
The Role of Nitroreductases in Resistance to Nitroimidazoles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Nitroimidazoles
3. Metronidazole
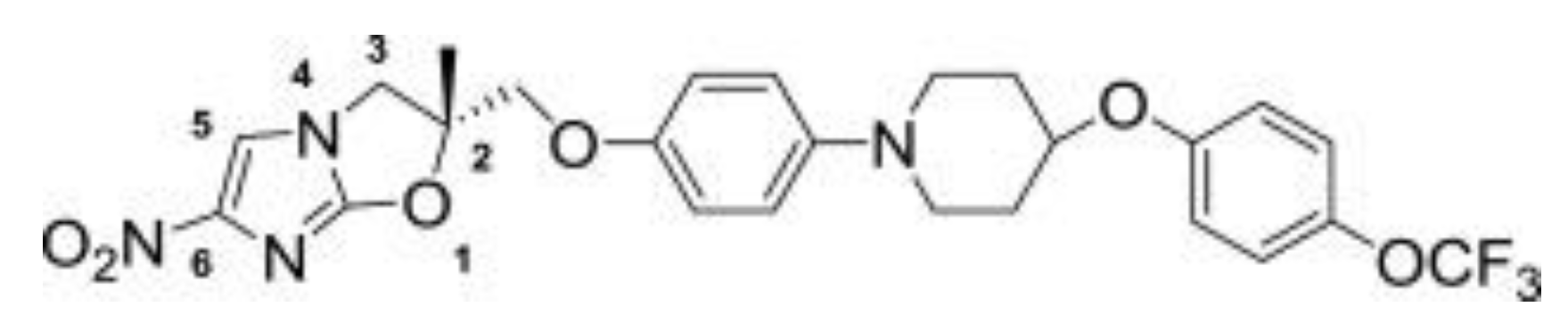
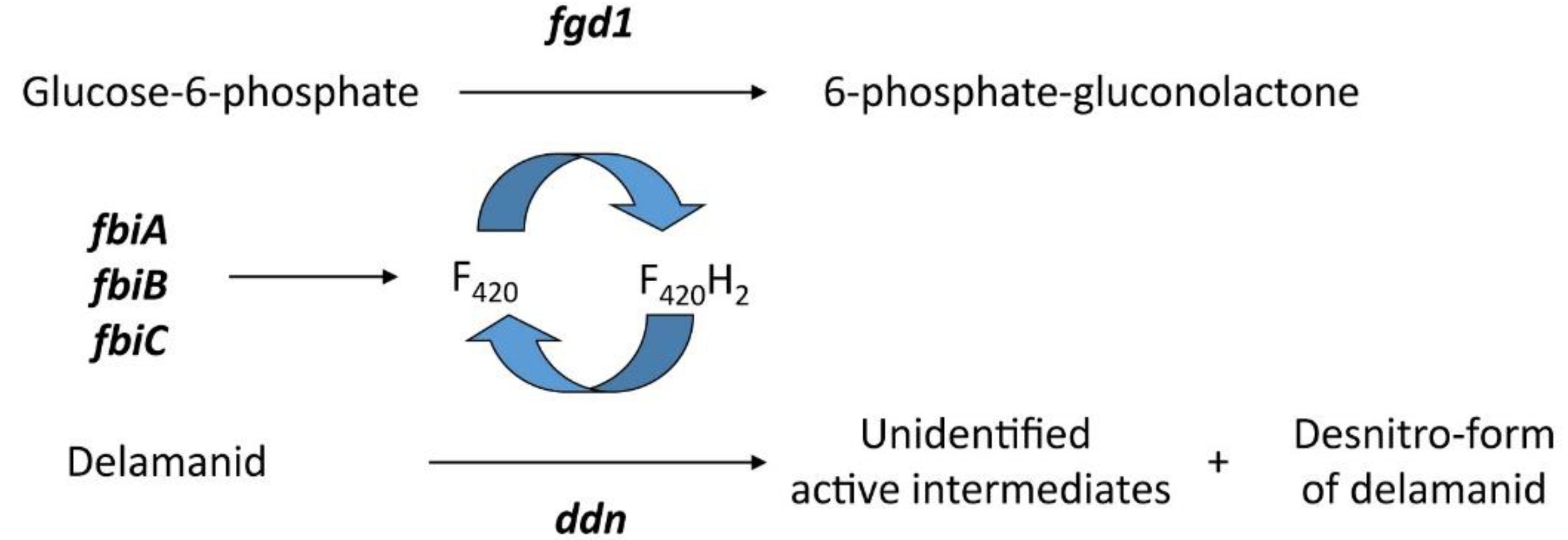
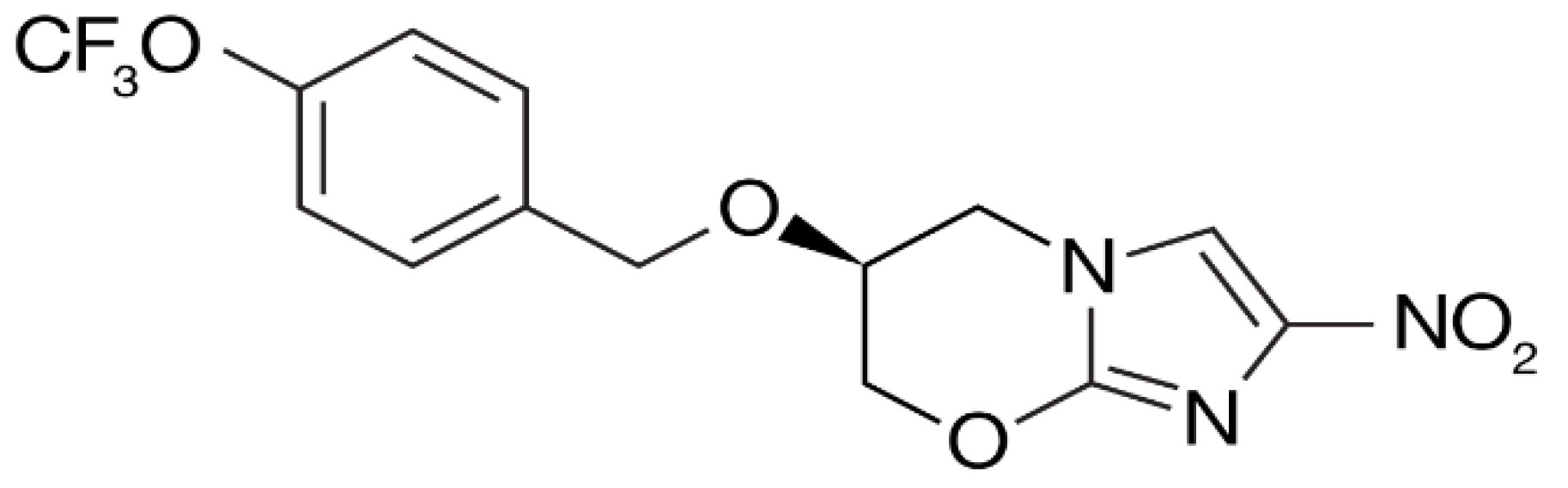
4. Delamanid and Pretomanid
5. Chloramphenicol
6. MT02
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gwenin, C.D.; Kalaji, M.; Williams, P.A.; Kay, C.M. A kinetic analysis of three modified novel nitroreductases. Biodegradation 2010, 22, 463–474. [Google Scholar] [CrossRef]
- Van Dillewijn, P.; Couselo, J.L.; Corredoira, E.; Delgado, A.; Wittich, R.-M.; Ballester, A.; Ramos, J.L. Bioremediation of 2,4,6-Trinitrotoluene by Bacterial Nitroreductase Expressing Transgenic Aspen. Environ. Sci. Technol. 2008, 42, 7405–7410. [Google Scholar] [CrossRef] [PubMed]
- Güngör, T.; Önder, F.C.; Tokay, E.; Gülhan, Ü.G.; Hacıoğlu, N.; Tok, T.T.; Çelik, A.; Köçkar, F.; Ay, M. Prodrugs for nitroreductase based cancer therapy-2: Novel amide/Ntr combinations targeting PC3 cancer cells. Eur. J. Med. Chem. 2019, 171, 383–400. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; You, X.; Zhang, X. Prodrug strategy for cancer cell-specific targeting: A recent overview. Eur. J. Med. Chem. 2017, 139, 542–563. [Google Scholar] [CrossRef]
- Chan-Hyams, J.V.E.; Copp, J.N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Evaluating the abilities of diverse ni-troaromatic prodrug metabolites to exit a model Gram negative vector for bacterial-directed enzyme-prodrug therapy. Biochem. Pharmacol. 2018, 158, 192–200. [Google Scholar] [CrossRef]
- Ball, P.; Thompson, E.; Anderson, S.; Gwenin, V.; Gwenin, C. Time dependent HPLC analysis of the product ratio of enzymatically reduced prodrug CB1954 by a modified and immobilised nitroreductase. Eur. J. Pharm. Sci. 2019, 127, 217–224. [Google Scholar] [CrossRef]
- Copp, J.N.; Mowday, A.M.; Williams, E.M.; Guise, C.P.; Ashoorzadeh, A.; Sharrock, A.V.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Engineering a multifunctional nitroreductase for improved activation of prodrugs and PET probes for cancer gene therapy. Cell Chem. Biol. 2017, 24, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Hu, F.; Huang, J.; Li, N.; Gu, Y.; Wang, P. A fluorescent turn-on probe for nitroreductase imaging in living cells and tissues under hypoxia conditions. Sens. Actuators B Chem. 2018, 268, 70–76. [Google Scholar] [CrossRef]
- Zhu, K.; Qin, T.; Zhao, C.; Luo, Z.; Huang, Y.; Liu, B.; Wang, L. A novel fluorescent turn-on probe for highly selective detection of nitroreductase in tumor cells. Sens. Actuators B Chem. 2018, 276, 397–403. [Google Scholar] [CrossRef]
- Li, Y.; Deng, Y.; Liu, J.; Fu, J.; Sun, Y.; Ouyang, R.; Miao, Y. A near-infrared frequency upconversion probe for nitroreductase detection and hypoxia tumor in vivo imaging. Sens. Actuators B Chem. 2019, 286, 337–345. [Google Scholar] [CrossRef]
- Kumari, R.; Sunil, D.; Ningthoujam, R.S. Naphthalimides in fluorescent imaging of tumor hypoxia—An up-to-date review. Bioorg. Chem. 2019, 88, 102979. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Bot, C.; Kelly, J.M.; Hall, B.S. Trypanocidal activity of nitroaromatic prodrugs: Current treatments and future perspectives. Curr. Top. Med. Chem. 2011, 11, 2072–2084. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Dey, N.; Negrette, O.S.; Parada, L.A.; Basombrio, M.A.; Garg, N.J. Hepatotoxicity in mice of a novel an-ti-parasite drug candidate hydroxymethylnitrofurazone: A comparison with Benznidazole. PLoS Negl. Trop. Dis. 2014, 8, e3231. [Google Scholar] [CrossRef] [PubMed]
- De la Calle, M.E.; Cabrera, G.; Cantero, D.; Valle, A.; Bolivar, J. A genetically engineered Escherichia coli strain overexpressing the ni-troreductase NfsB is capable of producing the herbicide D-DIBOA with 100% molar yield. Microb. Cell Fact. 2019, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Gwenin, C.; Kalaji, M.; Kay, C.M.; Williams, P.A.; Tito, D.N. An in situ amperometric biosensor for the detection of vapours from explosive compounds. Analyst 2008, 133, 621–625. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]
- Zenno, S.; Saigo, K.; Kanoh, H.; Inouye, S. Identification of the gene encoding the major NAD(P)H-flavin oxidoreductase of the bioluminescent bacterium Vibrio fischeri ATCC 7744. J. Bacteriol. 1994, 176, 3536–3543. [Google Scholar] [CrossRef][Green Version]
- Bryant, D.W.; McCalla, D.R.; Leeksma, M.; Laneuville, P. Type I nitroreductases of Escherichia coli. Can. J. Microbiol. 1981, 27, 81–86. [Google Scholar] [CrossRef]
- Roldán, M.D.; Pérez-Reinado, E.; Castillo, F.; Moreno-Vivián, C. Reduction of polynitroaromatic compounds: The bacterial nitroreductases. FEMS Microbiol. Rev. 2008, 32, 474–500. [Google Scholar] [CrossRef]
- Powell, N. Nitroreductase in Cancer Therapy. Available online: https://nitroreductaseincancertreatment.wordpress.com/nitroreductase/ (accessed on 17 December 2020).
- Whiteway, J.; Koziarz, P.; Veall, J.; Sandhu, N.; Kumar, P.; Hoecher, B.; Lambert, I.B. Oxygen-Insensitive Nitroreductases: Analysis of the Roles of nfsA and nfsB in Development of Resistance to 5-Nitrofuran Derivatives in Escherichia coli. J. Bacteriol. 1998, 180, 5529–5539. [Google Scholar] [CrossRef]
- Xiao, Y.; Wu, J.; Liu, H.; Wang, S.; Liu, S.; Zhou, N. Characterization of genes involved in the initial reactions of 4-chloronitrobenzene degradation in Pseudomonasputida ZWL73. Appl. Microbiol. Biotechnol. 2006, 73, 166–171. [Google Scholar] [CrossRef]
- Somerville, C.C.; Nishino, S.F.; Spain, J.C. Isolation and characterization of nitrobenzene nitroreductase from Pseudomonas pseudoalcaligenes JS45. J. Bacteriol. 1995, 177, 3837–3842. [Google Scholar] [CrossRef]
- Liochev, S.I.; Hausladen, A.; Beyer, W.F.; Fridovich, I. NADPH: Ferredoxin oxidoreductase acts as a paraquat diaphorase and is a member of the soxRS regulon. Proc. Natl. Acad. Sci. USA 1994, 91, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Hausladen, A.; Fridovich, I. Nitroreductase A is regulated as a member of the soxRS regulon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 3537–3539. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Liu, M.; Huang, S.; Tu, S.C. Vibrio harveyi NADPH-flavin oxidoreductase: Cloning, sequencing and overexpression of the gene and purification and characterization of the cloned enzyme. J. Bacteriol. 1994, 176, 3552–3558. [Google Scholar] [CrossRef]
- Ang, C.W.; Jarrad, A.M.; Cooper, M.; Blaskovich, M.A.T. Nitroimidazoles: Molecular Fireworks That Combat a Broad Spectrum of Infectious Diseases. J. Med. Chem. 2017, 60, 7636–7657. [Google Scholar] [CrossRef] [PubMed]
- WHO Factsheet. Available online: https://www.who.int/en/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 17 December 2020).
- Crofts, T.S.; Gasparrini, A.J.; Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017, 15, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic discovery: History, methods and perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef]
- Nakamura, S. Structure of Azomycin, a New Antibiotic. Pharm. Bull. 1955, 3, 379–383. [Google Scholar] [CrossRef]
- Edwards, D.I. Nitroimidazole drugs-action and resistance mechanisms I. Mechanism of action. J. Antimicrob. Chemother. 1993, 31, 9–20. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef]
- Edwards, D.I. Nitroimidazole drugs—Action and resistance mechanisms II. Mechanisms of resistance. J. Antimicrob. Chemother. 1993, 31, 201–210. [Google Scholar] [CrossRef]
- Samuelson, J. Why Metronidazole is Active against both Bacteria and Parasites. Antimicrob. Agents Chemother. 1999, 43, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; DeLuca, M. Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter cloacae. J. Biol. Chem. 1991, 266, 4119–4125. [Google Scholar] [CrossRef]
- Thaker, Y.; Moon, A.; Afzali, A. Helicobacter pylori: A Review of Epidemiology, Treatment, and Management. J. Clin. Gastroenterol. Treat. 2016, 2, 19. [Google Scholar] [CrossRef]
- Fischbach, W.; Goebeler-Kolve, M.E.; Dragosics, B.; Greiner, A.; Stolte, M. Long term outcome of patients with gastric marginal zone B cell lymphoma of mucosa associated lymphoid tissue (MALT) following exclusive Helicobacter pylori eradication therapy: Experience from a large prospective series. Gut 2004, 53, 34–37. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Schistosomes, Liver Flukes and Helicobacter pylori. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1994; p. 61. [Google Scholar]
- Thung, I.; Aramin, H.; Vavinskaya, V.; Gupta, S.; Park, J.Y.; Crowe, S.E.; Valasek, M.A. Review article: The global emergence of Helicobacter pylori antibiotic resistance. Aliment. Pharmacol. Ther. 2016, 43, 514–533. [Google Scholar] [CrossRef]
- Jeong, J.-Y.; Mukhopadhyay, A.K.; Dailidiene, D.; Wang, Y.; Velapatiño, B.; Gilman, R.H.; Parkinson, A.J.; Nair, G.B.; Wong, B.C.Y.; Lam, S.K.; et al. Sequential Inactivation of rdxA (HP0954) and frxA (HP0642) Nitroreductase Genes Causes Moderate and High-Level Metronidazole Resistance in Helicobacter pylori. J. Bacteriol. 2000, 182, 5082–5090. [Google Scholar] [CrossRef]
- Kwon, D.H.; Hulten, K.; Kato, M.; Kim, J.J.; Lee, M.; El-Zaatari, F.A.K.; Osato, M.S.; Graham, D.Y. DNA Sequence Analysis of rdxA andfrxA from 12 Pairs of Metronidazole-Sensitive and -Resistant Clinical Helicobacter pylori Isolates. Antimicrob. Agents Chemother. 2001, 45, 2609–2615. [Google Scholar] [CrossRef]
- Chua, E.-G.; Debowski, A.W.; Webberley, K.M.; Peters, F.; Lamichhane, B.; Loke, M.-F.; Vadivelu, J.; Tay, C.-Y.; Marshall, B.J.; Wise, M.J. Analysis of core protein clusters identifies candidate variable sites conferring metronidazole resistance in Helicobacter pylori. Gastroenterol. Rep. 2019, 7, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.-H.; El-Zaatari, F.A.K.; Kato, M.; Osato, M.S.; Reddy, R.; Yamaoka, Y.; Graham, D.Y. Analysis of rdxA and Involvement of Additional Genes Encoding NAD(P)H Flavin Oxidoreductase (FrxA) and Ferredoxin-Like Protein (FdxB) in Metronidazole Resistance of Helicobacter pylori. Antimicrob. Agents Chemother. 2000, 44, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Francesco, V.D.; Zullo, A.; Hassan, C.; Giorgio, F.; Rosania, R.; Ierardi, E. Mechanisms of Helicobacter pylori antibiotic resistance: An updated appraisal. World J. Gastrointest. Pathophysiol. 2011, 2, 35–41. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, N.; Kwon, Y.H.; Nam, R.H.; Kim, J.M.; Park, J.Y.; Lee, Y.S.; Lee, D.H. rdxA, frxA, and efflux pump in metronidazole-resistantHelicobacter pylori: Their relation to clinical outcomes. J. Gastroenterol. Hepatol. 2018, 33, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Reysset, G. Genetics of 5-Nitroimidazole Resistance inBacteroidesSpecies. Anaerobe 1996, 2, 59–69. [Google Scholar] [CrossRef]
- Haggoud, A.; Reysset, G.; Azeddoug, H.; Sebald, M. Nucleotide sequence analysis of two 5-nitroimidazole resistance determinants from Bacteroides strains and of a new insertion sequence upstream of the two genes. Antimicrob. Agents Chemother. 1994, 38, 1047–1051. [Google Scholar] [CrossRef]
- Carlier, J.P.; Sellier, N.; Rager, M.N.; Reysset, G. Metabolism of a 5-nitroimidazole in susceptible and resistant isogenic strains of Bacteroides fragilis. Antimicrob. Agents Chemother. 1997, 41, 1495–1499. [Google Scholar] [CrossRef]
- Alauzet, C.; Lozniewski, A.; Marchandin, H. Metronidazole resistance and nim genes in anaerobes: A review. Anaerobe 2019, 55, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Dingsdag, S.A.; Hunter, N. Metronidazole: An update on metabolism, structure–cytotoxicity and resistance mechanisms. J. Antimicrob. Chemother. 2018, 73, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Meggersee, R.; Abratt, V. The occurrence of antibiotic resistance genes in drug resistant Bacteroides fragilis isolates from Groote Schuur Hospital, South Africa. Anaerobe 2015, 32, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Husain, F.; Veeranagouda, Y.; Hsi, J.; Meggersee, R.; Abratt, V.; Wexler, H.M. Two Multidrug-Resistant Clinical Isolates of Bacteroides fragilis Carry a Novel Metronidazole ResistancenimGene (nimJ). Antimicrob. Agents Chemother. 2013, 57, 3767–3774. [Google Scholar] [CrossRef]
- Gal, M.; Brazier, J.S. Metronidazole resistance in Bacteroides spp. carrying nim genes and the selection of slow-growing metronidazole-resistant mutants. J. Antimicrob. Chemother. 2004, 54, 109–116. [Google Scholar] [CrossRef]
- Stanko, A.P.; Sóki, J.; Brkić, D.V.; Plečko, V. Lactate dehydrogenase activity in Bacteroides fragilis group strains with induced resistance to metronidazole. J. Glob. Antimicrob. Resist. 2016, 5, 11–14. [Google Scholar] [CrossRef]
- Theron, M.M.; Van Rensburg, M.N.J.; Chalkley, L.J. Nitroimidazole resistance genes (nimB) in anaerobic Gram-positive cocci (previously Peptostreptococcus spp.). J. Antimicrob. Chemother. 2004, 54, 240–242. [Google Scholar] [CrossRef]
- Kullin, B.; Brock, T.; Rajabally, N.; Anwar, F.; Vedantam, G.; Reid, S.; Abratt, V. Characterisation of Clostridium difficile strains isolated from Groote Schuur Hospital, Cape Town, South Africa. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1709–1718. [Google Scholar] [CrossRef]
- Sóki, J.; Hedberg, M.; Patrick, S.; Bálint, B.; Herczeg, R.; Nagy, I.; Hecht, D.W.; Nagy, E.; Urbán, E. Emergence and evolution of an international cluster of MDR Bacteroides fragilis isolates. J. Antimicrob. Chemother. 2016, 71, 2441–2448. [Google Scholar] [CrossRef]
- Sethi, S.; Shukla, R.; Bala, K.; Gautam, V.; Angrup, A.; Ray, P. Emerging metronidazole resistance in Bacteroides spp. and its association with the nim gene: A study from North India. J. Glob. Antimicrob. Resist. 2019, 16, 210–214. [Google Scholar] [CrossRef]
- Leitsch, D.; Sóki, J.; Kolarich, D.; Urbán, E.; Nagy, E. A study on Nim expression in Bacteroides fragilis. Microbiology 2014, 160, 616–622. [Google Scholar] [CrossRef]
- Ank, N.; Sydenham, T.V.; Iversen, L.H.; Justesen, U.S.; Wang, M. Characterisation of a multidrug-resistant Bacteroides fragilis isolate recovered from blood of a patient in Denmark using whole-genome sequencing. Int. J. Antimicrob. Agents 2015, 46, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, S.P.; Cunningham, S.A.; Jeraldo, P.R.; Wilson, J.W.; Khare, R.; Patel, R. Metronidazole- and car-bapenem-resistant bacteroides thetaiotaomicron isolated in Rochester, Minnesota, in 2014. Antimicrob. Agents Chemother. 2015, 59, 4157–4161. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M.; Spengler, G.; Urbán, E. Identification and Antimicrobial Susceptibility Testing of Anaerobic Bacteria: Ru-bik’s Cube of Clinical Microbiology? Antibiotics 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Boshoff, H. Nitroimidazoles for the treatment of TB: Past, present and future. Future Med. Chem. 2011, 3, 1427–1454. [Google Scholar] [CrossRef]
- WHO Global Tuberculosis Report. 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf?ua=1 (accessed on 17 December 2020).
- Boshoff, H.I.; Barry, C.E. Is the mycobacterial cell wall a hopeless drug target for latent tuberculosis? Drug Discov. Today Dis. Mech. 2006, 3, 237–245. [Google Scholar] [CrossRef]
- Singh, R.; Manjunatha, U.; Boshoff, H.I.M.; Ha, Y.H.; Niyomrattanakit, P.; Ledwidge, R.; Dowd, C.S.; Lee, I.Y.; Kim, P.; Zhang, L.; et al. PA-824 Kills Nonreplicating Mycobacterium tuberculosis by Intracellular NO Release. Science 2008, 322, 1392–1395. [Google Scholar] [CrossRef]
- Manjunatha, U.H.; Boshoff, H.; Dowd, C.S.; Zhang, L.; Albert, T.J.; Norton, J.E.; Daniels, L.; Dick, T.; Pang, S.S.; Barry, C.E. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2005, 103, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Matsumoto, M.; Ishida, H.; Ohguro, K.; Yoshitake, M.; Gupta, R.; Geiter, L.; Hafkin, J. Delamanid: From discovery to its use for pulmonary multidrug-resistant tuber-culosis (MDR-TB). Tuberculosis (Edinb.) 2018, 111, 20–30. [Google Scholar] [CrossRef]
- Xavier, A.S.; Lakshmanan, M. Delamanid: A new armor in combating drug-resistant tuberculosis. J. Pharmacol. Pharmacother. 2014, 5, 222–224. [Google Scholar] [CrossRef] [PubMed]
- WHO. Position Statement on the Use of Delamanid for Multidrug-Resistant Tuberculosis. Available online: https://www.who.int/tb/publications/2018/Position_Paper_Delamanid/en/ (accessed on 17 December 2020).
- Stinson, K.; Kurepina, N.; Venter, A.; Fujiwara, M.; Kawasaki, M.; Timm, J.; Shashkina, E.; Kreiswirth, B.N.; Liu, Y.; Matsumoto, M.; et al. MIC of Delamanid (OPC-67683) against Mycobacterium tuberculosis Clinical Isolates and a Proposed Critical Concentration. Antimicrob. Agents Chemother. 2016, 60, 3316–3322. [Google Scholar] [CrossRef] [PubMed]
- Hanaki, E.; Hayashi, M.; Matsumoto, M. Delamanid is not metabolized by Salmonella or human nitroreductases: A pos-sible mechanism for the lack of mutagenicity. Regul. Toxicol. Pharmacol. 2017, 84, 1–8. [Google Scholar] [CrossRef]
- Diacon, A.H.; Dawson, R.; Hanekom, M.; Narunsky, K.; Venter, A.; Hittel, N.; Geiter, L.J.; Wells, C.D.; Paccaly, A.J.; Donald, P.R. Early bactericidal activity of delamanid (OPC-67683) in smear-positive pulmonary tuberculosis patients. Int. J. Tuberc. Lung Dis. 2011, 15, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.J.; Lewis, J.M. The role of delamanid in the treatment of drug-resistant tuberculosis. Ther. Clin. Risk Manag. 2015, 11, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Skripconoka, V.; Danilovits, M.; Pehme, L.; Tomson, T.; Skenders, G.; Kummik, T.; Cirule, A.; Leimane, V.; Kurve, A.; Levina, K.; et al. Delamanid improves outcomes and reduces mortality in multi-drug-resistant tuberculosis. Eur. Respir. J. 2013, 41, 1393–1400. [Google Scholar] [CrossRef]
- Esposito, S.; Bosis, S.; Tadolini, M.; Bianchini, S.; Migliori, G.B.; Principi, N. Efficacy, safety, and tolerability of a 24-month treatment regimen including delamanid in a child with extensively drug-resistant tuberculosis. Medicine (Baltimore) 2016, 95, e5347. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996, 64, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G. In Vitro Model of Hypoxically Induced Nonreplicating Persistence of Mycobacterium tuberculosis. Methods Mol. Med. 2001, 54, 247–269. [Google Scholar] [CrossRef]
- Upton, A.M.; Cho, S.; Yang, T.J.; Kim, Y.; Wang, Y.; Lu, Y.; Wang, B.; Xu, J.; Mdluli, K.; Ma, Z.; et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hashizume, H.; Tomishige, T.; Nakamura, I.; Matsuba, M.; Fujiwara, M.; Kitamoto, R.; Hanaki, E.; Ohba, Y.; Matsumoto, M. Delamanid kills dormant myco-bacteria in vitro and in a Guinea pig model of tuberculosis. Antimicrob. Agents Chemother. 2017, 61, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with prom-ising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef]
- Zhang, Y.; Yew, W.-W. Mechanisms of drug resistance in Mycobacterium tuberculosis: Update 2015. Int. J. Tuberc. Lung Dis. 2015, 19, 1276–1289. [Google Scholar] [CrossRef]
- Bashiri, G.; Rehan, A.M.; Greenwood, D.R.; Dickson, J.M.J.; Baker, E.N. Metabolic Engineering of Cofactor F420 Production in Mycobacterium smegmatis. PLoS ONE 2010, 5, e15803. [Google Scholar] [CrossRef]
- Choi, K.-P.; Kendrick, N.; Daniels, L. Demonstration that fbiC Is Required by Mycobacterium bovis BCG for Coenzyme F420 and FO Biosynthesis. J. Bacteriol. 2002, 184, 2420–2428. [Google Scholar] [CrossRef]
- Fujiwara, M.; Kawasaki, M.; Hariguchi, N.; Liu, Y.; Matsumoto, M. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis 2018, 108, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Masto, B. Not-for-profit to launch antibiotic against drug-resistant tuberculosis. Nat. Biotechnol. 2019. [Google Scholar] [CrossRef]
- Haver, H.L.; Chua, A.; Ghode, P.; Lakshminarayana, S.B.; Singhal, A.; Mathema, B.; Wintjens, R.; Bifani, P. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 5316–5323. [Google Scholar] [CrossRef]
- Li, S.-Y.; Tasneen, R.; Tyagi, S.; Soni, H.; Converse, P.J.; Mdluli, K.; Nuermberger, E.L. Bactericidal and Sterilizing Activity of a Novel Regimen with Bedaquiline, Pretomanid, Moxifloxacin, and Pyrazinamide in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00913-17. [Google Scholar] [CrossRef]
- Dawson, R.; Diacon, A.H.; Everitt, D.; Van Niekerk, C.; Donald, P.R.; Burger, D.A.; Schall, R.; Spigelman, M.; Conradie, A.; Eisenach, K.; et al. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: A phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 2015, 385, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Warrener, P.; Van Devanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nat. Cell Biol. 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Greening, C.; Ahmed, F.H.; Mohamed, A.E.; Lee, B.M.; Pandey, G.; Warden, A.C.; Scott, C.; Oakeshott, J.G.; Taylor, M.C.; Jackson, C.J. Physiology, Biochemistry, and Applications of F420- and Fo-Dependent Redox Reactions. Microbiol. Mol. Biol. Rev. 2016, 80, 451–493. [Google Scholar] [CrossRef] [PubMed]
- Purwantini, E.; Mukhopadhyay, B. Conversion of NO2 to NO by reduced coenzyme F420 protects mycobacteria from nitrosative damage. Proc. Natl. Acad. Sci. USA 2009, 106, 6333–6338. [Google Scholar] [CrossRef]
- Gurumurthy, M.; Rao, M.; Mukherjee, T.; Rao, S.P.S.; Boshoff, H.I.; Dick, T.; Barry, C.E., 3rd; Manjunatha, U.H. A novel F(420)-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol. Microbiol. 2013, 87, 744–755. [Google Scholar] [CrossRef]
- Aharoni, A.; Gaidukov, L.; Khersonsky, O.; Gould, S.M.; Roodveldt, C.; Tawfik, D.S. The ’evolvability’ of promiscuous protein functions. Nat. Genet. 2004, 37, 73–76. [Google Scholar] [CrossRef]
- Lee, B.M.; Harold, L.K.; Almeida, D.V.; Afriat-Jurnou, L.; Aung, H.L.; Forde, B.M.; Hards, K.; Pidot, S.J.; Ahmed, F.H.; Mohamed, A.E.; et al. Predicting nitroimidazole antibiotic resistance mutations in Mycobacterium tuberculosis with protein engineering. PLoS Pathog. 2020, 16, e1008287. [Google Scholar] [CrossRef]
- Schena, E.; Nedialkova, L.; Borroni, E.; Battaglia, S.; Cabibbe, A.M.; Niemann, S.; Utpatel, C.; Merker, M.; Trovato, A.; Hofmann-Thiel, S.; et al. Delamanid susceptibility testing of Mycobacterium tuberculosis using the resazurin microtitre assay and the BACTEC™ MGIT™ 960 system. J. Antimicrob. Chemother. 2016, 71, 1532–1539. [Google Scholar] [CrossRef]
- Ryan, A. Azoreductases in drug metabolism. Br. J. Pharmacol. 2017, 174, 2161–2173. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Rahim, N.A.; Cheah, S.; Johnson, M.D.; Yu, H.; Sidjabat, H.E.; Boyce, J.; Butler, M.S.; Cooper, M.A.; Fu, J.; Paterson, D.L.; et al. Synergistic killing of NDM-producing MDR Klebsiella pneumoniae by two ’old’ antibiotics-polymyxin B and chloramphenicol. J. Antimicrob. Chemother. 2015, 70, 2589–2597. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Linet, M.; Gao, R.; Gao, Y.; Brinton, L.; Jin, F.; Fraumeni, J. Chloramphenicol use and childhood leukaemia in Shanghai. Lancet 1987, 330, 934–937. [Google Scholar] [CrossRef]
- Lim, C.; Takahashi, E.; Hongsuwan, M.; Wuthiekanun, V.; Thamlikitkul, V.; Hinjoy, S.; Day, N.P.; Peacock, S.J.; Limmathurotsakul, D. Epidemiology and burden of multidrug-resistant bacterial infection in a developing country. eLife 2016, 5, e18082. [Google Scholar] [CrossRef] [PubMed]
- Sood, S. Chloramphenicol—A Potent Armament Against Multi-Drug Resistant (MDR) Gram Negative Bacilli? J. Clin. Diagn. Res. 2016, 10, DC01-3. [Google Scholar] [CrossRef] [PubMed]
- Crofts, T.S.; Sontha, P.; King, A.O.; Wang, B.; Biddy, B.A.; Zanolli, N.; Gaumnitz, J.; Dantas, G. Discovery and character-isation of a nitroreductase capable of conferring bacterial resistance to chloramphenicol. Cell Chem. Biol. 2019, 26, 559–570. [Google Scholar] [CrossRef]
- Eliakim-Raz, N.; Lador, A.; Leibovici-Weissman, Y.; Elbaz, M.; Paul, M.; Leibovici, L. Efficacy and safety of chloramphenicol: Joining the revival of old antibiotics? Systematic review and meta-analysis of randomized controlled trials. J. Antimicrob. Chemother. 2015, 70, 979–996. [Google Scholar] [CrossRef] [PubMed]
- Menzel, T.M.; Tischer, M.; François, P.; Nickel, J.; Schrenzel, J.; Bruhn, H.; Albrecht, A.; Lehmann, L.; Holzgrabe, U.; Ohlsen, K. Mode-of-action studies of the novel bisquaternary bisnaphthalimide MT02 against Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 311–320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chatterjee, S.S.; Otto, M. Improved understanding of factors driving methicillin-resistant Staphylococcus aureus epidemic waves. Clin. Epidemiol. 2013, 5, 205–217. [Google Scholar]
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef]
- Shanson, D. Antibiotic-resistant Staphylococcus aureus. J. Hosp. Infect. 1981, 2, 11–36. [Google Scholar] [CrossRef]
- Smith, J.T.; Amyes, S.G.B. Bacterial resistance to antifolate chemotherapeutic agents mediated by Plasmids. Br. Med. Bull. 1984, 40, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Jevons, M.P. “Celbenin”—Resistant Staphylococci. BMJ 1961, 1, 124–125. [Google Scholar] [CrossRef]
- El-Hossary, E.M.; Förstner, K.U.; François, P.; Baud, D.; Streker, K.; Schrenzel, J.; Ohlsen, K.; Holzgrabe, U. A Novel Mechanism of Inactivating Antibacterial Nitro Compounds in the Human Pathogen Staphylococcus aureus by Overexpression of a NADH-Dependent Flavin Nitroreductase. Antimicrob. Agents Chemother. 2018, 62, e01510-17. [Google Scholar] [CrossRef]
- Tejman-Yarden, N.; Millman, M.; Lauwaet, T.; Davids, B.J.; Gillin, F.D.; Dunn, L.; Upcroft, J.A.; Miyamoto, Y.; Eckmann, L. Impaired parasite attachment as fitness cost of metronidazole re-sistance in Giardia lamblia. Antimicrob. Agents Chemother. 2011, 55, 4643–4651. [Google Scholar] [CrossRef]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development Against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef]
- Allarakhia, M. Open-source approaches for the repurposing of existing or failed candidate drugs: Learning from and applying the lessons across diseases. Drug Des. Dev. Ther. 2013, 7, 753–766. [Google Scholar] [CrossRef]
- Çelik, A.; Yetiş, G.; Ay, M.; Güngör, T. Modification of existing antibiotics in the form of precursor prodrugs that can be subsequently activated by nitroreductases of the target pathogen. Bioorg. Med. Chem. Lett. 2016, 26, 4057–4060. [Google Scholar] [CrossRef]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-Lactamase Inhibitors: A Therapeutic Renaissance in an MDR World. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef]
- Sander, C.; McShane, H. Translational Mini-Review Series on Vaccines: Development and evaluation of improved vaccines against tuberculosis. Clin. Exp. Immunol. 2007, 147, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Gengenbacher, M. Recombinant live vaccine candidates against tuberculosis. Curr. Opin. Biotechnol. 2012, 23, 900–907. [Google Scholar] [CrossRef]
- Kwon, K.W.; Kim, W.S.; Kim, H.; Han, S.J.; Hahn, M.-Y.; Lee, J.S.; Nam, K.T.; Cho, S.-N.; Shin, S.J. Novel vaccine potential of Rv3131, a DosR regulon-encoded putative nitroreductase, against hyper-virulent Mycobacterium tuberculosis strain K. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, C.; Gwenin, C.D. The Role of Nitroreductases in Resistance to Nitroimidazoles. Biology 2021, 10, 388. https://doi.org/10.3390/biology10050388
Thomas C, Gwenin CD. The Role of Nitroreductases in Resistance to Nitroimidazoles. Biology. 2021; 10(5):388. https://doi.org/10.3390/biology10050388
Chicago/Turabian StyleThomas, Carol, and Christopher D. Gwenin. 2021. "The Role of Nitroreductases in Resistance to Nitroimidazoles" Biology 10, no. 5: 388. https://doi.org/10.3390/biology10050388
APA StyleThomas, C., & Gwenin, C. D. (2021). The Role of Nitroreductases in Resistance to Nitroimidazoles. Biology, 10(5), 388. https://doi.org/10.3390/biology10050388