
Labelling Selective Sweeps Used in Durum Wheat Breeding from a Diverse and Structured Panel of Landraces and Cultivars




Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Genotyping
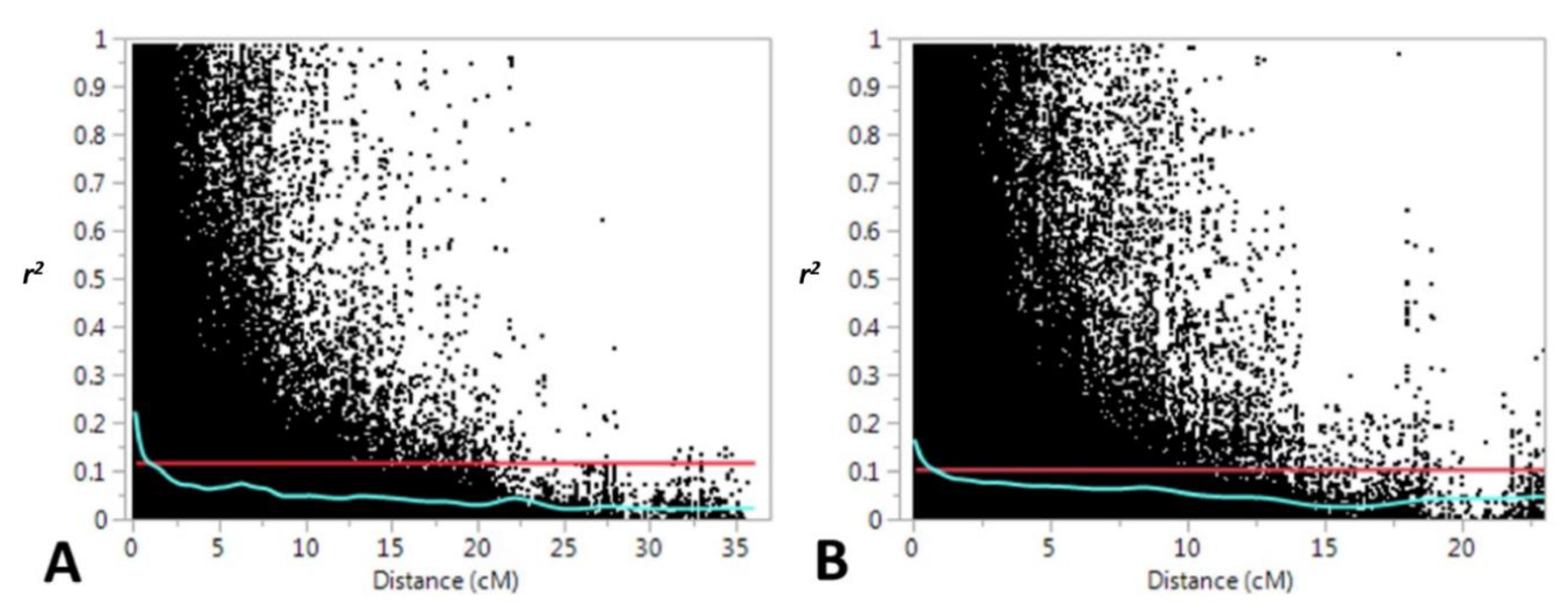
2.3. Data Analysis
2.4. Identification of Selective Sweeps
2.5. Gene Annotation
3. Results
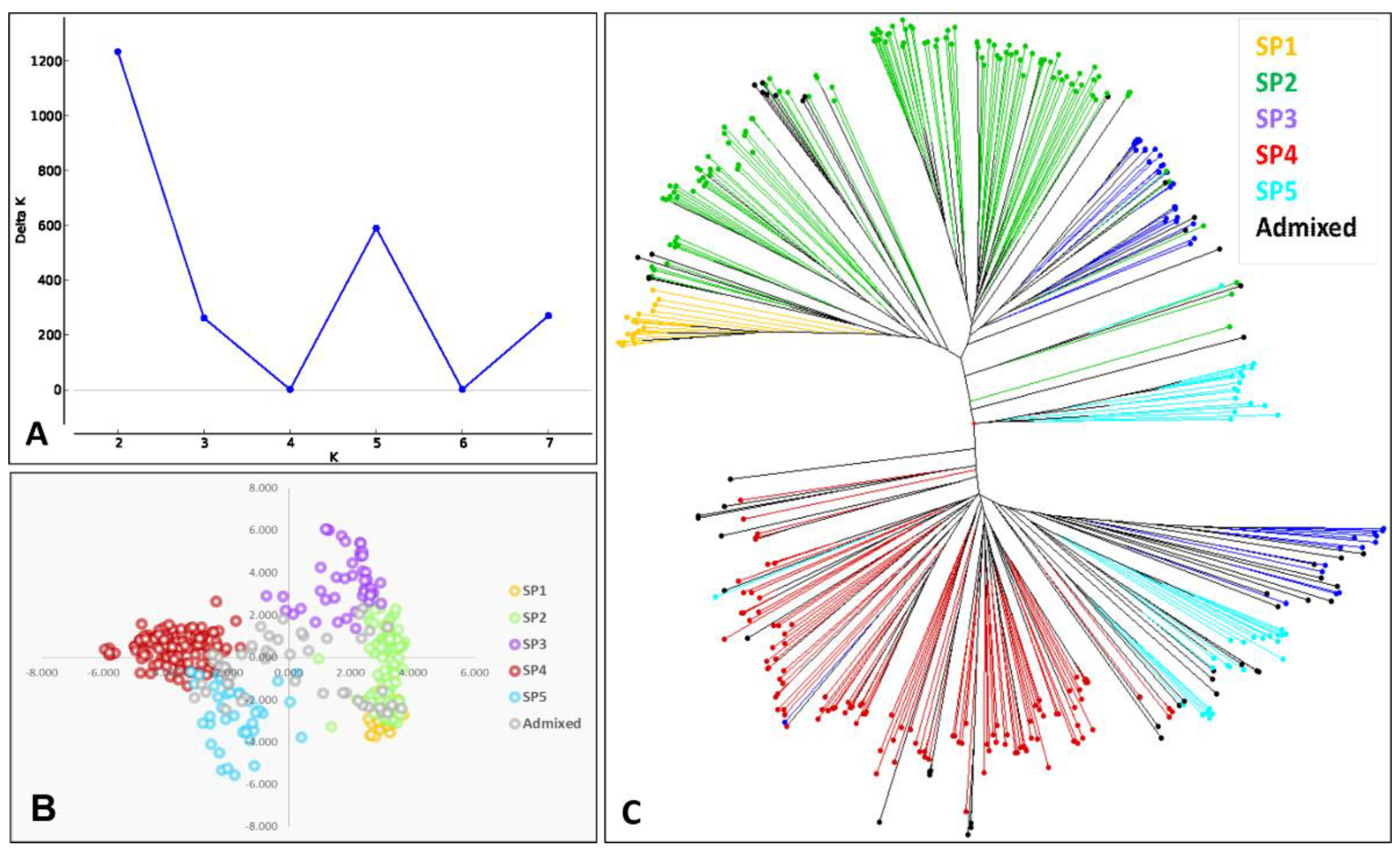
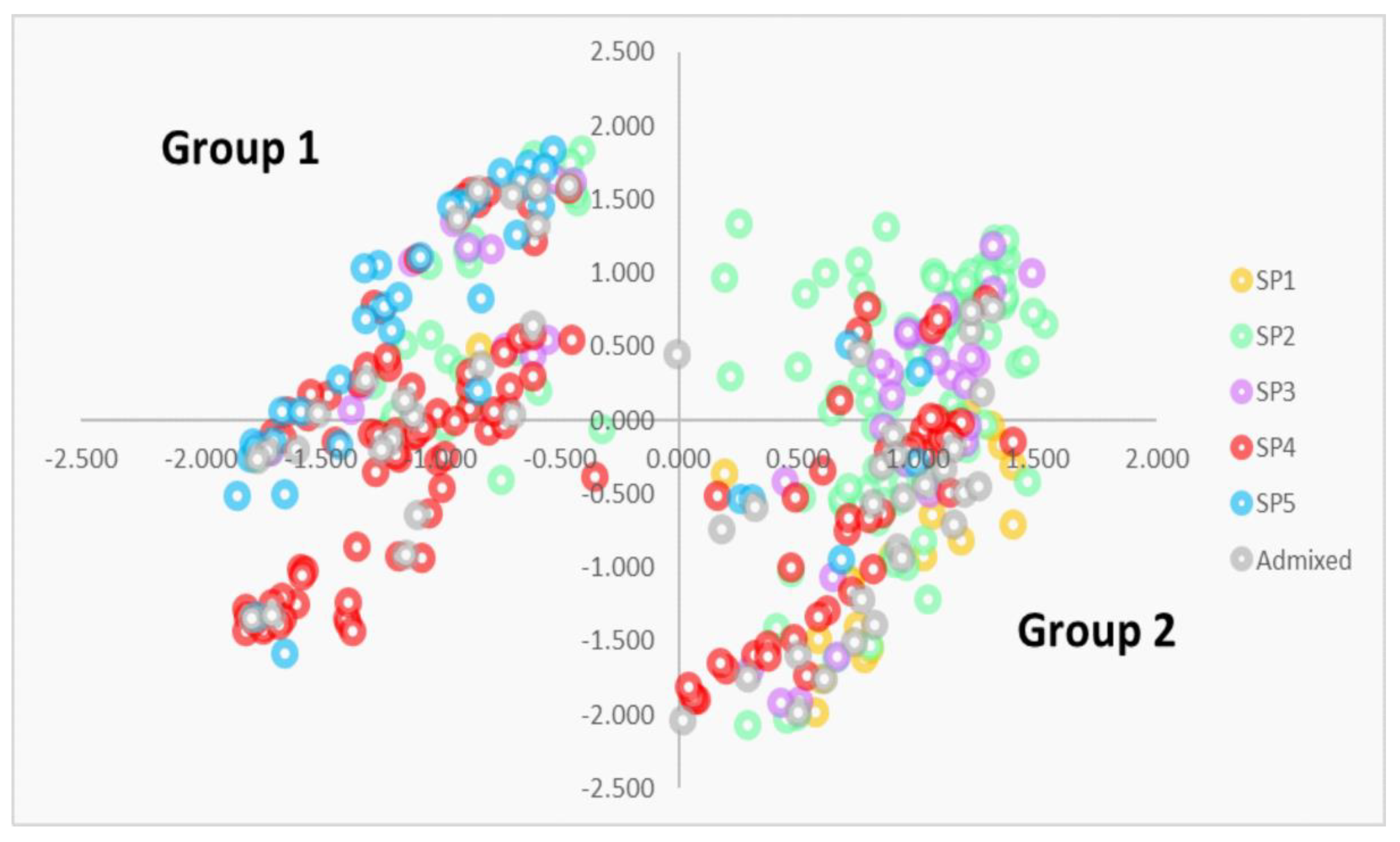
3.1. Genetic Diversity and Population Structure
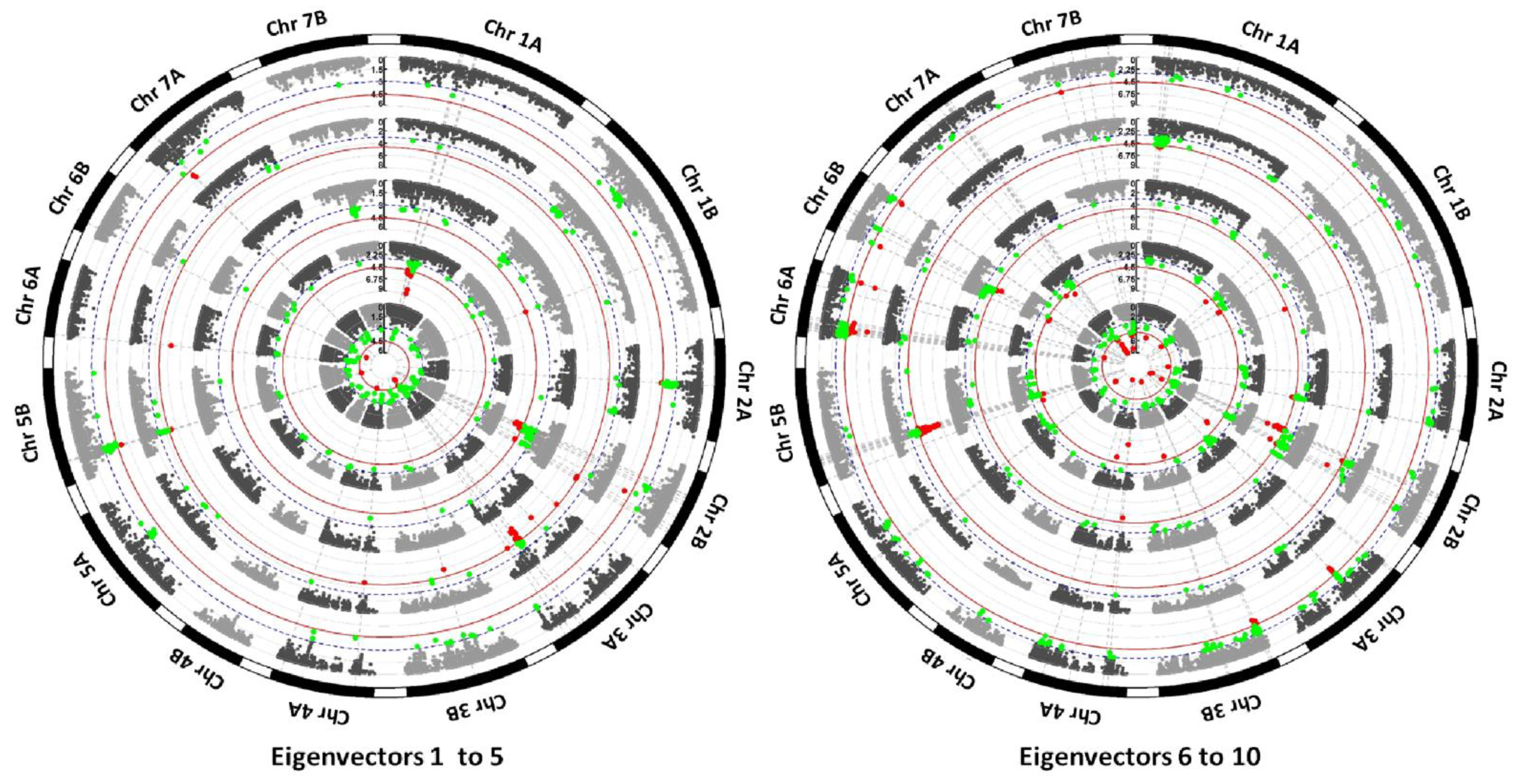
3.2. Identification of Loci under Selection by EigenGWAS
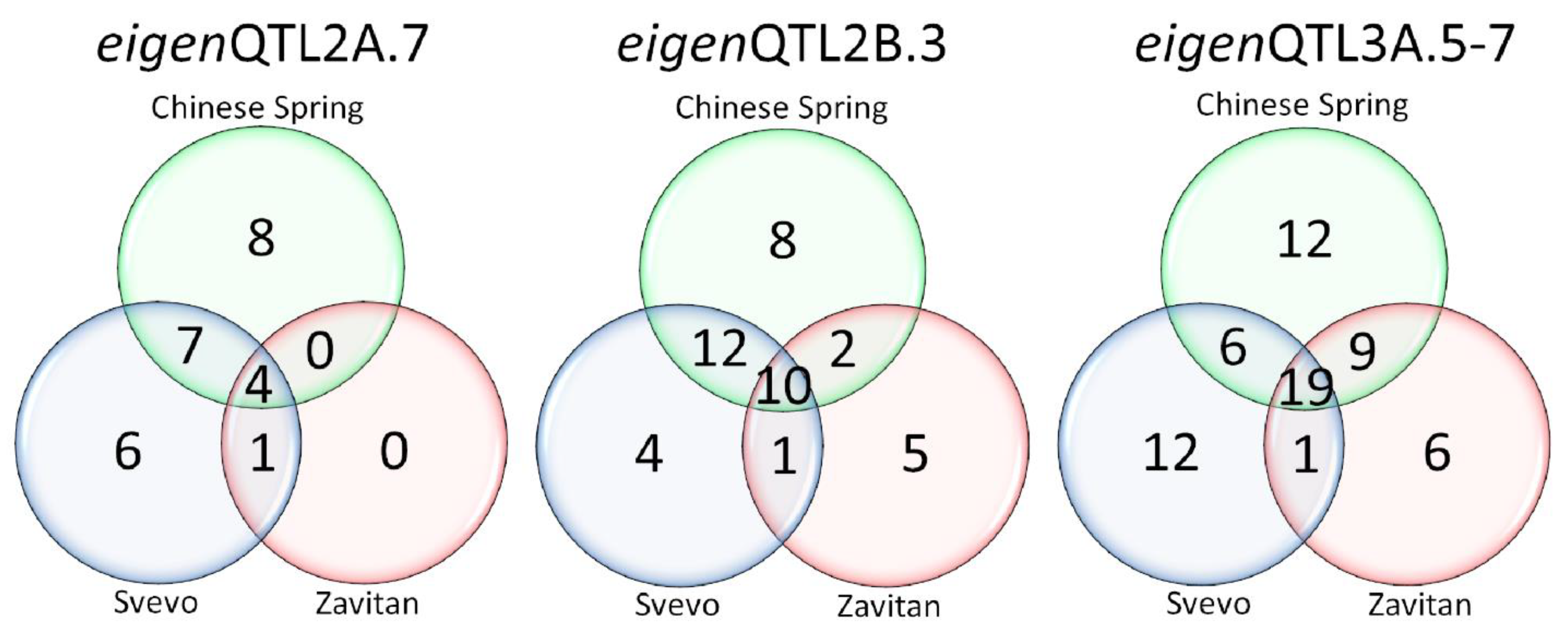
3.3. Identification of Selection Regions among SPs
3.4. Gene Annotation
4. Discussion
4.1. Genetic Diversity and Population Structure
4.2. Detection of Selective Sweeps by EigenGWAS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mac Key, J. Wheat: Its Concept, Evolution and Taxonomy. In Durum Wheat Breeding: Current Approaches and Future Strategies; Royo, C., Nachit, M., di Fonzo, N., Araus, J.L., Pfeiffer, W.H., Slafer, G.A., Eds.; Fodd Products Press: New York, NY, USA, 2005; pp. 3–62. [Google Scholar]
- Moragues, M.; García del Moral, L.; Moralejo, M.; Royo, C. Yield formation strategies of durum wheat landraces with distinct pattern of dispersal within the Mediterranean basin I: Yield components. Field Crop. Res. 2006, 95, 194–205. [Google Scholar] [CrossRef]
- Peng, J.H.; Sun, D.; Nevo, E. Domestication evolution, genetics and genomics in wheat. Mol. Breed. 2011, 28, 281–301. [Google Scholar] [CrossRef]
- Hammer, K.; Teklu, Y. Plant Genetic Resources: Selected Issues from Genetic Erosion to Genetic Engineering. J. Agric. Rural Dev. Trop. Subtrop. 2008, 109, 15–50. [Google Scholar]
- Nazco, R.; Villegas, D.; Ammar, K.; Peña, R.J.; Moragues, M.; Royo, C. Can Mediterranean durum wheat landraces contribute to improved grain quality attributes in modern cultivars? Euphytica 2012, 185, 1–17. [Google Scholar] [CrossRef]
- Nazco, R.; Peña, R.J.; Ammar, K.; Villegas, D.; Crossa, J.; Royo, C. Durum wheat (Triticum durum Desf.) Mediterranean landraces as sources of variability for allelic combinations at Glu-1/Glu-3 loci affecting gluten strength and pasta cooking quality. Genet. Resour. Crop Evol. 2014, 61, 1219–1236. [Google Scholar] [CrossRef]
- Kyzeridis, N.; Biesantz, A.; Limberg, P. Comparative trials with durum-wheat (Triticum turgidum var. durum) landraces and cultivars in different ecological environments in the mediterranean region. J. Agron. Crop Sci. 1995, 174, 133–144. [Google Scholar] [CrossRef]
- Du Toit, F. Components of Resistance in Three Bread Wheat Lines to Russian Wheat Aphid (Homoptera: Aphididae) in South Africa. J. Econ. Entomol. 1989, 82, 1779–1781. [Google Scholar] [CrossRef]
- Talas, F.; Longin, F.; Miedaner, T. Sources of resistance to Fusarium head blight within Syrian durum wheat landraces. Plant Breed. 2011, 130, 398–400. [Google Scholar] [CrossRef]
- Valdez, V.A.; Byrne, P.F.; Lapitan, N.L.V.; Pearis, F.B.; Bernardo, A.; Bai, G.; Haley, S.D. Inheritance and Genetic Mapping of Russian Wheat Aphid Resistance in Iranian Wheat Landrace Accession PI 626580. Crop Sci. 2012, 52, 676. [Google Scholar] [CrossRef]
- Stephan, W. Signatures of positive selection: From selective sweeps at individual loci to subtle allele frequency changes in polygenic adaptation. Mol. Ecol. 2016, 25, 79–88. [Google Scholar] [CrossRef]
- Lake, L.; Li, Y.; Casal, J.J.; Sadras, V.O. Negative association between chickpea response to competition and crop yield: Phenotypic and genetic analysis. Field Crop. Res. 2016, 196, 409–417. [Google Scholar] [CrossRef]
- Chen, G.B.; Lee, S.H.; Zhur, Z.X.; Benyamin, B.; Robinson, M.R. EigenGWAS: Finding loci under selection through genome-wide association studies of eigenvectors in structured populations. Heredity 2016, 117, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, G.B.; Rasheed, A.; Li, D.; Sonder, K.; Zavala Espinosa, C.; Li, H.; Hearne, S.J.; Schnable, P.S.; Costich, D.E.; et al. Identifying loci with breeding potential across temperate and tropical adaptation via EigenGWAS and EnvGWAS. Mol. Ecol. 2019, 28, 3544–3560. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rasheed, A.; He, Z.; Imtiaz, M.; Arif, A.; Mahmood, T.; Ghafoor, A.; Wen, W.; Gao, F.; Xie, C.; et al. Genome-wide variation patterns between landraces and cultivars uncover divergent selection during modern wheat breeding. Theor. Appl. Genet. 2019, 132, 2509–2523. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lhundrup, N.; Guo, G.; Dol, K.; Chen, P.; Gao, L.; Chemi, W.; Zhang, L.; Wang, L.; Li, H.; et al. Characterization of Genetic Diversity and Genome-Wide Association Mapping of Three Agronomic Traits in Qingke Barley (Hordeum Vulgare L.) in the Qinghai-Tibet Plateau. Front. Genet. 2020, 11, 638. [Google Scholar] [CrossRef]
- Mwadzingeni, L.; Shimelis, H.; Rees, D.J.G.; Tsilo, T.J. Genome-wide association analysis of agronomic traits in wheat under drought-stressed and non-stressed conditions. PLoS ONE 2017, 12, e0171692. [Google Scholar] [CrossRef]
- Wang, S.X.; Zhu, Y.L.; Zhang, D.X.; Shao, H.; Liu, P.; Hu, J.B.; Zhang, H.; Zhang, H.P.; Chang, C.; Lu, J.; et al. Genome-wide association study for grain yield and related traits in elite wheat varieties and advanced lines using SNP markers. PLoS ONE 2017, 12, e0188662. [Google Scholar] [CrossRef]
- Mangini, G.; Gadaleta, A.; Colasuonno, P.; Marcotuli, I.; Signorile, A.M.P.; Simenone, R.; de Vita, P.; Mastrangelo, A.M.; Laidò, G.; Pecchioni, N.; et al. Genetic dissection of the relationships between grain yield components by genome-wide association mapping in a collection of tetraploid wheats. PLoS ONE 2018, 13, e0190162. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Reynolds, M.P.; Sansaloni, C. Genome-Wide Association Analyses Identify QTL Hotspots for Yield and Component Traits in Durum Wheat Grown under Yield Potential, Drought, and Heat Stress Environments. Front. Plant Sci. 2018, 9, 81. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, C.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed]
- IWGSC. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef]
- Doyle, J.; Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, P54. [Google Scholar] [CrossRef]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.A.; McDermott, J.M. Population genetics of plant pathogenic fungi. Bioscience 1993, 43, 311–319. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, R.E.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goude, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Sokal, R.; Michener, C.A. Statistical method for evaluating systematic relationships. Sci. Bull. 1958, 8, 22. [Google Scholar]
- Perrier, X.; Flori, A.; Bonnot, F. Data analysis methods. In Genetic Diversity of Cultivated Tropical Plants; Hamon, P., Seguin, M., Perrier, X., Glaszmann, J.C., Eds.; Enfield Science Publishers: Montpellier, France, 2007; pp. 43–76. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pascual, L.; Ruiz, M.; López-Fernández, M.; Pérez-Peña, H.; Benavente, E.; Vázquez, J.F.; Sansaloni, C.; Giraldo, P. Genomic analysis of Spanish wheat landraces reveals their variability and potential for breeding. BMC Genom. 2020, 21, 122. [Google Scholar] [CrossRef]
- Lopes, M.S.; El-Basyoni, I.; Baenziger, P.S.; Sing, S.; Royo, C.; Ozbek, K.; Aktas, H.; Ozer, E.; Ozdemir, F.; Manickavelu, A.; et al. Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J. Exp. Bot. 2015, 66, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Royo, C.; Dreisigacker, S.; Ammar, K.; Villegas, D. Agronomic performance of durum wheat landraces and modern cultivars and its association with genotypic variation in vernalization response (Vrn-1) and photoperiod sensitivity (Ppd-1) genes. Eur. J. Agron. 2020, 120, 126129. [Google Scholar] [CrossRef]
- Baloch, F.S.; Alsaleh, A.; Shahid, M.Q.; Çiftçi, V.E.; Saenz de Miera, L.; Aasim, M.; Nadeem, M.A.; Aktas, H.; Özkan, H.; Hatipoğlu, R. A Whole Genome DArTseq and SNP Analysis for Genetic Diversity Assessment in Durum Wheat from Central Fertile Crescent. PLoS ONE 2017, 12, e0167821. [Google Scholar] [CrossRef]
- Kabbaj, H.; Sall, A.T.; Al-Abdallat, A.; Geleta, M.; Amri, A.; Filali-Maltouf, A.; Belkadi, B.; Ortiz, R.; Bassi, F.M. Genetic diversity within a global panel of durum wheat (Triticum durum) landraces and modern germplasm reveals the history of alleles exchange. Front. Plant Sci. 2017, 8, 1277. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; White, R.L.; Sholnick, M.; David, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Soriano, J.M.; Villegas, D.; Aranzana, M.; García del Moral, L.F.; Royo, C. Genetic Structure of Modern Durum Wheat Cultivars and Mediterranean Landraces Matches With Their Agronomic Performance. PLoS ONE 2016, 11, e0160983. [Google Scholar] [CrossRef] [PubMed]
- Rufo, R.; Alvaro, F.; Royo, C.; Soriano, J.M. From landraces to improved cultivars: Assessment of genetic diversity and population structure of Mediterranean wheat using SNP markers. PLoS ONE 2019, 14, e0219867. [Google Scholar] [CrossRef]
- Chesnokov, Y.V.; Artemyeva, A.M. Evaluation of the measure of polymorphism information of genetic diversity. Agric. Biol. 2015, 5, 571–578. [Google Scholar]
- Moragues, M.; Moralejo, M.A.; Sorrells, M.E.; Royo, C. Dispersal of durum wheat landraces across the Mediterranean basin assessed by AFLPs and microsatellites. Gen. Res. Crop Evol. 2007, 54, 1133–1144. [Google Scholar] [CrossRef]
- Royo, C.; Dreisigacker, S.; Soriano, J.M.; Lopes, M.S.; Ammar, K.; Villegas, D. Allelic variation at the vernalization response (Vrn-1) and photoperiod sensitivity (Ppd-1) genes and their association with the development of durum wheat landraces and modern cultivars. Front. Plant Sci. 2020, 11, 838. [Google Scholar] [CrossRef]
- Royo, C.; Elias, E.M.; Manthey, F.A. Durum Wheat Breeding. In Handbook of Plant Breeding: Cereals; Carena, M.J., Ed.; Springer Science + Business Media: Berlin, Germany, 2009; pp. 199–226. [Google Scholar]
- Parzies, H.K.; Spoor, W.; Ennos, R.A. Inferring seed exchange between farmers from population genetic structure of barley landrace Arabi Aswad from Northern Syria. Genet. Resour. Crop Evol. 2004, 51, 471–478. [Google Scholar] [CrossRef]
- Ben-Romdhane, M.; Riah, L.; Selmi, A.; Jardak, R.; Bouajila, A.; Ghorbel, A.; Zoghlami, N. Low genetic differentiation and evidence of gene flow among barley landrace populations in Tunisia. Crop Sci. 2017, 57, 1585–1593. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- McVean, G. A genealogical interpretation of principal compnents analysis. PLoS Genet. 2009, 5, e1000686. [Google Scholar] [CrossRef]
- Bryc, K.; Bryc, W.; Silverstein, J.W. Separation of the largest eigenvalues in eigenanalysis of genotype data from discrete subpopulations. Theor. Popul. Biol. 2013, 89, 34–43. [Google Scholar] [CrossRef]
- Cavanagh, C.R.; Chao, S.; Wang, S.; Huang, B.E.; Stephen, S.; Kiani, S.; Forrest, K.; Saintenac, C.; Brown-Guedira, G.L.; Akhunova, A.; et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. USA 2013, 110, 8057–8062. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Z.; Cheng, M.; Chen, J.; Zhu, T.T.; Wang, R.; Liu, Y.; Qi, P.; Chen, G.; Jiang, Q.; et al. Uncovering the dispersion history.; adaptive evolution and selection of wheat in China. Plant Biotechnol. J. 2018, 16, 280–291. [Google Scholar] [CrossRef]
- Sanchez-Garcia, M.; Álvaro, F.; Martín-Sánchez, J.A.; Sillero, J.C.; Escribano, J.; Royo, C. Breeding effects on the genotype×environment interaction for yield of bread wheat grown in Spain during the 20th century. Field Crop. Res. 2012, 126, 79–86. [Google Scholar] [CrossRef]
- De Vita, P.; Li Destri Nicosia, O.; Nigro, F.; Platani, C.; Riefolo, C.; Di Fonzo, N.; Cattivelli, L. Breeding progress in morpho-physiological.; agronomical and qualitative traits of durum wheat cultivars released in Italy during the 20th century. Eur. J. Agron. 2007, 26, 39–53. [Google Scholar] [CrossRef]
- Subirà, J.; Peña, R.J.; Álvaro, F.; Ammar, K.; Ramdani, A.; Royo, C. Breeding progress in the pasta-making quality of durum wheat cultivars released in Italy and Spain during the 20th Century. Crop. Pasture Sci. 2014, 65, 16–26. [Google Scholar] [CrossRef]
- Mangini, G.; Taranto, F.; Delvecchio, L.N.; Pasqualone, A.; Blanco, A. Development and validation of a new Ppo-A1 marker useful for marker-assisted selection in tetraploid wheats. Mol. Breed. 2014, 34, 385–392. [Google Scholar] [CrossRef]
- Taranto, F.; Mangini, G.; Pasqualone, A.; Gadaleta, A.; Blanco, A. Mapping and allelic variations of Ppo-B1 and Ppo-B2 gene-related polyphenol oxidase activity in durum wheat. Mol. Breed. 2015, 35, 80. [Google Scholar] [CrossRef]
- Wei, J.X.; Geng, H.W.; Zhang, Y.; Liu, J.D.; Wen, W.E.; Zhang, Y.; Xia, X.; Chen, X.; He, Z. Mapping quantitative loci for peroxidase activity and developing gene-specific markers for TaPod-A1 on wheat chromosome 3AL. Theor. Appl. Genet. 2015, 128, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Uauy, C.; Brevis, J.C.; Dubcovsky, C. The high grain protein content gene Gpc-B1 accelerates senescence and has pleiotropic effects on protein content in wheat. J. Exp. Bot. 2006, 57, 2785–2794. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, X.; Islam, S.; She, M.; Peng, Y.; Yu, Z.; Wylie, S.; Juhasz, A.; Dowla, M.; Yang, R.; et al. New insights into the evolution of wheat avenin-like proteins in wild emmer wheat (Triticum dicoccoides). Proc. Natl. Acad. Sci. USA 2018, 115, 13312–13317. [Google Scholar] [CrossRef]
- Parada, R.; Royo, C.; Gadaleta, A.; Colasuonno, P.; Marcotuli, I.; Matus, I.; Castillo, D.; Costa de Camargo, A.; Araya-Flores, J.; Villegas, D.; et al. Phytoene synthase (Psy-1) and lipoxygenase (Lpx-1) genes influence on semolina yellowness in wheat Mediterranean germplasm. Int. J. Mol. Sci. 2020, 21, 4669. [Google Scholar] [CrossRef]
- Jiang, Q.; Hou, J.; Hao, C.; Wang, L.; Ge, H.; Dong, Y.; Zhang, X. The wheat (T. aestivum) sucrose synthase 2 gene (TaSus2) active in endosperm development is associated with yield traits. Funct. Integr. Genom. 2011, 11, 49–61. [Google Scholar] [CrossRef]
- Zheng, J.; Liu, H.; Wang, Y.; Wang, L.; Chang, X.; Jing, R. TEF-7A.; a transcript elongation factor gene.; influences yield-related traits in bread wheat (Triticum aestivum L.). J. Exp. Bot. 2014, 65, 5351–5365. [Google Scholar] [CrossRef] [PubMed]
- Li, X.P.; Zhao, X.Q.; He, X.; Zhao, G.Y.; Li, B.; Li, D.C.; Zhang, A.M.; Zhang, X.Y.; Tong, Y.P.; Li, Z.S. Haplotype analysis of the genes encoding glutamine synthetase plastic isoforms and their association with nitrogen-use- and yield-related traits in bread wheat. New Phytol. 2011, 189, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Jia, H.; Yin, J.; Wang, B.Q.; Ma, Z.Q.; Shen, T. Development of an STS marker linked to powdery mildew resistance genes PmLK906 and Pm4a by gene chip hybridization. Agric. Sci. China 2010, 9, 331–336. [Google Scholar] [CrossRef]
- Larriba, E.; Jaime, M.D.; Carbonell-Caballero, J.; Conesa, A.; Dopazo, J.; Nislow, C.; Matin-Nieto, J.; López-Llorca, L.V. Sequencing and functional analysis of the genome of a nematode egg-parasitic fungus.; Pochonia chlamydosporia. Fungal Genet. Biol. 2014, 65, 69–80. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ahmad, D.; Zhang, X.; Zhang, Y.; Wu, L.; Jiang, P.; Alma, H. Genome-wide analysis of family-1 UDP glycosyltransferases (UGT) and identification of UGT genes for FHB resistance in wheat (Triticum aestivum L.). BMC Plant Biol. 2018, 18, 67. [Google Scholar] [CrossRef]
- Skirpan, A.L.; McCubbin, A.G.; Ishimizu, T.; Wang, X.; Hu, Y.; Dowd, P.E.; Alma, H.; Kao, T. Isolation and characterization of kinase interacting protein 1.; a pollen protein that interacts with the kinase domain of PRK1.; a receptor-like kinase of petunia. Plant Physiol. 2001, 26, 1480–1492. [Google Scholar] [CrossRef]
- Dufayard, J.F.; Bettembourg, M.; Fischer, I.; Droc, G.; Guiderdoni, E.; Périn, C.; Chantret, N.; Dievart, A. New Insights on Leucine-Rich Repeats Receptor-Like Kinase Orthologous Relationships in Angiosperms. Front. Plant Sci. 2017, 8, 381. [Google Scholar]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef]
- Salas-Muñoz, S.; Rodríguez-Hernández, A.A.; Ortega-Amaro, M.A.; Salazar-Badillo, F.B.; Jiménez-Bremont, J.F. Arabidopsis AtDjA3 Null Mutant Shows Increased Sensitivity to Abscisic Acid.; Salt.; and Osmotic Stress in Germination and Post-germination Stages. Front. Plant Sci. 2016, 7, 220. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, Y.; Luo, W.; Yang, C.; Ding, P.; Liu, Y.; Qiao, L.; Chang, Z.; Geng, H.; Wang, P.; et al. Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.). PLoS ONE 2017, 12, e0181443. [Google Scholar] [CrossRef]
- Guo, J.; Shi, X.X.; Zhang, J.S.; Duan, Y.H.; Bai, P.F.; Guan, X.N.; Kang, Z.S. A type I MADS-box gene is differentially expressed in wheat in response to infection by the stripe rust fungus. Biol. Plant. 2013, 57, 540–546. [Google Scholar] [CrossRef]
- Wei, B.; Zhang, R.Z.; Guo, J.J.; Liu, D.M.; Li, A.L.; Fan, R.C.; Mao, L.; Zhang, X.Q. Genome-wide analysis of the MADS-box gene family in Brachypodium distachyon. PLoS ONE 2014, 9, e84781. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Pandey, G.K. Expansion and Function of Repeat Domain Proteins During Stress and Development in Plants. Front. Plant Sci. 2016, 6, 1218. [Google Scholar] [CrossRef]
- Qiao, L.; Zhang, W.; Li, X.; Zhang, L.; Zhang, X.; Li, X.; Guo, H.; Ren, Y.; Zheng, J.; Chang, Z. Characterization and Expression Patterns of Auxin Response Factors in Wheat. Front. Plant Sci. 2018, 9, 1395. [Google Scholar] [CrossRef] [PubMed]
- Li, A.X.; Han, Y.Y.; Wang, X.; Chen, Y.H.; Zhao, M.R.; Zhou, S.M.; Wang, W. Root-specific expression of wheat expansin gene TaEXPB23 enhances root growth and water stress tolerance in tobacco. Environ. Exp. Bot. 2014, 110, 73–84. [Google Scholar] [CrossRef]
- Chang, H.; Chen, D.; Kam, J.; Richadson, T.; Drenth, J.; Guo, X.; McIntyre, L.; Chai, S.; Rae, A.L.; Xue, G.P. Abiotic stress upregulated TaZFP34 represses the expression of type-B response regulator and SHY2 genes and enhances root to shoot ratio in wheat. Plant Sci. 2016, 252, 88–102. [Google Scholar] [CrossRef]
- Lee, I.; Aukerman, M.J.; Gore, S.L.; Lohman, K.N.; Michaels, S.D.; Weaver, L.M.; John, M.C.; Feldmann, K.A.; Amasino, R.M. Isolation of LUMINIDEPENDENS—A gene involved in the control of flowering time in Arabidopsis. Plant Cell 1994, 6, 75–83. [Google Scholar] [PubMed]
- Li, M.; Tang, D.; Wang, K.; Wu, X.; Lu, W.; Yu, H.; Gu, M.; Yan, C.; Cheng, Z. Mutations in the F-box gene LARGER PANICLE improve the panicle architecture and enhance the grain yield in rice. Plant Biotechnol. J. 2011, 9, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.J.; Kim, D.Y.; Kang, S.Y.; Kim, D.S.; Kim, J.B.; Seo, Y.W. Wheat F-box protein recruits proteins and regulates their abundance during wheat spike development. Mol. Biol. Rep. 2012, 39, 9681–9696. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, C.; Xing, Y.; Jiang, Y.; Luo, L.; Sun, L.; Shao, D.; Xu, C.; Li, X.; Xiao, J.; et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011, 43, 1266–1269. [Google Scholar] [CrossRef]
- Lei, X.; Liu, B. Tapetum-Dependent Male Meiosis Progression in Plants: Increasing Evidence Emerges. Front. Plant Sci. 2020, 10, 1667. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subpopulation | N | HT | HS | DST | GST | Nm |
|---|---|---|---|---|---|---|
| Total | 388 | 0.40 | 0.37 | 0.03 | 0.08 | 6.02 |
| SP 1 | 19 | 0.36 | - | - | - | - |
| SP 2 | 119 | 0.36 | - | - | - | - |
| SP 3 | 43 | 0.35 | - | - | - | - |
| SP 4 | 116 | 0.40 | - | - | - | - |
| SP 5 | 39 | 0.38 | - | - | - | - |
| Admixed | 51 | 0.35 | - | - | - | - |
| SP 1–2 | 138 | 0.36 | 0.36 | 0.00 | 0.01 | 49.73 |
| SP 1–3 | 62 | 0.33 | 0.35 | 0.02 | 0.07 | 6.90 |
| SP 1–4 | 135 | 0.34 | 0.38 | 0.04 | 0.11 | 3.87 |
| SP 1–5 | 58 | 0.35 | 0.37 | 0.02 | 0.06 | 7.32 |
| SP 2–3 | 162 | 0.36 | 0.36 | 0.00 | 0.01 | 69.81 |
| SP 2–4 | 235 | 0.40 | 0.38 | 0.01 | 0.03 | 14.41 |
| SP 2–5 | 158 | 0.38 | 0.37 | 0.01 | 0.02 | 23.40 |
| SP 3–4 | 159 | 0.35 | 0.38 | 0.03 | 0.08 | 5.41 |
| SP 3–5 | 82 | 0.37 | 0.37 | 0.00 | 0.01 | 42.10 |
| SP 4–5 | 155 | 0.34 | 0.39 | 0.06 | 0.16 | 2.54 |
| Eigen Hotspot | CI Left | CI Right | N MTAs | FDR | Functional Genes |
|---|---|---|---|---|---|
| eigenQTL1A.1 | 12.90 | 40.37 | 58 | 6 | |
| eigenQTL1A.2 | 41.38 | 45.57 | 2 | 0 | |
| eigenQTL1A.3 | 75.97 | 86.96 | 13 | 4 | |
| eigenQTL1A.4 | 97.58 | 109.75 | 19 | 5 | |
| eigenQTL1A.5 | 116.31 | 119.74 | 3 | 1 | Glu-A1 |
| eigenQTL1A.6 | 242.61 | 254.18 | 11 | 0 | |
| eigenQTL1B.1 | 31.81 | 36.72 | 10 | 1 | |
| eigenQTL1B.2 | 37.42 | 41.32 | 2 | 0 | |
| eigenQTL1B.3 | 42.20 | 52.79 | 15 | 0 | |
| eigenQTL1B.4 | 70.87 | 96.28 | 29 | 1 | |
| eigenQTL1B.5 | 96.45 | 115.68 | 15 | 2 | |
| eigenQTL1B.6 | 137.22 | 140.22 | 12 | 0 | Glu-B1 |
| eigenQTL1B.7 | 195.66 | 202.99 | 4 | 2 | |
| eigenQTL1B.8 | 238.34 | 241.34 | 3 | 0 | |
| eigenQTL2A.1 | 10.07 | 14.84 | 2 | 0 | Ppd-A1 |
| eigenQTL2A.2 | 43.25 | 47.06 | 8 | 0 | |
| eigenQTL2A.3 | 57.36 | 73.45 | 48 | 1 | TaSus2-2A |
| eigenQTL2A.4 | 73.47 | 79.30 | 2 | 0 | |
| eigenQTL2A.5 | 85.85 | 91.05 | 4 | 0 | Ppo-A1 |
| eigenQTL2A.6 | 94.09 | 97.09 | 2 | 0 | |
| eigenQTL2A.7 | 112.04 | 126.00 | 101 | 12 | |
| eigenQTL2B.1 | 19.94 | 23.26 | 3 | 0 | Ppd-B1 |
| eigenQTL2B.2 | 24.75 | 29.36 | 4 | 0 | |
| eigenQTL2B.3 | 31.36 | 43.36 | 54 | 31 | |
| eigenQTL2B.4 | 44.56 | 58.73 | 70 | 5 | |
| eigenQTL2B.5 | 61.66 | 71.36 | 11 | 2 | |
| eigenQTL2B.6 | 72.96 | 90.37 | 119 | 6 | Ppo-B2, TaGS2-B1 |
| eigenQTL2B.7 | 105.36 | 106.86 | 16 | 0 | |
| eigenQTL3A.1 | 0.63 | 6.12 | 6 | 3 | |
| eigenQTL3A.2 | 10.95 | 14.70 | 2 | 0 | |
| eigenQTL3A.3 | 39.12 | 44.9 | 4 | 0 | |
| eigenQTL3A.4 | 45.25 | 53.20 | 8 | 0 | |
| eigenQTL3A.5 | 56.49 | 68.88 | 65 | 12 | |
| eigenQTL3A.6 | 101.40 | 106.34 | 47 | 13 | |
| eigenQTL3A.7 | 108.63 | 114.62 | 37 | 13 | |
| eigenQTL3A.8 | 132.17 | 135.20 | 3 | 1 | Pod-A1 |
| eigenQTL3A.9 | 145.36 | 149.24 | 16 | 0 | |
| eigenQTL3B.1 | 3.75 | 15.53 | 22 | 6 | |
| eigenQTL3B.2 | 23.22 | 28.73 | 9 | 0 | |
| eigenQTL3B.3 | 51.08 | 55.93 | 7 | 0 | |
| eigenQTL3B.4 | 63.71 | 70.46 | 9 | 3 | |
| eigenQTL3B.5 | 77.52 | 92.84 | 17 | 0 | |
| eigenQTL3B.6 | 93.68 | 102.94 | 18 | 0 | |
| eigenQTL3B.7 | 114.27 | 118.45 | 2 | 0 | |
| eigenQTL3B.8 | 136.49 | 141.10 | 3 | 0 | |
| eigenQTL4A.1 | 18.34 | 21.81 | 5 | 0 | |
| eigenQTL4A.2 | 23.50 | 27.20 | 7 | 2 | |
| eigenQTL4A.3 | 27.35 | 32.70 | 3 | 1 | |
| eigenQTL4A.4 | 74.67 | 77.67 | 3 | 3 | |
| eigenQTL4A.5 | 93.51 | 98.13 | 8 | 0 | |
| eigenQTL4A.6 | 110.56 | 117.64 | 7 | 1 | |
| eigenQTL4A.7 | 119.89 | 134.22 | 13 | 1 | TaALP-4A |
| eigenQTL4B.1 | 42.86 | 50.32 | 5 | 0 | Rht-B1 |
| eigenQTL4B.2 | 73.62 | 77.36 | 2 | 0 | |
| eigenQTL5A.1 | 12.37 | 18.03 | 3 | 0 | |
| eigenQTL5A.2 | 33.09 | 38.49 | 6 | 0 | |
| eigenQTL5A.3 | 46.29 | 50.38 | 4 | 0 | |
| eigenQTL5A.4 | 56.89 | 67.05 | 6 | 0 | |
| eigenQTL5A.5 | 75.81 | 88.74 | 21 | 2 | Vrn-A1, Rht12 |
| eigenQTL5A.6 | 104.85 | 116.96 | 10 | 0 | |
| eigenQTL5B.1 | 23.38 | 42.70 | 158 | 51 | |
| eigenQTL5B.2 | 51.84 | 56.56 | 2 | 0 | |
| eigenQTL5B.3 | 64.79 | 72.95 | 5 | 0 | |
| eigenQTL5B.4 | 83.39 | 86.39 | 3 | 0 | Vrn-B1 |
| eigenQTL5B.5 | 106.11 | 112.65 | 4 | 0 | |
| eigenQTL5B.6 | 112.84 | 115.99 | 4 | 0 | |
| eigenQTL5B.7 | 117.20 | 122.07 | 6 | 0 | |
| eigenQTL5B.8 | 149.77 | 152.77 | 2 | 0 | |
| eigenQTL6A.1 | 1.90 | 31.45 | 121 | 24 | |
| eigenQTL6A.1 | 1.90 | 31.45 | 121 | 24 | Rht25 |
| eigenQTL6A.1 | 1.90 | 31.45 | 121 | 24 | |
| eigenQTL6A.2 | 69.82 | 74.66 | 2 | 1 | |
| eigenQTL6A.3 | 88.40 | 99.66 | 8 | 2 | |
| eigenQTL6B.1 | 0.98 | 5.56 | 8 | 0 | |
| eigenQTL6B.2 | 6.69 | 9.69 | 4 | 1 | |
| eigenQTL6B.3 | 10.47 | 15.54 | 4 | 1 | |
| eigenQTL6B.4 | 20.52 | 40.79 | 27 | 4 | GPC-B1 |
| eigenQTL6B.5 | 59.52 | 62.86 | 2 | 0 | |
| eigenQTL6B.6 | 73.99 | 84.69 | 18 | 2 | |
| eigenQTL7A.1 | 28.87 | 34.42 | 7 | 4 | |
| eigenQTL7A.2 | 63.05 | 66.36 | 3 | 1 | TaTEF-7A |
| eigenQTL7A.3 | 70.42 | 83.26 | 21 | 0 | |
| eigenQTL7A.4 | 95.94 | 107.40 | 16 | 11 | |
| eigenQTL7A.5 | 147.09 | 160.29 | 11 | 0 | |
| eigenQTL7B.1 | 47.05 | 51.28 | 3 | 1 | |
| eigenQTL7B.2 | 73.10 | 79.67 | 8 | 2 | |
| eigenQTL7B.3 | 81.66 | 88.16 | 10 | 0 | |
| eigenQTL7B.4 | 92.07 | 97.38 | 4 | 2 | |
| eigenQTL7B.5 | 109.16 | 113.15 | 2 | 0 | |
| eigenQTL7B.6 | 123.88 | 132.06 | 10 | 1 | Psy-B1 |
| QTL Hotspot | Marker | Position (cM) | Genome Position (bp) | Allele Group 1 | Allele Group 2 | ||
|---|---|---|---|---|---|---|---|
| Zavitan | Svevo | Chinese Spring | (Frequency) | (Frequency) | |||
| eigenQTL2A.7 | 1089372 | 123.66 | 768,637,732 | 771,309,636 | 766,565,471 | 0 (0.81) | 1 (0.82) |
| 1096089 | 123.66 | 768,369,404 | 770,792,840 | 767,003,197 | 0 (0.81) | 1 (0.90) | |
| 1288584 | 123.66 | - | 772,466,381 | 765,605,244 | 1 (0.80) | 0 (0.90) | |
| eigenQTL2B.3 | 3935165 | 36.35 | 55,282,377 | 53,704,532 | 54,005,983 | 0 (0.89) | 1 (0.82) |
| 3946438 | 36.35 | 55,263,539 | - | 53,999,239 | 0 (0.84) | 1 (0.87) | |
| 3955840 | 36.35 | 55,263,539 | - | 53,999,239 | 0 (0.84) | 1 (0.87) | |
| 4404794 | 36.35 | - | 53,704,524 | 54,005,983 | 1 (1.00) | 0 (0.85) | |
| 4404891 | 36.35 | - | 53,704,524 | 54,005,983 | 1 (1.00) | 0 (0.85) | |
| 4409154 | 36.35 | - | 53,703,534 | - | 1 (1.00) | 0 (0.85) | |
| 3022498 | 37.15 | 56,411,136 | 54,740,047 | 55,031,700 | 0 (0.84) | 1 (0.80) | |
| 1125733 | 38.57 | 59,371,071 | 57,490,889 | 57,917,326 | 0 (0.89) | 1 (0.80) | |
| 1353553 | 40.74 | 55,744,579 | 54,098,441 | 54,443,978 | C (0.84) | T (0.87) | |
| 3021610 | 40.74 | 55,523,159 | 53,972,355 | 54,272,933 | C (0.89) | T (0.87) | |
| 4004228 | 40.74 | 57,503,553 | 56,011,661 | 55,991,662 | 1 (0.95) | 0 (0.85) | |
| 4004312 | 40.99 | 56,411,136 | 54,740,047 | 55,031,700 | 1 (0.95) | 0 (0.82) | |
| 986135 | 40.99 | 56,166,013 | 54,516,891 | 54,786,611 | A (0.89) | C (0.85) | |
| 1124640 | 41.86 | 56,147,572 | 54,468,610 | 54,770,824 | A (0.84) | G (0.85) | |
| eigenQTL3A.6 | 2257732 | 103.85 | 693,610,895 | 688,415,545 | 697,202,220 | 0 (0.98) | 1 (0.98) |
| 1007286 | 103.92 | 687,773,343 | 682,345,965 | 691,736,154 | 0 (0.98) | 1 (0.95) | |
| 1061286 | 103.92 | 687,959,611 | 682,871,589 | 692,054,958 | 0 (0.99) | 1 (0.88) | |
| 1099726 | 103.92 | 693,660,065 | - | 697,248,312 | 0 (0.98) | 1 (0.97) | |
| 2257138 | 103.92 | 688,886,018 | 683,409,098 | 692,987,209 | 0 (0.98) | 1 (0.99) | |
| 3033940 | 103.92 | 690,079,348 | 684,307,722 | 694,092,980 | 0 (0.96) | 1 (0.99) | |
| 3940178 | 103.92 | 691,844,961 | 686,017,151 | 695,739,301 | 0 (0.97) | 1 (0.99) | |
| 3945420 | 103.92 | 688,521,622 | 682,907,278 | 692,471,812 | 0 (0.98) | 1 (0.96) | |
| 3952975 | 103.92 | 688,369,820 | 685,647,294 | 692,316,203 | 0 (0.98) | 1 (0.98) | |
| 3957848 | 103.92 | 691,844,961 | 686,017,151 | 695,739,301 | 0 (0.97) | 1 (0.99) | |
| 4005072 | 103.92 | 688,885,643 | 683,409,473 | 692,987,584 | 0 (0.97) | 1 (0.99) | |
| eigenQTL3A.7 | 1062254 | 110.13 | 691,603,242 | 685,647,297 | 695,515,284 | T (0.98) | G (0.98) |
| 1120615 | 110.13 | 687,953,731 | 682,789,499 | 692,048,455 | 1 (0.95) | 0 (0.96) | |
| 1127998 | 110.13 | 691,772,662 | 685,990,476 | 695,671,629 | T (0.93) | C (0.96) | |
| 1755023 | 110.13 | 692,894,801 | 687,945,767 | - | 1 (0.99) | 0 (0.96) | |
| 2275425 | 110.13 | 690,565,280 | 684,664,645 | 694,538,535 | A (0.98) | G (0.97) | |
| 4003435 | 110.13 | 689,966,914 | 684,172,863 | 693,979,130 | 1 (0.99) | 0 (0.96) | |
| 4004625 | 110.13 | - | 682,650,634 | - | 1 (0.98) | 0 (0.97) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soriano, J.M.; Sansaloni, C.; Ammar, K.; Royo, C. Labelling Selective Sweeps Used in Durum Wheat Breeding from a Diverse and Structured Panel of Landraces and Cultivars. Biology 2021, 10, 258. https://doi.org/10.3390/biology10040258
Soriano JM, Sansaloni C, Ammar K, Royo C. Labelling Selective Sweeps Used in Durum Wheat Breeding from a Diverse and Structured Panel of Landraces and Cultivars. Biology. 2021; 10(4):258. https://doi.org/10.3390/biology10040258
Chicago/Turabian StyleSoriano, Jose Miguel, Carolina Sansaloni, Karim Ammar, and Conxita Royo. 2021. "Labelling Selective Sweeps Used in Durum Wheat Breeding from a Diverse and Structured Panel of Landraces and Cultivars" Biology 10, no. 4: 258. https://doi.org/10.3390/biology10040258
APA StyleSoriano, J. M., Sansaloni, C., Ammar, K., & Royo, C. (2021). Labelling Selective Sweeps Used in Durum Wheat Breeding from a Diverse and Structured Panel of Landraces and Cultivars. Biology, 10(4), 258. https://doi.org/10.3390/biology10040258