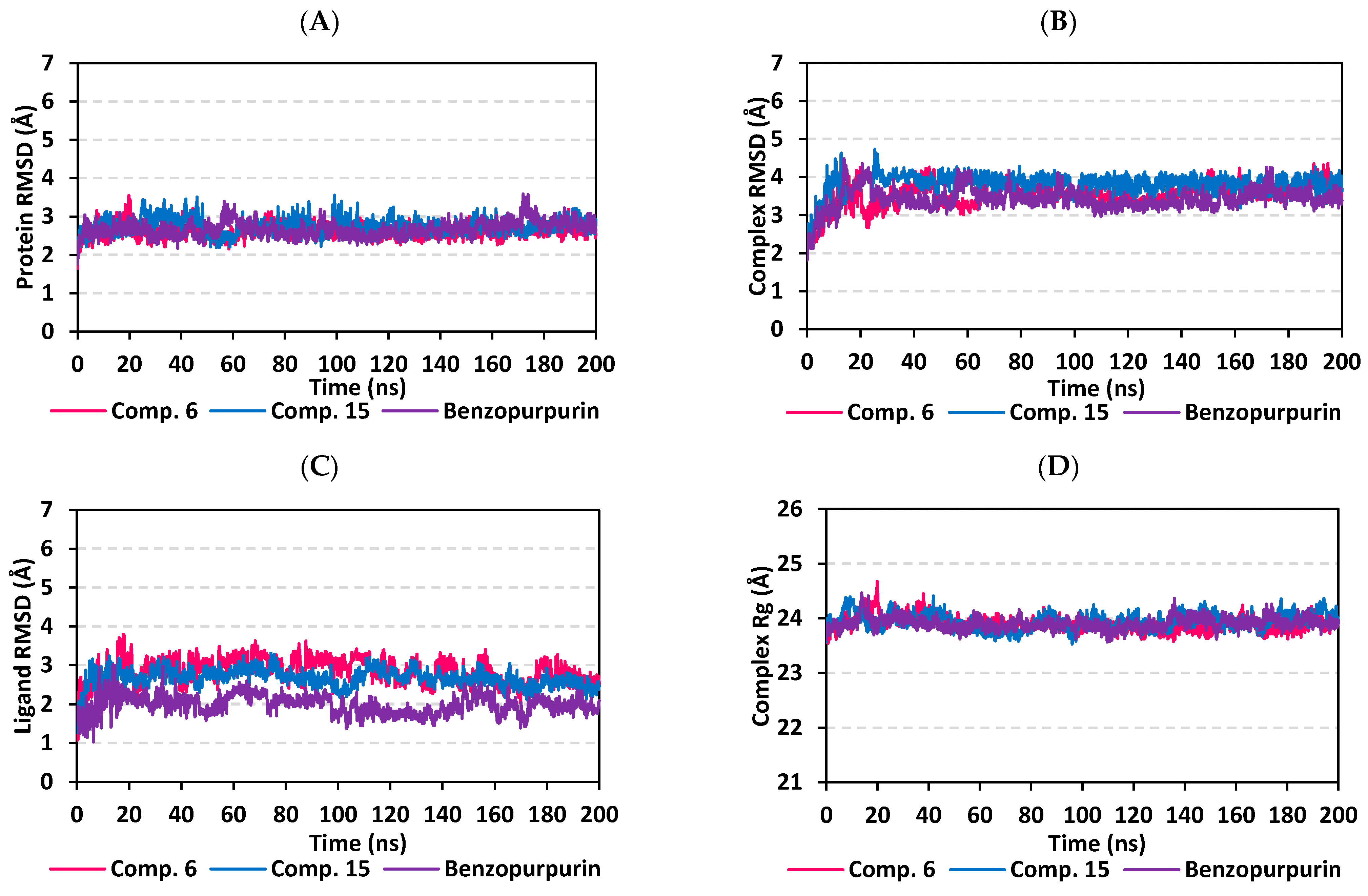
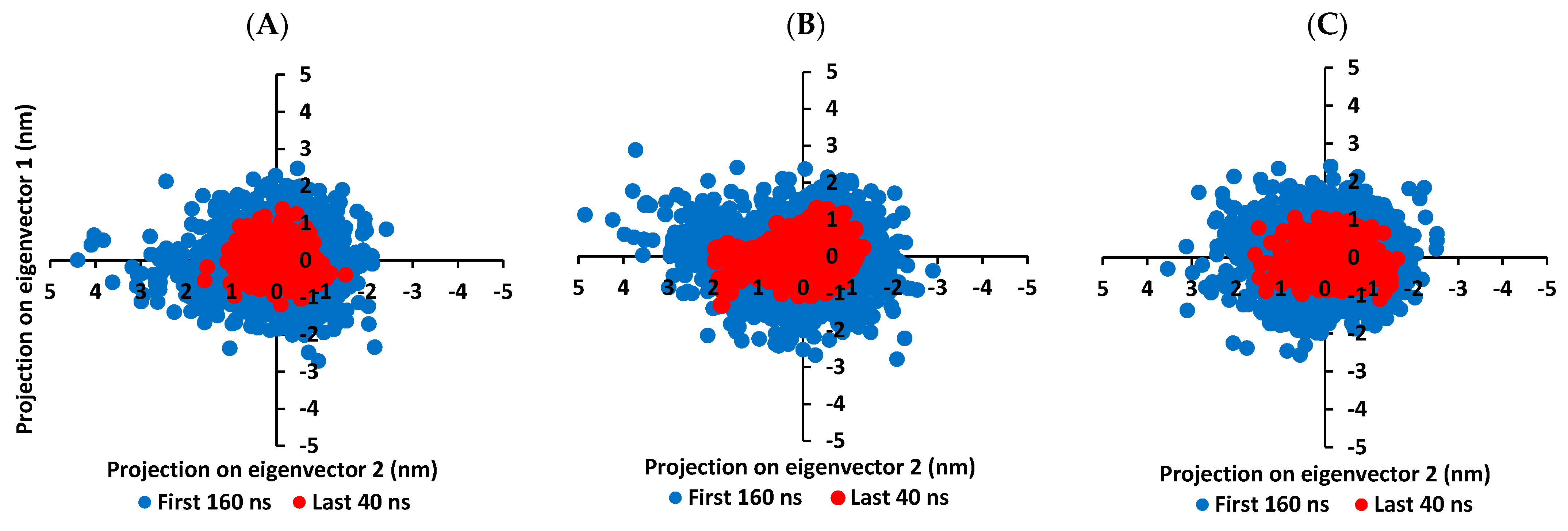
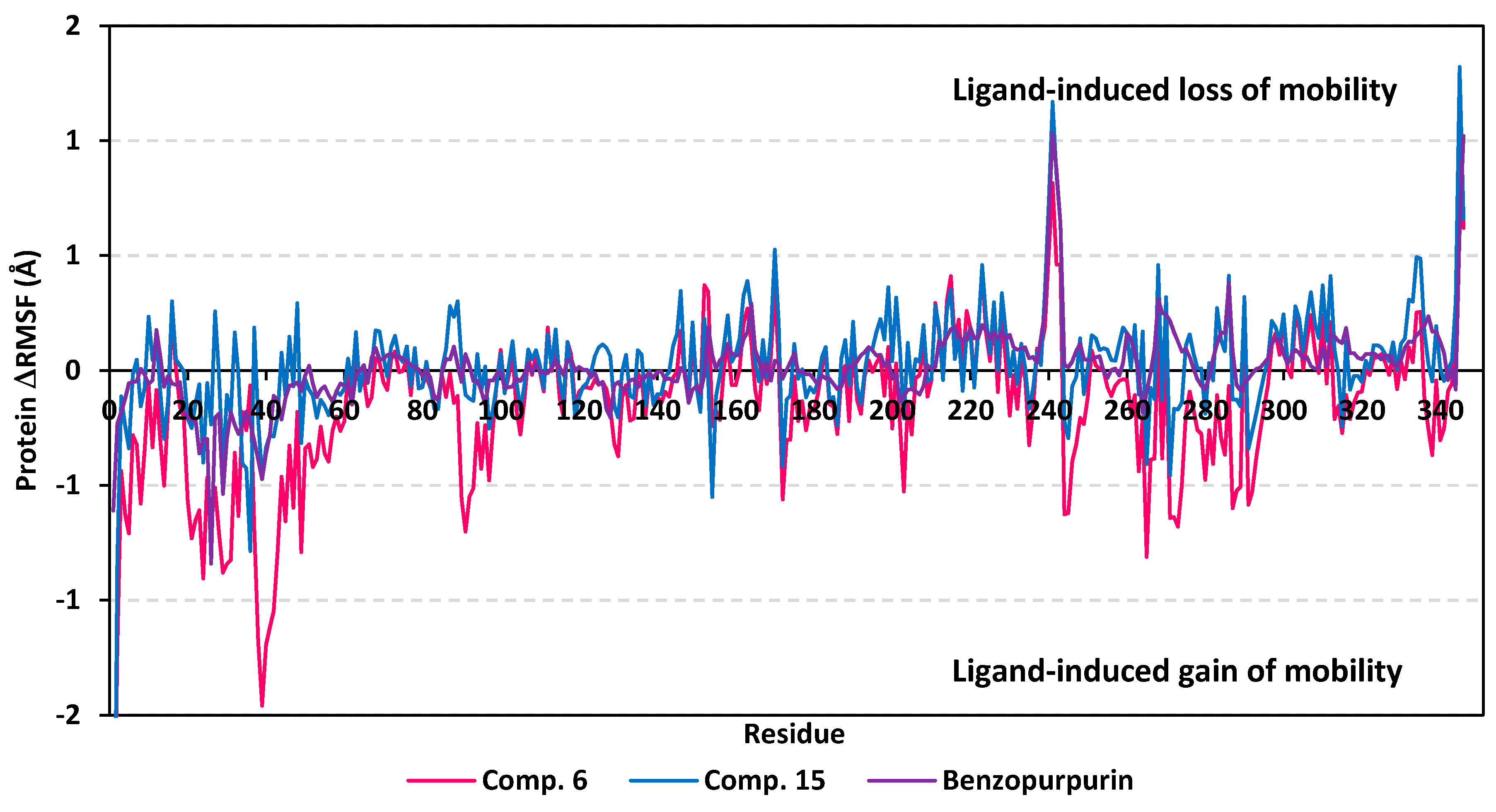
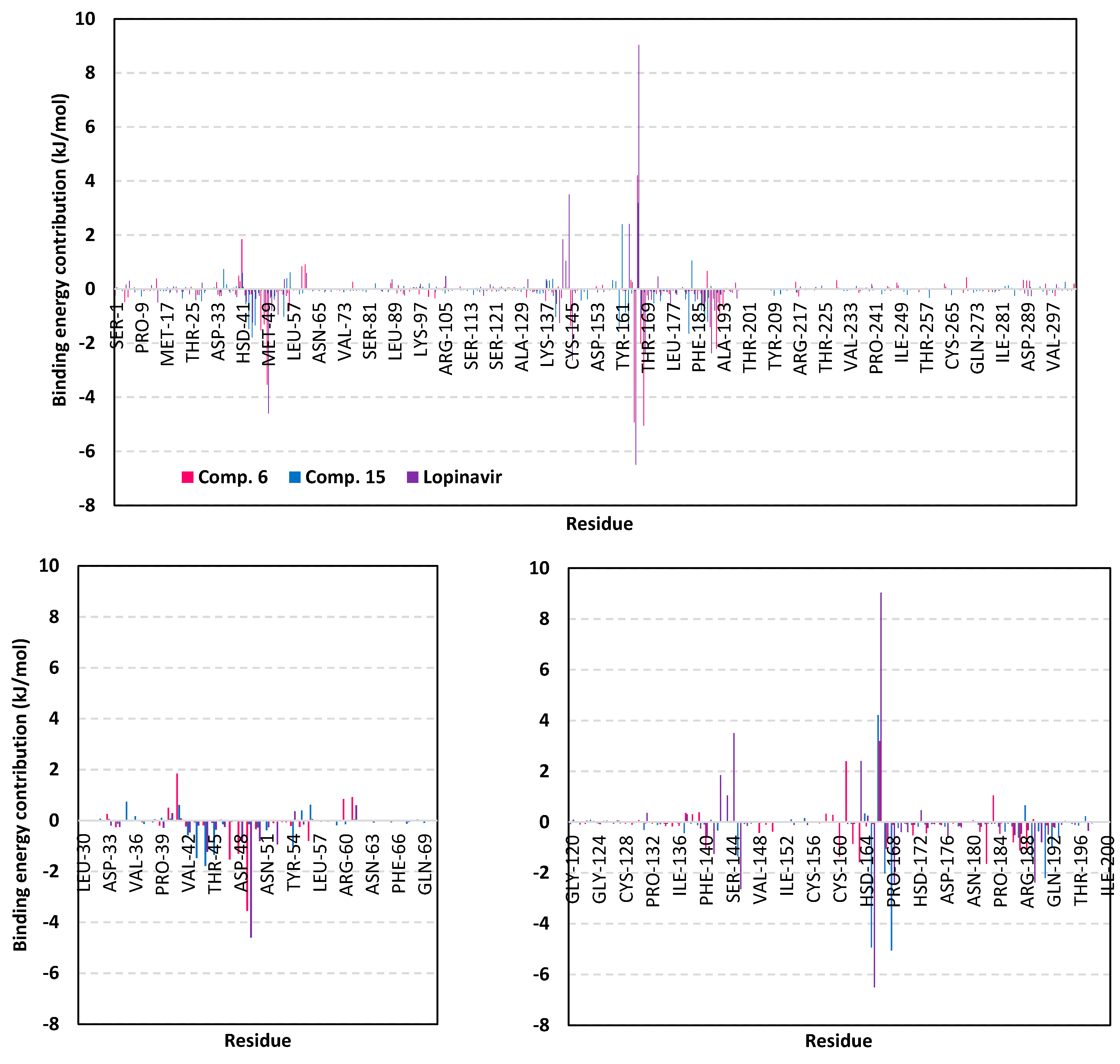
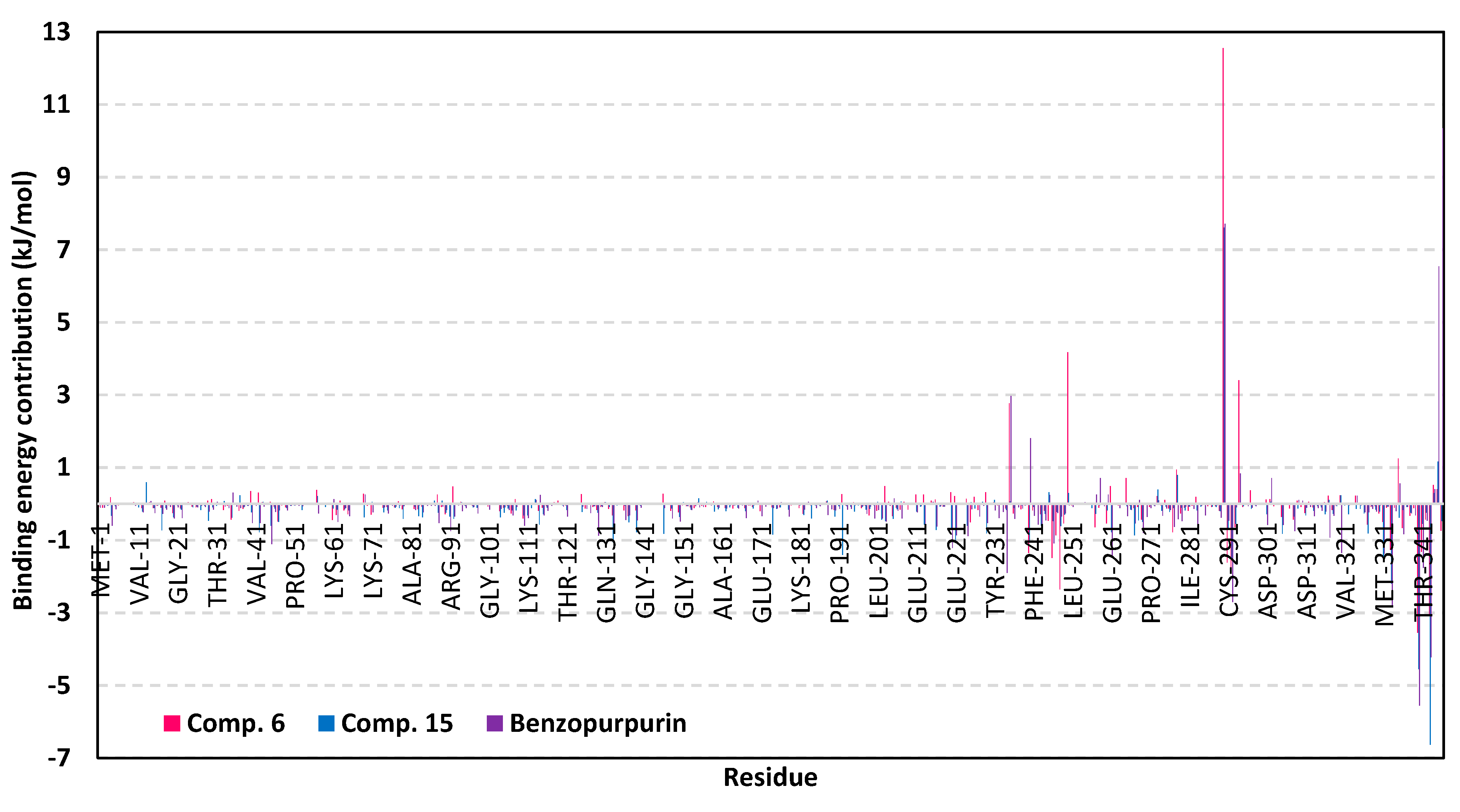
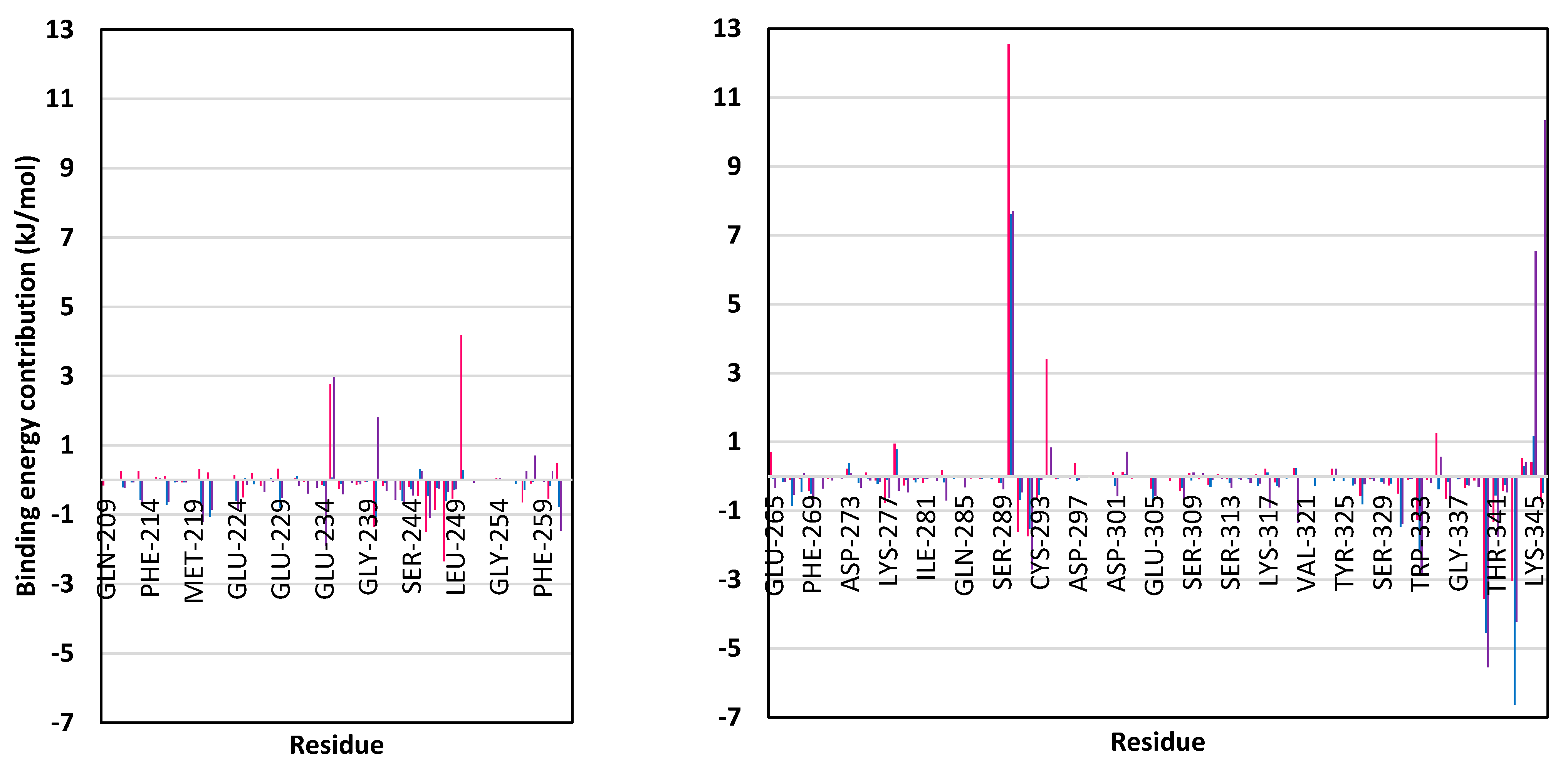
3.5. MD Simulation Analysis of Promising COVID-19 Multi-Target Inhibitor
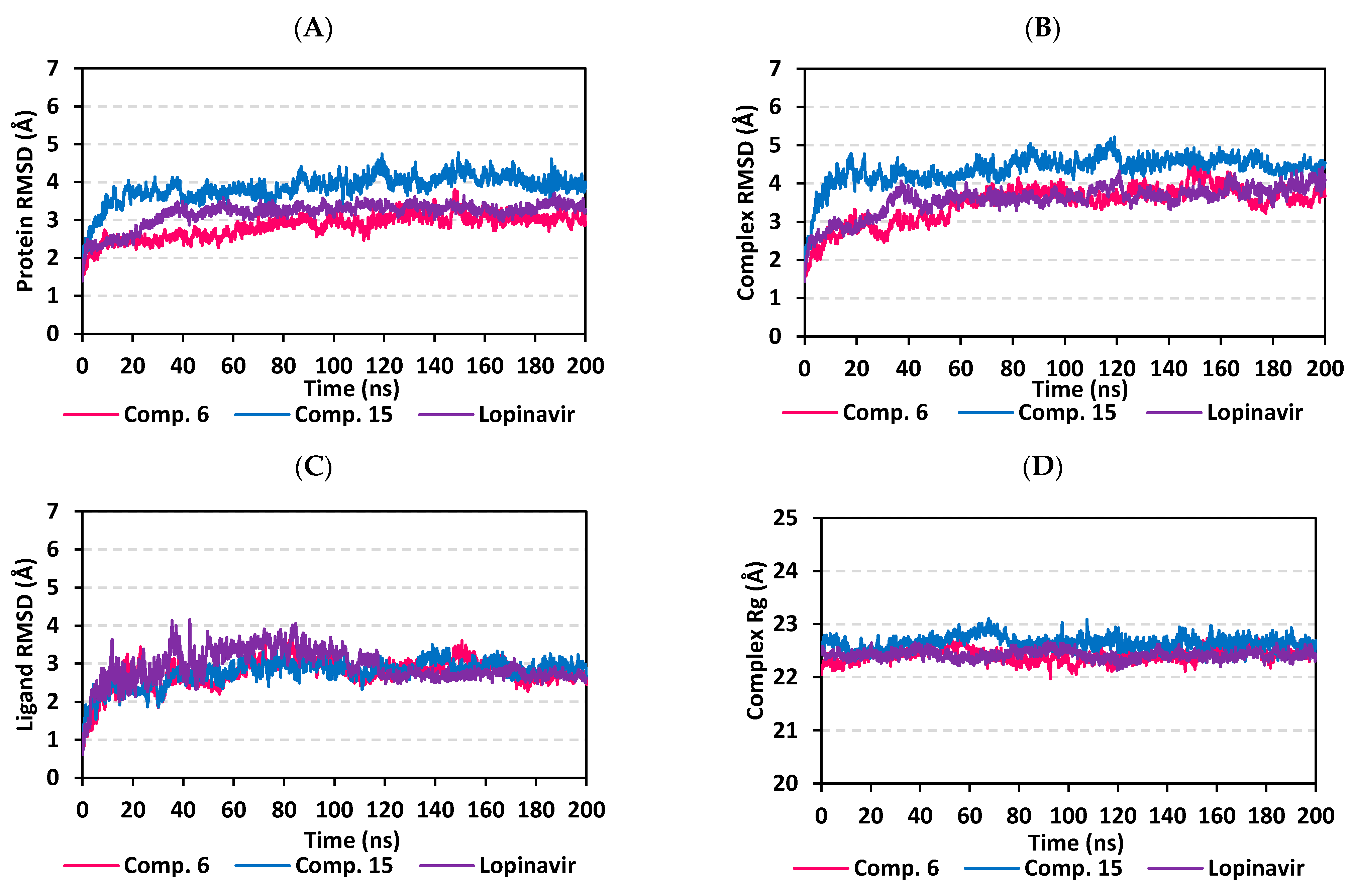
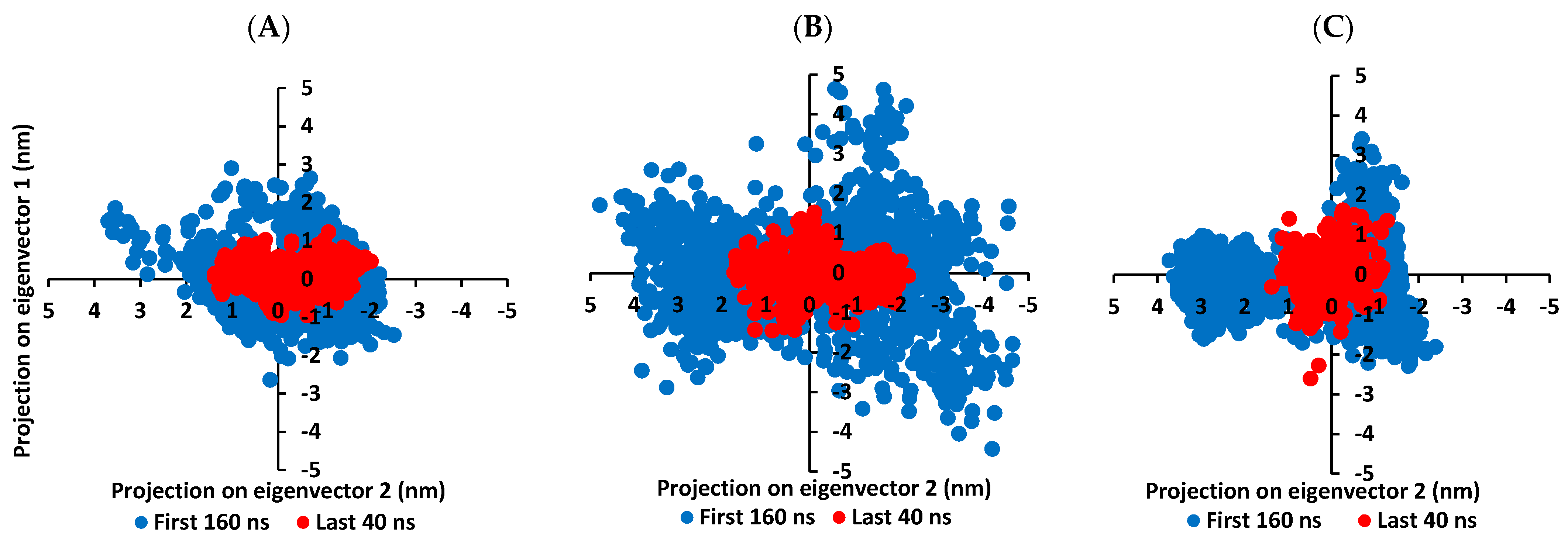
3.5.1. MD Analysis on M-Pro Target
3.5.2. MD Analysis on Nsp15 NendoU Target
3.5.3. Binding-Free Energy
3.6. ADME/Tox Analysis and Values of Principal Molecular Descriptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stanworth, S.J.; New, H.V.; Apelseth, T.O.; Brunskill, S.; Cardigan, R.; Doree, C.; Germain, M.; Goldman, M.; Massey, E.; Prati, D.; et al. Effects of the COVID-19 pandemic on supply and use of blood for transfusion. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72,314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Salazar, E.; Perez, K.K.; Ashraf, M.; Chen, J.; Castillo, B.; Christensen, P.A.; Eubank, T.; Bernard, D.W.; Eagar, T.N.; Long, S.W.; et al. Treatment of Coronavirus Disease 2019 (COVID-19) Patients with Convalescent Plasma. Am. J. Pathol. 2020, 190, 1680–1690. [Google Scholar] [CrossRef]
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control. J. Clin. Med. 2020, 9, 1225. [Google Scholar] [CrossRef]
- Brown, B.L.; McCullough, J. Treatment for emerging viruses: Convalescent plasma and COVID-19. Transfus. Apher. Sci. 2020, 59, 102790. [Google Scholar] [CrossRef]
- Lim, J.; Jeon, S.; Shin, H.Y.; Kim, M.J.; Seong, Y.M.; Lee, W.J.; Choe, K.W.; Kang, Y.M.; Lee, B.; Park, S.J. Case of the Index Patient Who Caused Tertiary Transmission of COVID-19 Infection in Korea: The Application of Lopinavir/Ritonavir for the Treatment of COVID-19 Infected Pneumonia Monitored by Quantitative RT-PCR. J. Korean Med. Sci. 2020, 35, e79. [Google Scholar] [CrossRef]
- Huang, X.; Xu, Y.; Yang, Q.; Chen, J.; Zhang, T.; Li, Z.; Guo, C.; Chen, H.; Wu, H.; Li, N. Efficacy and biological safety of lopinavir/ritonavir based anti-retroviral therapy in HIV-1-infected patients: A meta-analysis of randomized controlled trials. Sci. Rep. 2015, 5, 8528. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Shalhoub, S.; Mandourah, Y.; Al-Hameed, F.; Al-Omari, A.; Al Qasim, E.; Jose, J.; Alraddadi, B.; Almotairi, A.; Al Khatib, K.; et al. Ribavirin and Interferon Therapy for Critically Ill Patients With Middle East Respiratory Syndrome: A Multicenter Observational Study. Clin. Infect. Dis. 2020, 70, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Al-Tawfiq, J.A.; Momattin, H.; Dib, J.; Memish, Z.A. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: An observational study. Int. J. Infect. Dis 2014, 20, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Q.; Tang, S.-Q.; Xu, X.-L.; Zeng, Y.-M.; He, X.-Q.; Li, Y.; Harypursat, V.; Lu, Y.-Q.; Wan, Y.; Zhang, L.; et al. No Statistically Apparent Difference in Antiviral Effectiveness Observed Among Ribavirin Plus Interferon-Alpha, Lopinavir/Ritonavir Plus Interferon-Alpha, and Ribavirin Plus Lopinavir/Ritonavir Plus Interferon-Alpha in Patients With Mild to Moderate Coronavirus Disease 2019: Results of a Randomized, Open-Labeled Prospective Study. Front. Pharmacol. 2020, 11, 1071. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef]
- Peng, H.; Chen, Z.; Wang, Y.; Ren, S.; Xu, T.; Lai, X.; Wen, J.; Zhao, M.; Zeng, C.; Du, L.; et al. Systematic Review and Pharmacological Considerations for Chloroquine and Its Analogs in the Treatment for COVID-19. Front. Pharmacol. 2020, 11, 554172. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Pundir, H.; Mathpal, S.; Chandra, S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2020. [Google Scholar] [CrossRef] [PubMed]
- Naik, B.; Gupta, N.; Ojha, R.; Singh, S.; Prajapati, V.K.; Prusty, D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int. J. Biol. Macromol. 2020, 160, 1–17. [Google Scholar] [CrossRef]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Owis, A.I.; El-Hawary, M.S.; El Amir, D.; Aly, O.M.; Abdelmohsen, U.R.; Kamel, M.S. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020, 10, 19570–19575. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL. J. Pharm. Anal. 2020. [Google Scholar] [CrossRef]
- Krishnan, D.A.; Sangeetha, G.; Vajravijayan, S.; Nandhagopal, N.; Gunasekaran, K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Inform. Med. Unlocked 2020. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.K.; Prasad, S.K.; Islam, M.A.; Gurav, S.S.; Patil, R.B.; AlFaris, N.A.; Aldayel, T.S.; AlKehayez, N.M.; Wabaidur, S.M.; Shakya, A. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Kong, R.; Yang, G.; Xue, R.; Liu, M.; Wang, F.; Hu, J.; Guo, X.; Chang, S. COVID-19 Docking Server: A meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef]
- Ulferts, R.; Ziebuhr, J. Nidovirus ribonucleases: Structures and functions in viral replication. RNA Biol. 2011, 8, 295–304. [Google Scholar] [CrossRef]
- Sayed, A.M.; Khattab, A.R.; AboulMagd, A.M.; Hassan, H.M.; Rateb, M.E.; Zaid, H.; Abdelmohsen, U.R. Nature as a treasure trove of potential anti-SARS-CoV drug leads: A structural/mechanistic rationale. RSC Adv. 2020, 10, 19790–19802. [Google Scholar] [CrossRef]
- Ma, J.; Huo, X.Q.; Chen, X.; Zhu, W.X.; Yao, M.C.; Qiao, Y.J.; Zhang, Y.L. Study on screening potential traditional Chinese medicines against 2019-nCoV based on Mpro and PLP. Zhongguo Zhong Yao Za Zhi 2020, 45, 1219–1224. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.N.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct Dyn. 2020. [Google Scholar] [CrossRef]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct Dyn. 2020. [Google Scholar] [CrossRef]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Saeed, M.; Greten, H.; Efferth, T. Identification of Novel Compounds against Three Targets of SARS CoV-2 Coronavirus by Combined Virtual Screening and Supervised Machine Learning. Comput. Biol. Med. 2021, 133, 104359. [Google Scholar] [CrossRef]
- Elhady, S.S.; Al-Abd, A.M.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. Antiproliferative Scalarane-Based Metabolites from the Red Sea Sponge Hyrtios erectus. Mar. Drugs 2016, 14, 130. [Google Scholar] [CrossRef]
- Elhady, S.S.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. A New Bioactive Metabolite Isolated from the Red Sea Marine Sponge Hyrtios erectus. Molecules 2016, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Alahdal, A.M.; Asfour, H.Z.; Ahmed, S.A.; Noor, A.O.; Al-Abd, A.M.; Elfaky, M.A.; Elhady, S.S. Anti-Helicobacter, Antitubercular and Cytotoxic Activities of Scalaranes from the Red Sea Sponge Hyrtios erectus. Molecules 2018, 23, 978. [Google Scholar] [CrossRef]
- Youssef, D.T.; Shaala, L.A.; Emara, S. Antimycobacterial scalarane-based sesterterpenes from the Red Sea sponge Hyrtios erecta. J. Nat. Prod. 2005, 68, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Crews, P.; Bescansa, P. Sesterterpenes from a common marine sponge, Hyrtios erecta. J. Nat. Prod. 1986, 49, 1041–1052. [Google Scholar] [CrossRef]
- Kikuchi, H.; Tsukitani, Y.; Shimizu, I.; Kobayashi, M.; Kitagawa, I. Marine Natural Products. XI. An Antiinflammatory Scalarane-type Bishomosesterterpene, Foliaspongin, from the Okinawan Marine Sponge Phyllospongia foliascens (PALLAS). Chem. Pharm. Bull. 1983, 31, 552–556. [Google Scholar] [CrossRef]
- Baekman, J.C.; Daloze, D.; Kaisin, M.; Moussiaux, B. Ichthyotoxic sesterterpenoids from the neo guinean sponge carteriospongia foliascens. Tetrahedron 1985, 41, 4603–4614. [Google Scholar] [CrossRef]
- Walker, R.P.; Thompson, J.E.; Faulkner, D.J. Sesterterpenes from Spongia idia. J. Org. Chem. 1980, 45, 4976–4979. [Google Scholar] [CrossRef]
- Terem, B.; Scheuer, P.J. Scalaradial derivatives from the nudibranch chromodoris youngbleuthi and the sponge spongia oceania. Tetrahedron 1986, 42, 4409–4412. [Google Scholar] [CrossRef]
- Hochlowski, J.E.; Faulkner, D.J.; Bass, L.S.; Clardy, J. Metabolites of the dorid nudibranch Chromodoris sedna. J. Org. Chem. 1983, 48, 1738–1740. [Google Scholar] [CrossRef]
- Bergquist, P.R.; Cambie, R.C.; Kernan, M.R. Scalarane sesterterpenes from Collospongia auris, a new thorectid sponge. Biochem. Syst. Ecol. 1990, 18, 349–357. [Google Scholar] [CrossRef]
- Nakagawa, M.; Hamamoto, Y.; Ishihama, M.; Hamasaki, S.; Endo, M. Pharmacologically active homosesterterpenes from palauan sponges. Tetrahedron Lett. 1987, 28, 431–434. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Wells, R.J. Five new C26 tetracyclic terpenes from a sponge (Lendenfeldia sp.). Aust. J. Chem. 1982, 35, 51–59. [Google Scholar] [CrossRef]
- Youssef, D.T.; Yamaki, R.K.; Kelly, M.; Scheuer, P.J. Salmahyrtisol A, a novel cytotoxic sesterterpene from the Red Sea sponge Hyrtios erecta. J. Nat. Prod. 2002, 65, 2–6. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Cichacz, Z.A. Antineoplastic Agents. 542. Isolation and Structure of Sesterstatin 6 from the Indian Ocean Sponge Hyrtios erecta. J. Nat. Prod. 2005, 68, 1253–1255. [Google Scholar] [CrossRef]
- Ryu, G.; Matsunaga, S.; Fusetani, N. Three New Cytotoxic Sesterterpenes from the Marine Sponge Hyrtios cf. erectus. J. Nat. Prod. 1996, 59, 515–517. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, A.; Tan, R.; Hoard, M.S.; Melody, N.; Pettit, R. Antineoplastic agents. 386. Isolation of sesterstatins 1-3 from the marine sponge Hyrtios erecta. J. Nat. Prod. 1998, 61, 13–16. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Tan, R.; Herald, D.L.; Melody, N.; Hoard, M.S.; Doubek, D.L.; Hooper, J.N. Antineoplastic Agents 385. The Isolation and Structure of a Scalarane-Type Sesterterpene from the Indian Ocean Porifera Hyrtios erecta. Collect. Czechoslov. Chem. Commun. 1998, 63, 1671–1677. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Melody, N.; Cichacz, Z.A.; Herald, D.L.; Hoard, M.S.; Pettit, R.K.; Chapuis, J.-C. Antineoplastic agents 397: Isolation and structure of sesterstatins 4 and 5 from hyrtios erecta (the republic of maldives). Bioorganic Med. Chem. Lett. 1998, 8, 2093–2098. [Google Scholar] [CrossRef]
- Nasu, S.S.; Yeung, B.K.S.; Hamann, M.T.; Scheuer, P.J.; Kelly-Borges, M.; Goins, K. Puupehenone-related metabolites from two Hawaiian sponges, Hyrtios spp. J. Org. Chem. 1995, 60, 7290–7292. [Google Scholar] [CrossRef]
- Antonio, E.; Alexander, K.; Florence, L.; Alessio, C.; Ramesh, D.; Marco, E.; Véronique, M.; Robert, K. Sesterterpenoids with Anticancer Activity. Curr. Med. Chem. 2015, 22, 3502–3522. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC 1010 Sherbooke St. West, Suite #910: Montreal, QC, Canada, 2020. [Google Scholar]
- Han, W.; Quan, B.; Guo, Y.; Zhang, J.; Lu, Y.; Feng, G.; Wu, Q.; Fang, F.; Cheng, L.; Jiao, N.; et al. The course of clinical diagnosis and treatment of a case infected with coronavirus disease 2019. J. Med. Virol. 2020, 92, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Cheng, V.C.; Hung, I.F.; Wong, M.M.; Chan, K.H.; Chan, K.S.; Kao, R.Y.; Poon, L.L.; Wong, C.L.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Ortiz-Alcantara, J.; Bhardwaj, K.; Palaninathan, S.; Frieman, M.; Baric, R.C.K. small molecule inhibitors of the sArs-coV nsp15 endoribonuclease. Virus Adapt. Treat. 2010, 2, 125–133. [Google Scholar]
- El Raey, M.A.; El-Hagrassi, A.M.; Osman, A.F.; Darwish, K.M.; Emam, M. Acalypha wilkesiana flowers: Phenolic profiling, cytotoxic activity of their biosynthesized silver nanoparticles and molecular docking study for its constituents as Topoisomerase-I inhibitors. Biocatal. Agric. Biotechnol. 2019, 20, 101243. [Google Scholar] [CrossRef]
- Wadie, M.A.; Kishk, S.M.; Darwish, K.M.; Mostafa, S.M.; Elgawish, M.S. Simultaneous Determination of Losartan and Rosuvastatin in Rat Plasma Using Liquid Chromatography–Tandem Mass Spectrometric Technique for Application into Pharmacokinetic and Drug–Drug Interaction Studies. Chromatographia 2020, 83, 1477–1494. [Google Scholar] [CrossRef]
- Malebari, A.M.; Ibrahim, T.S.; Salem, I.M.; Salama, I.; Khayyat, A.N.; Mostafa, S.M.; El-Sabbagh, O.I.; Darwish, K.M. The Anticancer Activity for the Bumetanide-Based Analogs via Targeting the Tumor-Associated Membrane-Bound Human Carbonic Anhydrase-IX Enzyme. Pharmaceuticals 2020, 13, 252. [Google Scholar] [CrossRef]
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. A Publ. Protein Soc. 1998, 7, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, 2.0.6; Schrödinger, LLC: New York, NY, USA, 2020.
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Golo, V.L.; Shaĭtan, K.V. Dynamic attractor for the Berendsen thermostat an the slow dynamics of biomacromolecules. Biofizika 2002, 47, 611–617. [Google Scholar] [PubMed]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from Monte Carlo simulations. Bioorg Med. Chem. Lett. 2000, 10, 1155–1158. [Google Scholar] [CrossRef]
- Colmenarejo, G.; Alvarez-Pedraglio, A.; Lavandera, J.-L. Cheminformatic Models To Predict Binding Affinities to Human Serum Albumin. J. Med. Chem. 2001, 44, 4370–4378. [Google Scholar] [CrossRef]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef]
- Duffy, E.M.; Jorgensen, W.L. Prediction of Properties from Simulations: Free Energies of Solvation in Hexadecane, Octanol, and Water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yiu, C.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease 3CL. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wower, J.; Maltseva, N.; Chang, C.; Jedrzejczak, R.; Wilamowski, M.; Kang, S.; Nicolaescu, V.; Randall, G.; Michalska, K.; et al. Tipiracil Binds to Uridine Site and Inhibits Nsp15 Endoribonuclease NendoU from SARS-CoV-2. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Yan, L.; Ming, Z.; Jia, Z.; Lou, Z.; Rao, Z. Structural and Biochemical Characterization of Endoribonuclease Nsp15 Encoded by Middle East Respiratory Syndrome Coronavirus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Guarino, L.A.; Bhardwaj, K.; Dong, W.; Sun, J.; Holzenburg, A.; Kao, C. Mutational analysis of the SARS virus Nsp15 endoribonuclease: Identification of residues affecting hexamer formation. J. Mol. Biol. 2005, 353, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure Of Biomolecules Through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Likic, V.A.; Gooley, P.R.; Speed, T.P.; Strehler, E.E. A statistical approach to the interpretation of molecular dynamics simulations of calmodulin equilibrium dynamics. Protein Sci. A Publ. Protein Soc. 2005, 14, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Srikumar, P.S.; Rohini, K.; Rajesh, P.K. Molecular dynamics simulations and principal component analysis on human laforin mutation W32G and W32G/K87A. Protein J. 2014, 33, 289–295. [Google Scholar] [CrossRef]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef]
- Pandey, B.; Grover, S.; Goyal, S.; Kumari, A.; Singh, A.; Jamal, S.; Kaur, J.; Grover, A. Alanine mutation of the catalytic sites of Pantothenate Synthetase causes distinct conformational changes in the ATP binding region. Sci. Rep. 2018, 8, 903. [Google Scholar] [CrossRef] [PubMed]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.S.; Pacheco, B.D.C.; Pinheiro, S.; Muri, E.M.F.; Dias, L.R.S.; Lima, C.H.S.; Garrett, R.; de Moraes, M.B.M.; de Souza, B.E.G.; Puzer, L. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorganic Med. Chem. Lett. 2019, 29, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol. Biol. 2020, 2114, 257–268. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Lipinski, C.A. Chris Lipinski discusses life and chemistry after the Rule of Five. Drug Discov. Today. 2003, 8, 12–16. [Google Scholar] [PubMed]
- Lotz, S.D.; Dickson, A. Unbiased Molecular Dynamics of 11 min Timescale Drug Unbinding Reveals Transition State Stabilizing Interactions. J. Am. Chem. Soc. 2018, 140, 618–628. [Google Scholar] [CrossRef]
- Dixon, T.; Lotz, S.D.; Dickson, A. Predicting ligand binding affinity using on- and off-rates for the SAMPL6 SAMPLing challenge. J. Comput. Aided Mol. Des. 2018, 32, 1001–1012. [Google Scholar] [CrossRef]
- Deb, I.; Frank, A.T. Accelerating Rare Dissociative Processes in Biomolecules Using Selectively Scaled MD Simulations. J. Chem. Theory Comput. 2019, 15, 5817–5828. [Google Scholar] [CrossRef] [PubMed]
- Kokh, D.B.; Amaral, M.; Bomke, J.; Grädler, U.; Musil, D.; Buchstaller, H.-P.; Dreyer, M.K.; Frech, M.; Lowinski, M.; Vallee, F.; et al. Estimation of Drug-Target Residence Times by τ-Random Acceleration Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 3859–3869. [Google Scholar] [CrossRef] [PubMed]
- Mollica, L.; Decherchi, S.; Zia, S.R.; Gaspari, R.; Cavalli, A.; Rocchia, W. Kinetics of protein-ligand unbinding via smoothed potential molecular dynamics simulations. Sci. Rep. 2015, 5, 11539. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Andricioaei, I. Weighted ensemble milestoning (WEM): A combined approach for rare event simulations. J. Chem. Phys. 2020, 152, 234114. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name | Binding Energy (Kcal/mol) a | |
|---|---|---|---|
| 5r7z | 6w01 | ||
| 1 | Sesterstatin 7 | −6.4842 | −10.6901 |
| 2 | Heteronemin | −10.2889 | −10.3251 |
| 3 | Scalarolide | −8.7294 | −9.5520 |
| 4 | 12-epi-24-deoxyscalarin | −10.3702 | −10.6610 |
| 5 | Scalarolide acetate | −8.0121 | −9.3353 |
| 6 | 19 acetylsesterstatin 3 | −10.8712 | −10.3133 |
| 7 | 12-deacetyl-12,18-di-epi-scalaradial | −7.0172 | −7.8822 |
| 8 | 12-deacetyl-12-epi-scalaradial | −9.5947 | −6.5206 |
| 9 | Sesterstatin 3 | −6.6789 | −9.4241 |
| 10 | 12β,20α-dihydroxy-16β-acetoxy-17-scalaren-19,20-olide | −8.3400 | −10.6001 |
| 11 | 12-O-acetyl-16-O-methylhyrtiolide | −10.0880 | −10.2102 |
| 12 | 12β-acetoxy,16β-methoxy,20α-hydroxy-17-scalaren-19,20-olide | −10.3784 | −11.3058 |
| 13 | 24α-methoxypetrosaspongia C | −6.0879 | −8.2669 |
| 14 | 12-O-deacetyl-12,19-di-epi-scalarin | −8.1694 | −8.6984 |
| 15 | 12β-acetoxy,16-epi-hyrtiolide | −10.4134 | −10.9658 |
| M-pro Reference | Lopinavir | −9.1319 | – |
| NendoU Reference | Benzopurpurin 4B | – | −10.0317 |
| Compound | Docking Energy (Kcal/mol) a | H-Bond Interactions | Hydrophobic Interactions | Sulphur-Dipole | π-Interaction | van der Waal with Side Chain Carbons | |
|---|---|---|---|---|---|---|---|
| Preliminary | Directed | ||||||
| 2 | −10.2889 | −12.0270 | Glu166, Thr190, Gln192 | Thr25, His41, Met49, Met165, Pro168 | Met165 19α-acetyloxy | — | Gln189 (Cβ,Cδ) |
| 4 | −10.3702 | −10.1306 | Glu166 | Thr25, Thr26, His41, Met49, Phe140, Leu141, Met165 | — | — | Gln189 (Cβ,Cδ) |
| 6 | −10.8712 | −12.7685 | Cys145, Glu166 | His41, Met49, Met165, Leu167, Pro168, Ala191 | — | His41 (π-H) | Gln189 (Cβ,Cδ) |
| 8 | −9.5947 | −10.8893 | His41, Glu166 | Met49, Met165, Leu167, Pro168 | Met165 18β-aldehyde | — | Gln189 (Cβ,Cδ) |
| 11 | −10.0880 | −11.5470 | Glu166, Thr190, Gln192 | Thr25, His41, Met49, Met165, Leu167, Pro168, Ala191 | Met165 12β-acetyloxy | — | Gln189 (Cβ,Cδ) Asp187 (Cβ) |
| 12 | −10.3784 | −11.0882 | Thr190, Ala191, Gln192 | Thr25, His41, Met49, Met165, Pro168, Ala191 | — | — | Gln189 (Cβ,Cδ) |
| 15 | −10.4134 | −12.4339 | Arg188, Gln192 | His41, Met49, Phe140, Leu141, Met165, Leu167, Pro168 | — | — | Gln189 (Cβ,Cδ) Asp187 (Cβ) |
| Lopinavir | −9.1319 | −10.0396 | Glu166, Gln189 | His41, Met49, Phe140, Leu141, Asn142, His163, Met165, Leu167, Pro168 | — | — | Gln189 (Cβ,Cδ) Asp187 (Cβ) |
| Compound | Docking Energy (Kcal/mol) a | H-Bond Interactions | Hydrophobic Interactions | π-Interactions | |
|---|---|---|---|---|---|
| Preliminary | Directed | ||||
| 1 | −10.6901 | −10.7543 | His235, Gly248, Lys290, Try343 | Val292, Trp333, Thr341, Tyr343, Lys345 | — |
| 2 | −10.3251 | −11.7468 | Leu246, Gly248, Lys290 | Val292, Trp333, Thr341, Tyr343 | — |
| 4 | −10.6610 | −10.4221 | Gly248, His250, Lys290, Ser294 | Val292, Trp333, Thr341, Tyr343, Pro344 | Trp333 (π-H) |
| 6 | −10.3133 | −10.1971 | Gln245, Leu246, Gly248, Lys290 | Val292, Met331, Thr341, Trp333, Tyr343 | — |
| 10 | −10.6001 | −10.8170 | Gln245, Gly248, His250, Lys290, Thr341 | Ile236, His243, Leu246, Thr341, Trp333 | — |
| 11 | −10.2102 | −10.4656 | Gln245, Gly248, Lys290, Tyr343 | His243, Leu246, Trp333, Thr341, Lys335 | — |
| 12 | −11.3058 | −10.9415 | Gly248, Lys290, Ser294 | Val292, Trp333, Thr341, Pro344, Lys345, Leu346 | Tyr343 (π-H) |
| 15 | −10.9658 | −11.2535 | Gln245, Gly248, Lys290 | Val292, Trp333, Thr341, Leu246 | Tyr343 (π-H) |
| Benzopurpurin 4B | −10.0317 | −10.0997 | Asp240, Asn278, Glu340, Leu346 | His235, Val292, Tyr343, Thr341, Lys345 | Tyr343 (H-π) |
| Energy (kJ/mol ± SE) | M-Pro Complex | Nsp15 NendoU Complex | ||||
|---|---|---|---|---|---|---|
| Comp. 6 | Comp. 15 | Lopinavir | Comp. 6 | Comp. 15 | Benzopurpurin 4B | |
| ΔGvan der Waal | −132.92 ± 10.58 | −106.70 ± 10.75 | −189.57 ± 12.57 | −98.73 ± 7.84 | −85.26 ± 12.10 | −111.03 ± 14.65 |
| ΔGelectrostatic | −9.347 ± 9.51 | −14.39 ± 9.37 | −31.56 ± 9.60 | −51.96 ± 7.96 | −7.71 ± 5.32 | −102.46 ± 14.83 |
| ΔGsolvation; Polar | 81.32 ± 10.38 | 63.83 ± 18.13 | 148.85 ± 16.24 | 113.71 ± 12.92 | 25.36 ± 26.30 | 156.85 ± 24.82 |
| ΔGsolvation; SASA | −15.41 ± 0.77 | −11.09 ± 1.47 | −20.65 ± 1.11 | −12.76 ± 0.80 | −10.25 ± 1.35 | −16.18 ± 1.66 |
| ΔGbinding energy | −76.37 ± 9.69 | −68.3 ± 13.49 | −92.93 ± 14.34 | −49.73 ± 11.53 | −77.87 ± 19.49 | −72.82 ± 17.77 |
| Drug-Likeness/Predicted ADME Descriptors | Compound 6 | Compound 15 |
|---|---|---|
| QPlogPo/w (−2.0 to 6.5) | 3.72 | 3.16 |
| QPlogS mol/dm3 (−6.5 to 0.5) | −5.73 | −5.77 |
| QPPCaco nm/sec (<25 poor >500 great) | 732.21 | 344.90 |
| QPPMDCK nm/sec (<25 poor >500 great) | 353.21 | 156.55 |
| QPlogBB (−3.0 to 1.2) | −0.69 | −0.95 |
| QPlogKHSA (−1.5 to 1.5) | 0.67 | 0.52 |
| % HOA (<25% poor >80% great) | 100 | 90.84 |
| QPlogHERG (above −5.0) | −3.78 | −3.52 |
| Oral rat LD50 mg/Kg | 193.16 | 628.19 |
| AMES Mutagenicity test | Negative | Negative |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elhady, S.S.; Abdelhameed, R.F.A.; Malatani, R.T.; Alahdal, A.M.; Bogari, H.A.; Almalki, A.J.; Mohammad, K.A.; Ahmed, S.A.; Khedr, A.I.M.; Darwish, K.M. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology 2021, 10, 389. https://doi.org/10.3390/biology10050389
Elhady SS, Abdelhameed RFA, Malatani RT, Alahdal AM, Bogari HA, Almalki AJ, Mohammad KA, Ahmed SA, Khedr AIM, Darwish KM. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology. 2021; 10(5):389. https://doi.org/10.3390/biology10050389
Chicago/Turabian StyleElhady, Sameh S., Reda F. A. Abdelhameed, Rania T. Malatani, Abdulrahman M. Alahdal, Hanin A. Bogari, Ahmad J. Almalki, Khadijah A. Mohammad, Safwat A. Ahmed, Amgad I. M. Khedr, and Khaled M. Darwish. 2021. "Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors" Biology 10, no. 5: 389. https://doi.org/10.3390/biology10050389
APA StyleElhady, S. S., Abdelhameed, R. F. A., Malatani, R. T., Alahdal, A. M., Bogari, H. A., Almalki, A. J., Mohammad, K. A., Ahmed, S. A., Khedr, A. I. M., & Darwish, K. M. (2021). Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology, 10(5), 389. https://doi.org/10.3390/biology10050389