
Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci

 ,
, 
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. D. eres and Related Species Isolates Used
2.2. Selection of Suitable Markers for Genetic Diversity Analysis
2.3. Sequence Alignment and Phylogenetic Analyses
2.4. Genealogical Concordance Phylogenetic Species Recognition (GCPSR) Analysis
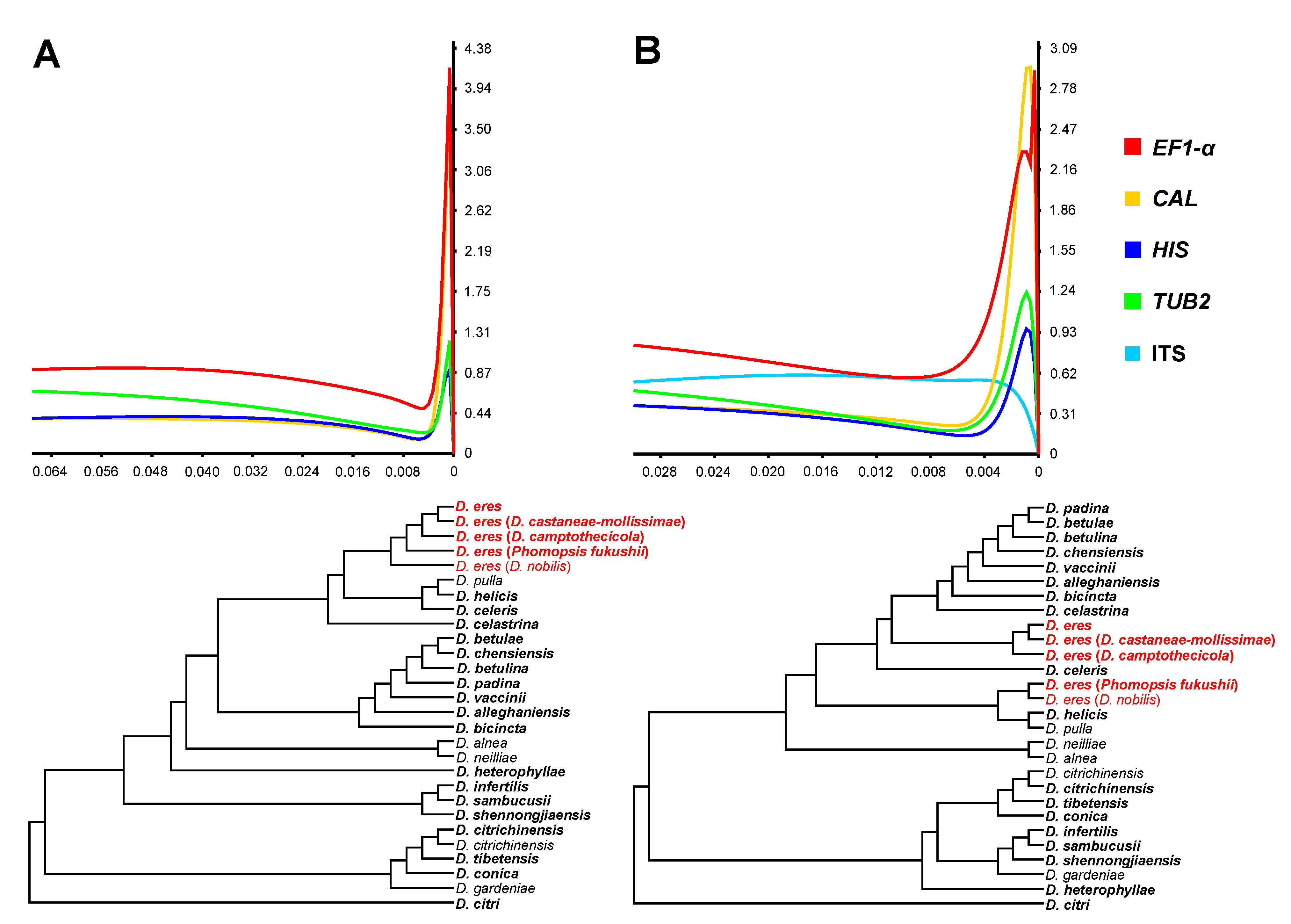
2.5. Phylogenetic Informativeness Analysis
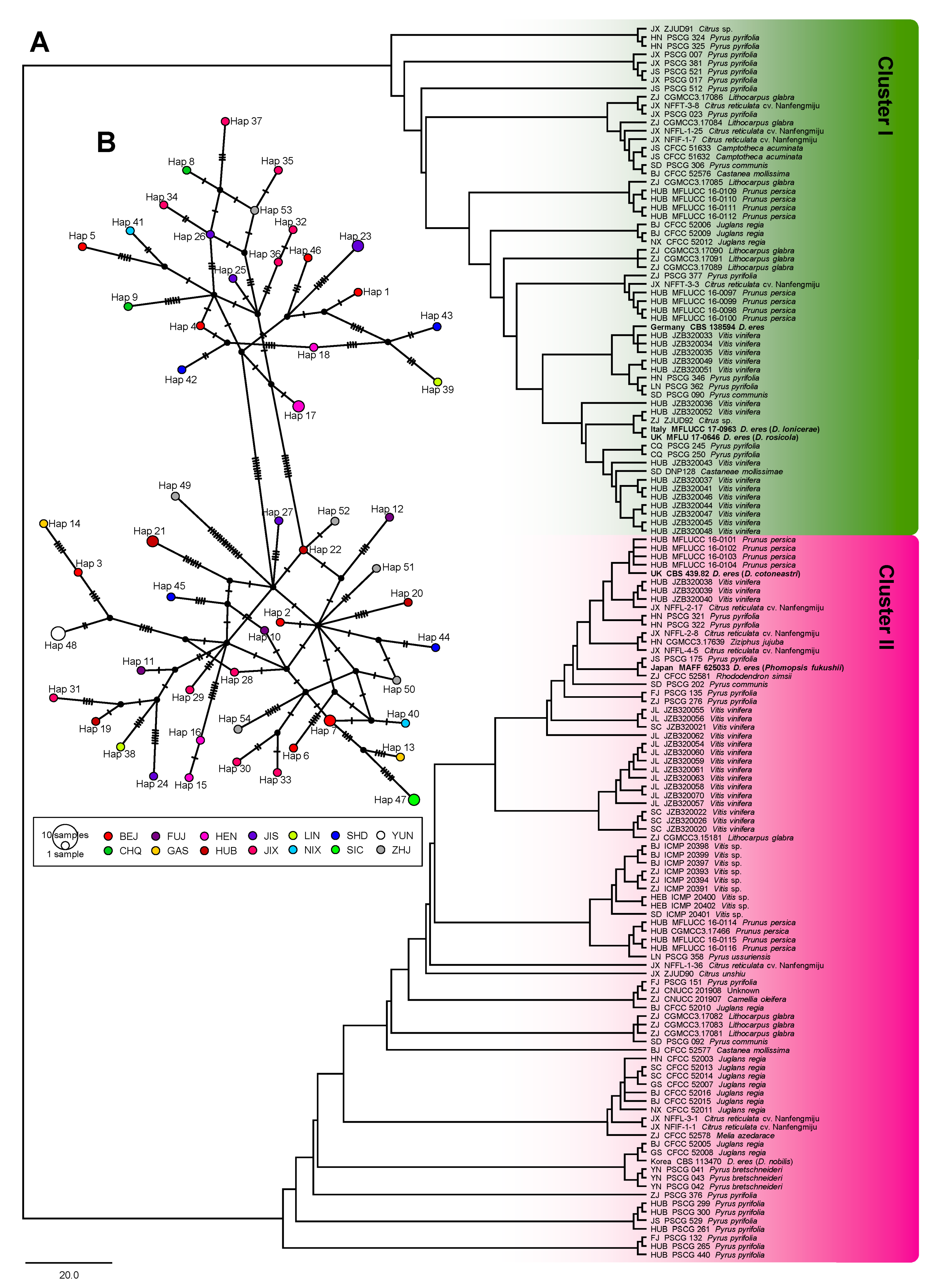
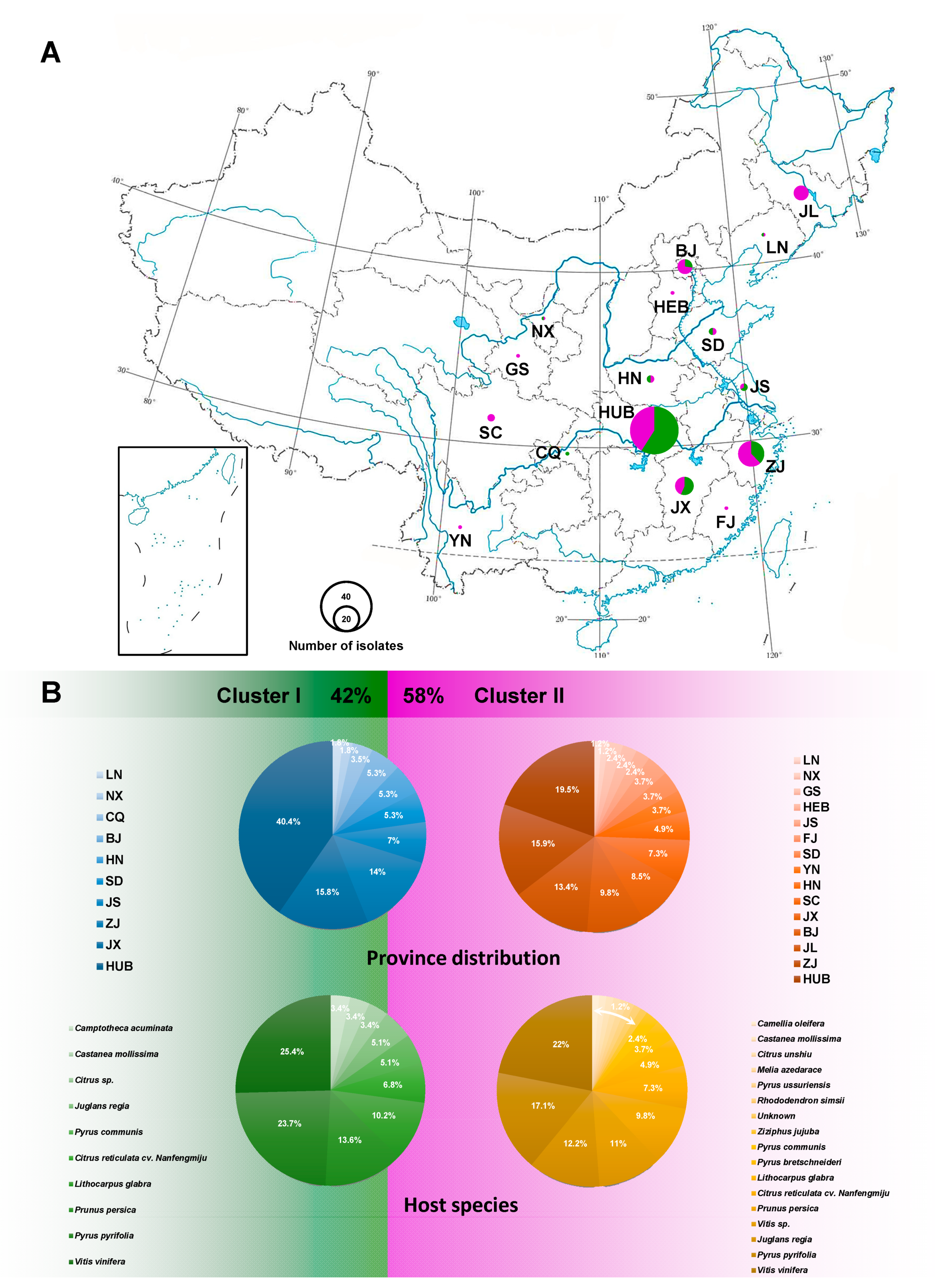
2.6. Population Aggregation and Haplotype Network Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diaporthe Species a | Isolate Number b | Origin | GenBank Accession Numbers c | References | ||||
|---|---|---|---|---|---|---|---|---|
| ITS | EF1-α | TUB2 | CAL | HIS | ||||
| D. acerigenaT | CFCC 52554 | China | MH121489 | MH121531 | – | MH121413 | MH121449 | [20] |
| D. alleghaniensisT | CBS 495.72 | Canada | KC343007 | KC343733 | KC343975 | KC343249 | KC343491 | [46] |
| D. alnea | CBS 146.46 | Netherlands | KC343008 | KC343734 | KC343976 | KC343250 | KC343492 | [46] |
| D. apiculatumT | CGMCC3.17533 | China | KP267896 | KP267970 | KP293476 | – | – | [15] |
| D. betulaeT | CFCC 50469 | China | KT732950 | KT733016 | KT733020 | KT732997 | KT732999 | [70] |
| D. betulinaT | CFCC 52562 | China | MH121497 | MH121539 | MH121579 | MH121421 | MH121457 | [20] |
| D. bicinctaEP | CBS 121004 | USA | KC343134 | KC343860 | KC344102 | KC343376 | KC343618 | [43,46] |
| D. celastrinaEP | CBS 139.27 | USA | KC343047 | KC343773 | KC344015 | KC343289 | KC343531 | [43,46] |
| D. celerisT | CBS 143349 | UK | MG281017 | MG281538 | MG281190 | MG281712 | MG281363 | [4] |
| D. charlesworthiiT | BRIP 54884m | Australia | KJ197288 | KJ197250 | KJ197268 | – | – | [6] |
| D. chensiensisT | CFCC 52567 | China | MH121502 | MH121544 | MH121584 | MH121426 | MH121462 | [20] |
| D. citriT | CBS 135422 | USA | KC843311 | KC843187 | KC843071 | KC843157 | MF418281 | [3,7] |
| D. citrichinensisT | CGMCC3.15225 | China | JQ954648 | JQ954666 | MF418524 | KC357494 | KJ490516 | [3,17,18] |
| D. citrichinensis | ZJUD034B | China | KJ210539 | KJ210562 | KJ420829 | KJ435042 | KJ420879 | [43] |
| D. collarianaT | MFLUCC 17-2636 | Thailand | MG806115 | MG783040 | MG783041 | MG783042 | – | [71] |
| D. conicaT | CFCC 52571 | China | MH121506 | MH121548 | MH121588 | MH121428 | MH121466 | [20] |
| D. eresEP | CBS 138594 | Germany | KJ210529 | KJ210550 | KJ420799 | KJ434999 | KJ420850 | [43] |
| D. eres (D. biguttusis) T | CGMCC3.17081 | Unknown | KF576282 | KF576257 | KF576306 | – | – | [39] |
| D. eres (D. camptothecicola) T | CFCC 51632 | China | KY203726 | KY228887 | KY228893 | KY228877 | KY228881 | [37] |
| D. eres (D. castaneae-mollissimae) T | DNP128 | China | JF957786 | KJ210561 | KJ420801 | KJ435040 | KJ420852 | [43,72] |
| D. eres (D. cotoneastri) T | CBS 439.82 | UK | FJ889450 | GQ250341 | JX275437 | JX197429 | – | [72] |
| D. eres (D. ellipicola) T | CGMCC3.17084 | China | KF576270 | KF576245 | KF576291 | – | – | [39] |
| D. eres (D. henanensis) T | CGMCC3.17639 | China | KC898258 | – | KF600608 | – | KF600609 | [73] |
| D. eres (D. longicicola) T | CGMCC3.17089 | Unknown | KF576267 | KF576242 | KF576291 | – | – | [39] |
| D. eres (D. lonicerae) T | MFLUCC 17-0963 | Italy | KY964190 | KY964146 | KY964073 | KY964116 | – | [74] |
| D. eres (D. mahothocarpus) T | CGMCC3.15181 | China | KC153096 | KC153087 | KF576312 | – | – | [39,40] |
| D. eres (D. momicola) T | CGMCC3.17466 | China | KU557563 | KU557631 | KU557587 | KU557611 | – | [13] |
| D. eres (D. nobilis) | CBS 113470 | Korea | KC343146 | KC343872 | KC344114 | KC343388 | KC343630 | [46] |
| D. eres (D. rosicola) T | MFLU 17-0646 | UK | MG828895 | MG829270 | MG843877 | – | – | [75] |
| D. eres (Phomopsis fukushii) NE | MAFF 625033 | Japan | JQ807468 | JQ807417 | KJ420814 | KJ435017 | KJ420865 | [43] |
| D. eucommiicolaH | SCHM 3607 | China | AY578071 | – | – | – | – | [76] |
| D. fraxinicolaT | CFCC 52582 | China | MH121517 | MH121559 | – | MH121435 | – | [20] |
| D. gardeniae | CBS 288.56 | Italy | KC343113 | KC343839 | KC344081 | KC343355 | KC343597 | [46] |
| D. helicisEP | CBS 138596 | France | KJ210538 | KJ210559 | KJ420828 | KJ435043 | KJ420875 | [43] |
| D. heterophyllaeT | CBS 143769 | France | MG600222 | MG600224 | MG600226 | MG600218 | MG600220 | [77] |
| D. infertilisT | CBS 230.52 | Suriname | KC343052 | KC343778 | KC344020 | KC343294 | KC343536 | [3,46] |
| D. maritimaT | DAOMC 250563 | Canada | KU552025 | KU552023 | KU574615 | – | – | [78] |
| D. neilliae | CBS 144.27 | Unknown | KC343144 | KC343870 | KC344112 | KC343386 | KC343628 | [46] |
| D. oracciniiT | CGMCC3.17531 | China | KP267863 | KP267937 | KP293443 | – | KP293517 | [15] |
| D. padinaT | CFCC 52590 | China | MH121525 | MH121567 | MH121604 | MH121443 | MH121483 | [20] |
| D. penetriteumT | CGMCC3.17532 | China | KP714505 | KP714517 | KP714529 | – | KP714493 | [15] |
| D. phragmitisT | CBS 138897 | China | KP004445 | – | KP004507 | – | KP004503 | [79] |
| D. pulla | CBS 338.89 | Yugoslavia | KC343152 | KC343878 | KC344120 | KC343394 | KC343636 | [43] |
| D. sambucusiiT | CFCC 51986 | China | KY852495 | KY852507 | KY852511 | KY852499 | KY852503 | [80] |
| D. sennicolaT | CFCC 51634 | China | KY203722 | KY228883 | KY228889 | KY228873 | – | [81] |
| D. shennongjiaensisT | CNUCC 201905 | China | MN216229 | MN224672 | MN227012 | MN224551 | MN224559 | [35] |
| D. subclavataT | CGMCC3.17257 | China | KJ490630 | KJ490509 | KJ490451 | – | KJ490572 | [18] |
| D. tibetensisT | CFCC 51999 | China | MF279843 | MF279858 | MF279873 | MF279888 | MF279828 | [14] |
| D. ukurunduensisT | CFCC 52592 | China | MH121527 | MH121569 | – | MH121445 | MH121485 | [20] |
| D. vacciniiT | CBS 160.32 | USA | KC343228 | KC343954 | KC344196 | KC343470 | KC343712 | [46] |
| D. virgiliaeT | CBS 138788 | South Africa | KP247573 | – | KP247582 | – | – | [82] |
3. Results
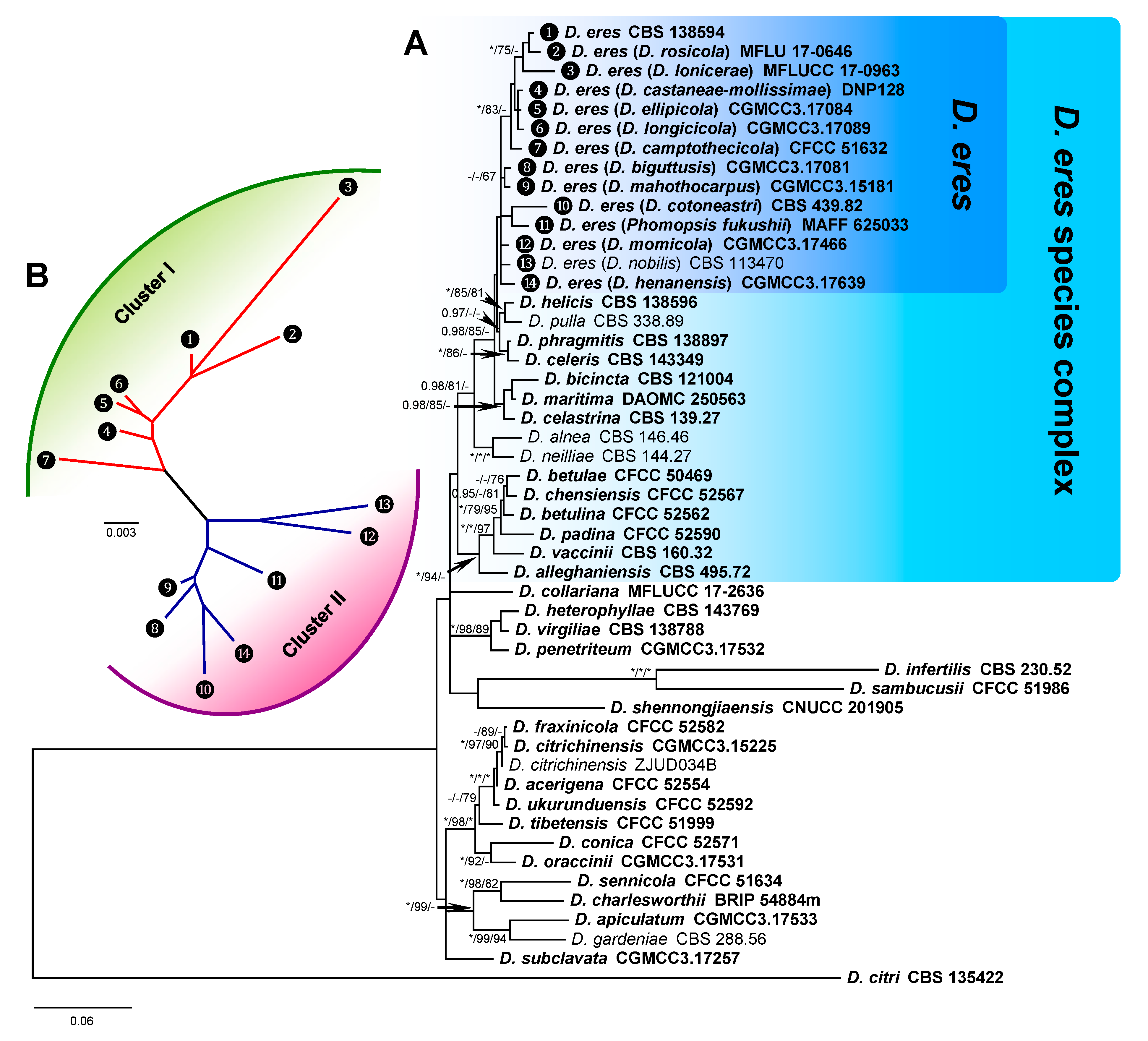
3.1. Phylogenetic Analysis of D. eres
3.2. Phylogenetic Informative Analysis
3.3. D. eres Species Boundaries
3.4. Population Aggregation and Haplotype Network Analysis
3.5. Phylogenetic Informative Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senanayake, I.C.; Crous, P.W.; Groenewald, J.Z.; Maharachchikumbura, S.S.N.; Jeewon, R.; Phillips, A.J.L.; Bhat, J.D.; Perera, R.H.; Li, Q.R.; Li, W.J.; et al. Families of Diaporthales based on morphological and phylogenetic evidence. Stud. Mycol. 2017, 86, 217–296. [Google Scholar] [CrossRef]
- Wehmeyer, L.E. The genus Diaporthe Nitschke and its segregates. Univ. Mich. Stud. Sci. Ser. 1933, 9, 1–349. [Google Scholar]
- Guarnaccia, V.; Crous, P.W. Emerging citrus diseases in Europe caused by species of Diaporthe. IMA Fungus 2017, 8, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, V.; Groenewald, J.Z.; Woodhall, J.; Armengol, J.; Cinelli, T.; Eichmeier, A.; Ezra, D.; Fontaine, F.; Gramaje, D.; Gutierrez-Aguirregabiria, A.; et al. Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 2018, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Phillips, A.J.L. Resolving the complex of Diaporthe (Phomopsis) species occurring on Foeniculum vulgare in Portugal. Fungal Divers. 2009, 34, 111–125. [Google Scholar]
- Thompson, S.M.; Tan, Y.P.; Shivas, R.G.; Neate, S.M.; Morin, L.; Bissett, A.; Aitken, E.A.B. Green and brown bridges between weeds and crops reveal novel Diaporthe species in Australia. Persoonia 2015, 35, 39–49. [Google Scholar] [CrossRef]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Hyde, K.D. Species limits in Diaporthe: Molecular re-assessment of D. citri, D. cytosporella, D. foeniculina and D. rudis. Persoonia 2014, 32, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Reyne, A.; López-Medrano, F.; Morales, J.M.; Esteban, C.G.; Martín, I.; Eraña, I.; Meije, Y.; Lalueza, A.; Alastruey-Izquierdo, A.; Rodríguez-Tudela, J.L.; et al. Cutaneous infection by Phomopsis longicolla in a renal transplant recipient from guinea: First report of human infection by this fungus. Transpl. Infect. Dis. 2011, 13, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Laosakul, K.; Youngchim, S.; Chuamanochan, M.; Rujiwetpongstorn, R.; Tovanabutra, N.; Chiewchanvit, S. Phaeohyphomycosis caused by Diaporthe phaseolorum in an immunocompetent patient in Thailand: A case report. Access Microbiol. 2020, 2. [Google Scholar] [CrossRef]
- Warmelo, K.T.; Marasas, W.F.O. Phomopsis leptostromiformis: The causal fungus of lupinosis, a mycotoxicosis, in sheep. Mycologia 1972, 64, 316–324. [Google Scholar] [CrossRef]
- Webber, J.F.; Gibbs, J.N. Colonization of elm bark by Phomopsis oblonga. Trans. Br. Mycol. Soc. 1984, 82, 348–352. [Google Scholar] [CrossRef]
- Chi, P.K.; Jiang, Z.D.; Xiang, M.M. Flora Fungorum Sinicorum; Science Press: Beijing, China, 2007; Volume 34. (In Chinese) [Google Scholar]
- Dissanayake, A.J.; Zhang, W.; Liu, M.; Hyde, K.D.; Zhao, W.S.; Li, X.H.; Yan, J.Y. Diaporthe species associated with peach tree dieback in Hubei, China. Mycosphere 2017, 8, 533–549. [Google Scholar] [CrossRef]
- Fan, X.L.; Yang, Q.; Bezerra, J.D.P.; Alvarez, L.V.; Tian, C.M. Diaporthe from walnut tree (Juglans regia) in China, with insight of the Diaporthe eres complex. Mycol. Progress 2018, 17, 841–853. [Google Scholar] [CrossRef]
- Gao, Y.H.; Liu, F.; Duan, W.J.; Crous, P.W.; Cai, L. Diaporthe is paraphyletic. IMA Fungus 2017, 8, 153–187. [Google Scholar] [CrossRef]
- Guo, Y.S.; Crous, P.W.; Bai, Q.; Fu, M.; Yang, M.M.; Wang, X.H.; Du, Y.M.; Hong, N.; Xu, W.X.; Wang, G.P. High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 2020, 45, 132–162. [Google Scholar] [CrossRef]
- Huang, F.; Hou, X.; Dewdney, M.M.; Fu, Y.S.; Chen, G.Q.; Hyde, K.D.; Li, H.Y. Diaporthe species occurring on citrus in China. Fungal Divers. 2013, 61, 237–250. [Google Scholar] [CrossRef]
- Huang, F.; Udayanga, D.; Wang, X.H.; Hou, X.; Mei, X.F.; Fu, Y.S.; Hyde, K.D.; Li, H.Y. Endophytic Diaporthe associated with citrus: A phylogenetic reassessment with seven new species from China. Fungal Biol. 2015, 119, 331–347. [Google Scholar] [CrossRef]
- Manawasighe, I.S.; Dissanayake, A.J.; Li, X.H.; Liu, M.; Wanasinghe, D.N.; Xu, J.P.; Zhao, W.S.; Zhang, W.; Zhou, Y.Y.; Hyde, K.D.; et al. High genetic diversity and species complexity of Diaporthe associated with grapevine dieback in China. Front. Microbiol. 2019, 10, 1936. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fan, X.L.; Guarnaccia, V.; Tian, C.M. High diversity of Diaporthe species associated with dieback diseases in China, with twelve new species described. MycoKeys 2018, 39, 97–149. [Google Scholar] [CrossRef]
- Yang, Q.; Jiang, N.; Tian, C.M. Three new Diaporthe species from Shaanxi province, China. MycoKeys 2020, 67, 1–18. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Chen, Y.Y.; Liu, J.K. Unravelling Diaporthe species associated with woody hosts from Karst Formations (Guizhou) in China. J. Fungi 2020, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Anagnostakis, S.L. Diaporthe eres (Phomopsis oblonga) as a pathogen of butternut (Juglans cinerea) in connecticut. Plant Dis. 2007, 91, 1198. [Google Scholar] [CrossRef] [PubMed]
- Quaroni, S.; Sardi, P.; Locci, R. Apical blight in Acer pseudoplatanus associated with Diaporthe eres Nits. (Phomopsis acerina Pir. et Cart). Riv. Patol. Veg. 1980, 16, 109. [Google Scholar]
- Bastidea, F.; Sérandat, I.; Gombertc, J.; Laurentc, E.; Moreld, E.; Koloppe, J.; Guillerminf, P.L.; Hamona, B.; Simoneaua, P.; Berruyera, R.; et al. Characterization of fungal pathogens (Diaporthe angelicae and D. eres) responsible for umbel browning and stem necrosis on carrot in France. Plant Pathol. 2016, 66, 221–227. [Google Scholar]
- Meepagala, K.M.; Briscoe, W.E.; Techen, N.; Johnson, R.D.; Clausen, B.M.; Duke, S.O. Isolation of a phytotoxic isocoumarin from Diaporthe eres-infected Hedera helix (english ivy) and synthesis of its phytotoxic analogs. Pest Manag. Sci. 2018, 74, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, M.; Zapparoli, G. Identification of Pestalotiopsis bicilita, Diplodia seriata and Diaporthe eres causing fruit rot in withered grapes in Italy. Eur. J. Plant Pathol. 2018, 151, 1089–1093. [Google Scholar] [CrossRef]
- Ali, S.; Renderos, W.; Bevis, E.; Hebb, J.; Abbasi, P.A. Diaporthe eres causes stem cankers and death of young apple rootstocks in Canada. Can. J. Plant Pathol. 2019, 42, 218–227. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.Y.; Song, Q.N.; Liu, J.; Yang, Q.P.; Luan, F.G.; Li, D. First of Diaporthe eres causing branch canker on Cinnamomum camphora (camphor tree) in Jiangxi province, China. Plant Dis. 2021. First Look. [Google Scholar]
- Tao, H.; Wang, H.; Huang, S.X.; Zhang, Y.; Zhang, Z.H.; Liu, W.; Shi, N.X.; Zhu, F.; Ji, Z.L.; Chen, X.R. Identification and characterization of Diaporthe eres causing leaf blight disease on the medicinal herb Polygonatum sibiricum. J. Gen. Plant Pathol. 2020, 86, 468–476. [Google Scholar] [CrossRef]
- Song, L.M.; Wang, X.; Chen, T.G.; Liang, W.X.; Yang, Q.Q. First report of Diaporthe eres leaf spot on Photinia x fraseri ‘red robin’ in Qingdao, China. Plant Dis. 2019, 103, 159. [Google Scholar] [CrossRef]
- Mei, P.Y.; Song, X.H.; Zhu, Z.Y.; Li, L.Y. First report of Diaporthe eres causing root rot of Coptis chinensis Franchet. Plant Dis. 2021. First Look. [Google Scholar] [CrossRef]
- Zhao, Y.Q.; Geng, G.M.; Tian, Y.L.; Hu, B.S. First report of shoot blight of Japanese maple caused by Diaporthe eres in China. J. Plant Pathol. 2019, 101, 179. [Google Scholar] [CrossRef]
- Sun, Z.Q.; Xue, C.Y.; Wang, Y.F.; Xu, C.; Zang, R.; Geng, Y.H.; Miao, Y.C.; Wu, H.Y.; Zhang, M. First report of Diaporthe eres causing twig dieback of white bark pine in China. Plant Dis. 2020, 104, 1862. [Google Scholar] [CrossRef]
- Zhou, H.; Hou, C.L. Three new species of Diaporthe from China based on morphological characters and DNA sequence data analyses. Phytotaxa 2019, 422, 157–174. [Google Scholar] [CrossRef]
- Li, Y.; Tan, P.; Zhao, D.G. Diaporthe nobilis, a new record on Camellia sinensis in Guizhou province, China. Mycosphere 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Yang, Q.; Fan, X.L.; Du, Z.; Liang, Y.M.; Tian, C.M. Diaporthe camptothecicola sp. nov. on Camptotheca acuminata in China. Mycotaxon 2017, 132, 591–601. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Liu, M.; Zhang, W.; Chen, Z.; Udayanga, D.; Chukeatirote, E.; Li, X.H.; Yan, J.Y.; Hyde, K.D. Morphological and molecular characterisation of Diaporthe species associated with grapevine trunk disease in China. Fungal Biol. 2015, 119, 283–294. [Google Scholar] [CrossRef]
- Gao, Y.H.; Su, Y.Y.; Sun, W.; Cai, L. Diaporthe species occurring on Lithocarpus glabra in China, with descriptions of five new species. Fungal Biol. 2015, 119, 295–309. [Google Scholar] [CrossRef]
- Gao, Y.H.; Sun, W.; Su, Y.Y.; Cai, L. Three new species of Phomopsis in Gutianshan nature reserve in China. Mycol. Progress 2014, 13, 111–121. [Google Scholar] [CrossRef]
- Li, L.; Pan, H.; Chen, M.Y.; Zhang, S.J.; Zhong, C.H. Isolation and identification of pathogenic fungi causing postharvest fruits rot kiwifruit (Actinidia chinensis) in China. J. Phytopathol. 2017, 165, 782–790. [Google Scholar] [CrossRef]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. Insights into the genus Diaporthe: Phylogenetic species delimitation in the D. eres species complex. Fungal Divers. 2014, 67, 203–229. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 2009, 6. [Google Scholar] [CrossRef] [PubMed]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef]
- Gomes, R.R.; Glienke, C.; Videira, S.I.R.; Lombard, L.; Groenewald, J.Z.; Crous, P.W. Diaporthe: A genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 2013, 31, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Alves, A.; Alves, R. Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. PeerJ 2017, 5, 1–26. [Google Scholar] [CrossRef]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. The Diaporthe sojae species complex: Phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol. 2015, 119, 383–407. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Carbone, I.; Kohn, L.M. A method for desianing primer sets for speciation studies in filamentous ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microb. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Crous, P.W.; Groenewald, J.Z.; Risède, J.M.; Simoneau, P.; Hywel-Jones, N.L. Calonectria species and their Cylindrocladium anamorphs: Species with clavate vesicles. Stud. Mycol. 2004, 50, 415–430. [Google Scholar] [CrossRef]
- Chaisiri, C.; Liu, X.Y.; Lin, Y.; Li, J.B.; Xiong, B.; Luo, C.X. Phylogenetic analysis and development of molecular tool for detection of Diaporthe citri causing melanose disease of citrus. Plants 2020, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Hall, A.T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucleic Acids Res 1999, 41, 95–98. [Google Scholar]
- Glez-Peña, D.; Gómez-Blanco, D.; Reboiro-Jato, M.; Fdez-Riverola, F.; Posada, D. ALTER: Program-oriented conversion of DNA and protein alignments. Nucleic Acids Res. 2010, 38, W14–W18. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. In TeraGrid Conference: Extreme Digital Discovery; San Diego Supercomputer Center: La Jolla, San Diego, CA, USA, 2011; Volume 41, pp. 1–8. [Google Scholar]
- Swofford, D.L. PAUP* Phylogenetic Analysis Using Parsimony, (*and Other Methods); Version 4.0 b10; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J.A.A. MrModeltest v.2. Program Distributed by the Author; Evolitionary Biology Centre, Uppsala Univeristy: Uppsala, Sweden, 2004. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetic using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef]
- Townsend, J.P. Profiling phylogenetic informativeness. Syst. Biol. 2007, 56, 222–231. [Google Scholar] [CrossRef]
- Lopez-Giraldez, F.; Townsend, J.P. PhyDesign: An online application for profiling phylogenetic informativeness. BMC Evol. Biol. 2011, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. PopArt: Full-feature softwere for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Du, Z.; Fan, X.L.; Hyde, K.D.; Yang, Q.; Liang, Y.M.; Tian, C.M. Phylogeny and morphology reveal two new species of Diaporthe from Betula spp. in China. Phytotaxa 2016, 269, 90–102. [Google Scholar] [CrossRef][Green Version]
- Perera, R.H.; Hyde, K.D.; Dissanayake, A.J.; Jones, E.B.G.; Liu, J.K.; Wei, D.; Liu, Z.Y. Diaporthe collariana sp. nov., with prominent collarettes associated with Magnolia champaca fruits in Thailand. Stud. Fungi 2018, 3, 141–151. [Google Scholar] [CrossRef]
- Udayanga, D.; Liu, X.Z.; Crous, P.W.; McKenzie, E.H.C.; Chukeatirote, E.; Hyde, K.D. A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers. 2012, 56, 157–171. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y.X.; Zhang, Y.K.; Wu, H.Y.; Zhang, M. Diaporthe henanensis sp. nov., an endophytic fungus in Ziziphus jujuba from China. Mycotaxon 2016, 131, 645–652. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Camporesi, E.; Hyde, K.D.; Zhang, W.; Yan, J.Y.; Li, X.H. Molecular phylogenetic analysis reveals seven new Diaporthe species from Italy. Mycosphere 2017, 8, 853–877. [Google Scholar] [CrossRef]
- Wanasinghe, D.N.; Phukhamsakda, C.; Hyde, K.D.; Jeewon, R.; Lee, H.B.; Jones, E.B.G.; Tibpromma, S.; Tennakoon, D.S.; Dissanayake, A.J.; Jayasiri, S.C.; et al. Fungal diversity notes 709–839: Taxonomic and phylogenetic contributions to fungal taxa with an emphasis on fungi on Rosaceae. Fungal Divers. 2018, 89, 1–60. [Google Scholar] [CrossRef]
- Chang, C.Q.; Xi, P.G.; Xiang, M.M.; Jiang, Z.D.; Chi, P.K. New species of Phomopsis on woody plants in Hunan province. Mycosystema 2005, 24, 145–154. [Google Scholar]
- Marin-Felix, Y.; Hernández-Restrepo, M.; Wingfield, M.J.; Akulov, A.; Carnegie, A.J.; Cheewangkoon, R.; Gramaje, D.; Groenewald, J.Z.; Guarnaccia, V.; Halleen, F.; et al. Genera of phytopathogenic fungi: GOPHY 2. Stud. Mycol. 2019, 92, 47–133. [Google Scholar] [CrossRef] [PubMed]
- Tanney, J.B.; McMullin, D.R.; Green, B.D.; Miller, J.D.; Seifert, K.A. Production of antifungal and antiinsectan metabolites by the Picea endophyte Diaporthe maritima sp. nov. Fungal Biol. 2016, 120, 1448–1457. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Schumacher, R.K.; Summerell, B.A.; Giraldo, A.; Gené, J.; Guarro, J.; Wanasinghe, D.N.; Hyde, K.D.; Camporesi, E.; et al. Fungal planet description sheets: 281–319. Persoonia 2014, 33, 212–289. [Google Scholar] [CrossRef]
- Yang, Q.; Du, Z.; Tian, C.M. Phylogeny and morphology reveal two new species of Diaporthe from traditional Chinese medicine in Northeast China. Phytotaxa 2018, 336, 159–170. [Google Scholar] [CrossRef]
- Yang, Q.; Fan, X.L.; Du, Z.; Tian, C.M. Diaporthe species occurring on Senna bicapsularis in Southern China, with descriptions of two new species. Phytotaxa 2017, 302, 145–155. [Google Scholar] [CrossRef]
- Machingambi, N.M.; Dreyer, L.L.; Oberlander, K.C.; Roux, J.; Roets, F. Death of endemic Virgilia oroboides trees in South Africa caused by Diaporthe virgiliae sp. nov. Plant Pathol. 2015, 64, 1149–1156. [Google Scholar] [CrossRef]
- Nitschke, T.R.J. Pyrenomycetes germanici. In Die Kernpilze Deutschlands Bearbeitet von Dr. Th. Nitschke; Eduard Trewendt: Breslau, Germany, 1870; Volume 2, pp. 161–320. [Google Scholar]
- Castlebury, L.A.; Farr, D.F.; Rossman, A.Y.; Jaklitsch, W. Diaporthe angelicae comb. nov., a modern description and placement of Diaporthopsis in Diaporthe. Mycoscience 2003, 44, 203–208. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z. Hosts, species and genotypes: Opinions versus data. Australasian Plant Pathol. 2005, 34, 463–470. [Google Scholar] [CrossRef]
- Crous, P.W.; Hawksworth, D.L.; Wingfield, M.J. Identifying and naming plant-pathogenic fungi: Past, present, and future. Annu. Rev. Phytopathol. 2015, 53, 247–267. [Google Scholar] [CrossRef]
- Hibbett, D.; Abarenkov, K.; Kõljalg, U.; Öpik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.W.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar]
- Hyde, K.D.; Nilsson, R.H.; Alias, S.A.; Ariyawansa, H.A.; Blair, J.E.; Cai, L.; de Cock, A.W.A.M.; Dissanayake, A.J.; Glockling, S.L.; Goonasekara, I.D.; et al. One stop shop: Backbones trees for important phytopathogenic genera: I (2014). Fungal Divers. 2014, 67, 21–125. [Google Scholar] [CrossRef]
- van Rensburg, J.C.J.; Lamprecht, S.C.; Groenewald, J.Z.; Castlebury, L.A.; Crous, P.W. Characterisation of Phomopsis spp. associated with die-back of rooibos (Aspalathus linearis) in South Africa. Stud. Mycol. 2006, 55, 65–74. [Google Scholar] [CrossRef]
- Farr, D.F.; Castlebury, L.A.; Rossman, A.Y.; Putnam, M.L. A new species of Phomopsis causing twig dieback of Vaccinium vitis-idaea (lingonberry). Mycol. Res. 2002, 106, 745–752. [Google Scholar] [CrossRef]
- Santos, J.M.; Correia, V.G.; Phillips, A.J.L.; Spatafora, J.W. Primers for mating-type diagnosis in Diaporthe and Phomopsis: Their use in teleomorph induction in vitro and biological species definition. Fungal Biol. 2010, 114, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Mostert, L.; Crous, P.W.; Kang, J.C.; Phillips, A.J.L. Species of Phomopsis and a Libertella sp. occurring on grapevines with specific reference to South Africa: Morphological, cultural, molecular and pathological characterization. Mycologia 2001, 93, 146–167. [Google Scholar] [CrossRef]
- Uecker, F.A. A world list of Phomopsis names with notes on nomenclature, morphology and biology. Mycol. Memoirs. 1988, 13, 1–231. [Google Scholar]
- Brayford, D. Variation in Phomopsis isolates from Ulmus species in the British Isles and Italy. Mycol Res 1990, 94, 691–697. [Google Scholar] [CrossRef]
- van Niekerk, J.M.; Groenewald, J.Z.; Farr, D.F.; Fourie, P.H.; Halleen, F.; Crous, P.W. Reassessment of Phomopsis species on grapevine. Australasian Plant Pathol. 2005, 34, 27–39. [Google Scholar] [CrossRef]
- Rossman, A.Y.; Adams, G.C.; Cannon, P.F.; Castlebury, L.A.; Crous, P.W.; Gryzenhout, M.; Jaklitsch, W.M.; Mejia, L.C.; Stoykov, D.; Udayanga, D.; et al. Recommendations of generic names in Diaporthales competing for protection or use. IMA Fungus 2015, 6, 145–154. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Phillips, A.J.L.; Hyde, K.D.; Yan, J.Y.; Li, X.H. The current status of species in Diaporthe. Mycosphere 2017, 8, 1106–1156. [Google Scholar] [CrossRef]
- Castlebury, L.A.; Farr, D.F.; Rossman, A.Y. Phylogenetic distinction of Phomopsis isolates from cucurbits. Inoculum 2001, 52, 25. [Google Scholar]





| Locus a | Individual Locus | Combined Loci | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 + 2 | 1 + 3 | 1 + 4 | 2 + 3 | 2 + 4 | 3 + 4 | 1 + 2 + 3 | 1 + 2 + 4 | 1 + 2 + 3 + 4 | 1 + 2 + 3 + 4 + 5 | |
| No. of taxa analyzed | 47 | 36 | 47 | 35 | 51 | 47 | 50 | 49 | 50 | 41 | 49 | 50 | 49 | 50 | 51 |
| Aligned length (with gap) | 592 | 542 | 828 | 502 | 598 | 1134 | 1420 | 1094 | 1370 | 1044 | 1330 | 1962 | 1636 | 2464 | 3062 |
| Invariable characters | 289 | 319 | 449 | 355 | 466 | 608 | 738 | 644 | 768 | 674 | 804 | 1057 | 963 | 1412 | 1878 |
| Number of parsimony-informative characters | 171 | 105 | 136 | 83 | 74 | 276 | 307 | 254 | 241 | 188 | 219 | 412 | 359 | 495 | 569 |
| Number of parsimony-uninformative characters | 132 | 118 | 243 | 64 | 58 | 250 | 375 | 196 | 361 | 674 | 307 | 493 | 314 | 557 | 615 |
| Tree length (TL) | 746 | 371 | 597 | 281 | 348 | 1150 | 1379 | 1072 | 995 | 670 | 918 | 1786 | 1474 | 2124 | 2592 |
| Consistency index (CI) | 0.635 | 0.801 | 0.782 | 0.698 | 0.511 | 0.670 | 0.682 | 0.625 | 0.768 | 0.736 | 0.722 | 0.693 | 0.656 | 0.675 | 0.622 |
| Retention index (RI) | 0.698 | 0.793 | 0.712 | 0.767 | 0.739 | 0.699 | 0.676 | 0.681 | 0.714 | 0.755 | 0.688 | 0.680 | 0.688 | 0.667 | 0.640 |
| Rescaled consistency index (RC) | 0.444 | 0.635 | 0.557 | 0.535 | 0.378 | 0.468 | 0.461 | 0.426 | 0.549 | 0.555 | 0.497 | 0.471 | 0.451 | 0.451 | 0.398 |
| Homoplasy index (HI) | 0.365 | 0.199 | 0.218 | 0.302 | 0.489 | 0.330 | 0.318 | 0.375 | 0.232 | 0.264 | 0.278 | 0.307 | 0.344 | 0.325 | 0.378 |
| Gene/Locus a,b | Individual Locus | Combined Loci | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 + 2 + 3 + 4 | |
| Aligned length (with gap) | 278 | 492 | 355 | 421 | 450 | 1429 |
| No. of taxa analyzed | 132 | 118 | 137 | 70 | 137 | 61 |
| No. of sites | 382 | 323 | 794 | 479 | 596 | 1464 |
| %GC | 0.548 | 0.56 | 0.567 | 0.62 | 0.53 | 0.579 |
| No. of polymorphic (segregating) sites (S) | 130 | 27 | 58 | 34 | 83 | 115 |
| Nei’s nucleotide diversity (π) | 0.026051 | 0.00558 | 0.02233 | 0.00593 | 0.33040 | 0.01056 |
| Haplotype numeric (Hap) | 43 | 24 | 36 | 19 | 59 | 54 |
| Haplotype diversity (Hd) | 0.91579 | 0.79500 | 0.92486 | 0.83520 | 0.97639 | 0.99562 |
| Nucleotide diversity from S (θw) | 0.09955 | 0.01603 | 0.03179 | 0.01676 | 0.04126 | 0.01794 |
| Tajima’s D | −2.39947 * | −1.87578 *** | −0.92870 **** | −2.07658 *** | −0.63716 **** | −1.43591 **** |
| Fu and Li’s D | −1.53703 **** | −2.58454 *** | −0.33757 **** | −3.48112 ** | −3.88613 ** | −1.67899 **** |
| Fu’s Fs | −14.6930 | −16.3734 | −6.25117 | −8.43211 | −14.1927 | −36.71492 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaisiri, C.; Liu, X.; Lin, Y.; Fu, Y.; Zhu, F.; Luo, C. Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci. Biology 2021, 10, 179. https://doi.org/10.3390/biology10030179
Chaisiri C, Liu X, Lin Y, Fu Y, Zhu F, Luo C. Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci. Biology. 2021; 10(3):179. https://doi.org/10.3390/biology10030179
Chicago/Turabian StyleChaisiri, Chingchai, Xiangyu Liu, Yang Lin, Yanping Fu, Fuxing Zhu, and Chaoxi Luo. 2021. "Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci" Biology 10, no. 3: 179. https://doi.org/10.3390/biology10030179
APA StyleChaisiri, C., Liu, X., Lin, Y., Fu, Y., Zhu, F., & Luo, C. (2021). Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci. Biology, 10(3), 179. https://doi.org/10.3390/biology10030179