
Exploring the Gut Microbiome Alteration of the European Hare (Lepus europaeus) after Short-Term Diet Modifications

 , , ,
, , ,  , , , , ,
, , , , ,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. DNA Extraction and Amplification
2.3. Amplicon Sequence Variants Detection
2.4. Statistical Analysis
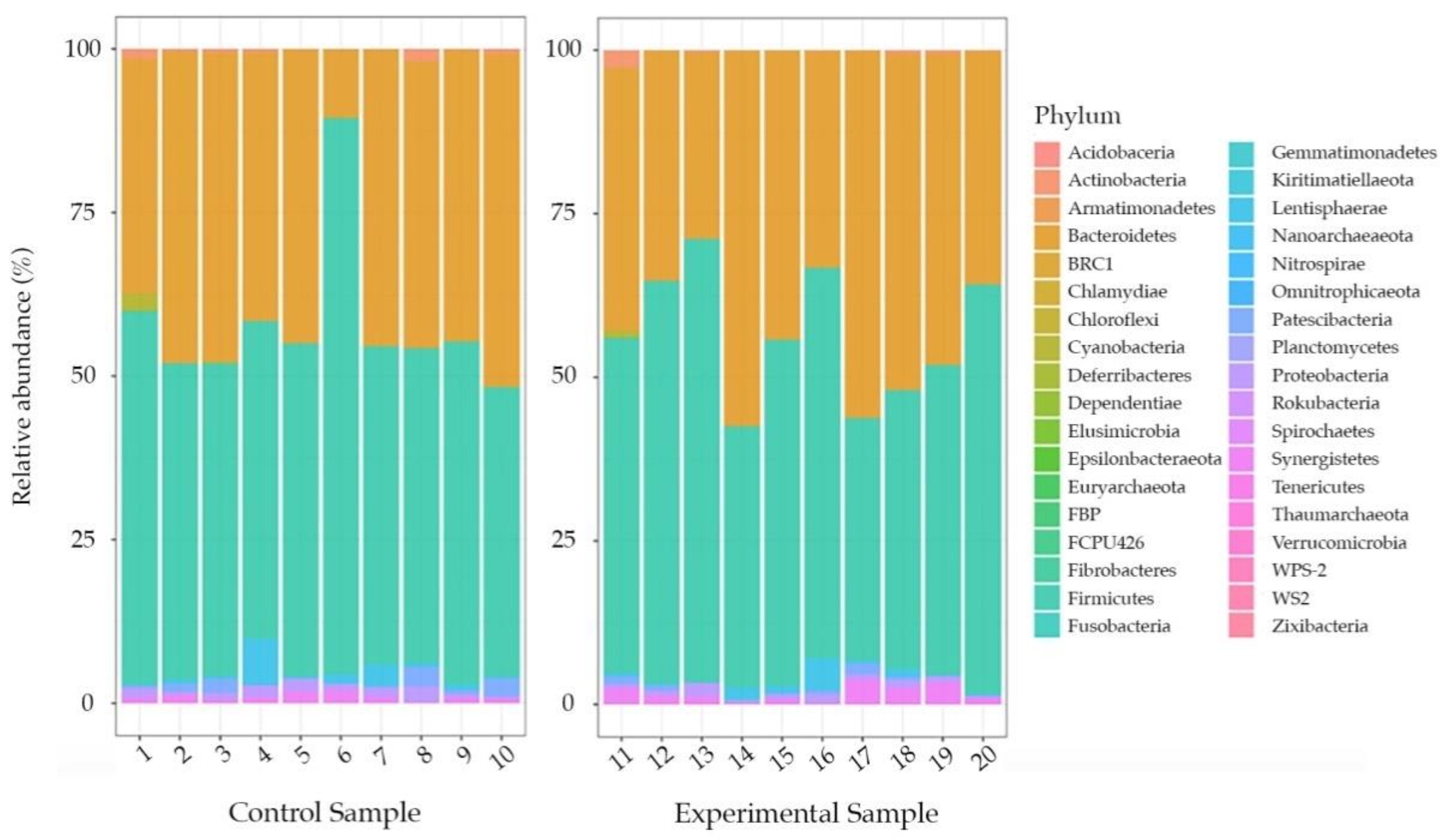
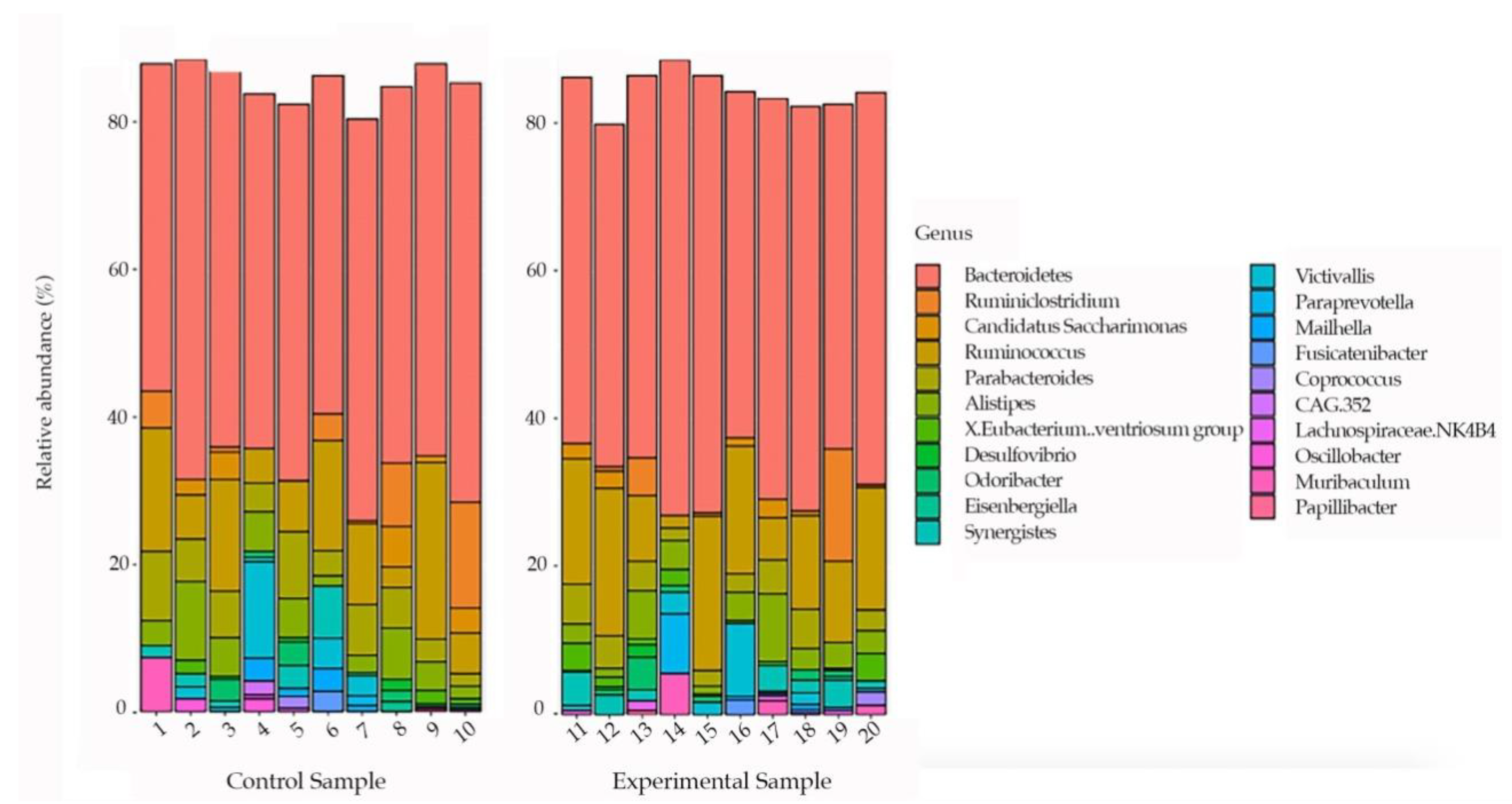
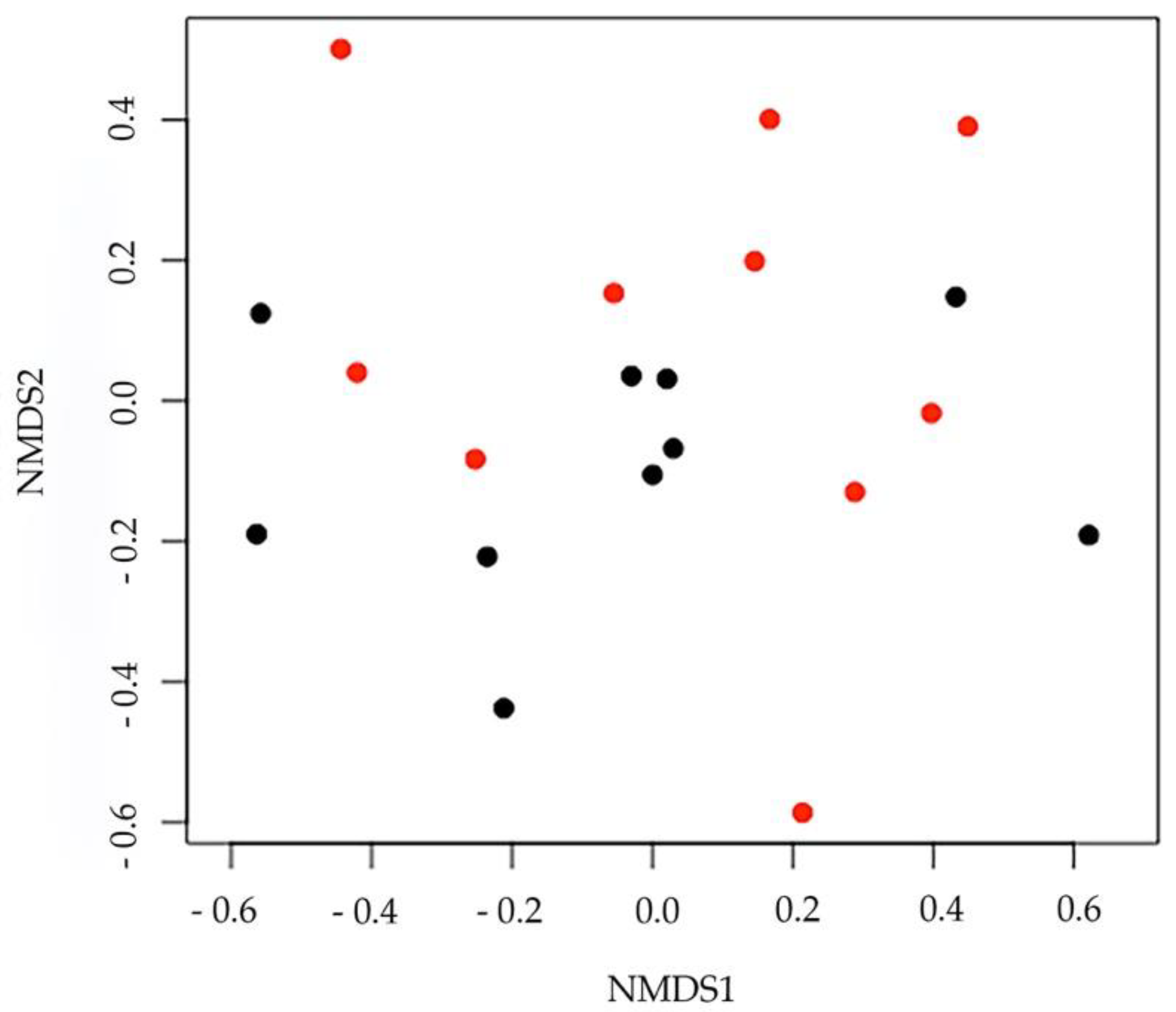
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Smith, R.K.; Vaughan Jennings, N.; Harris, S. A quantitative analysis of the abundance and demography of European hares Lepus europaeus in relation to habitat type, intensity of agriculture and climate. Mammal Rev. 2005, 35, 1–24. [Google Scholar] [CrossRef]
- Popescu, F.D.; Hackländer, K.; Arnold, W.; Ruf, T. Effects of season and reproductive state on lipid intake and fatty acid composition of gastrointestinal tract contents in the European hare. J. Comp. Physiol. B 2011, 181, 681–689. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Naldi, L.; Greco, I.; Ferretti, M.; Zaccaroni, M. Density Estimates and Habitat Preferences of the European Hare (Lepus europaeus) on Mountainous Areas in Italy. Mammal Study 2020, 45, 123–131. [Google Scholar] [CrossRef]
- Rigo, N.; Trocino, A.; Poppi, L.; Giacomelli, M.; Grilli, G.; Piccirillo, A. Performance and mortality of farmed hares. Animal 2015, 9, 1025–1031. [Google Scholar] [CrossRef][Green Version]
- Shetty, S.A.; Marathe, N.P.; Shouche, Y.S. Opportunities and challenges for gut microbiome studies in the Indian population. Microbiome 2013, 1, 24. [Google Scholar] [CrossRef]
- Stalder, G.L.; Pinior, B.; Zwirzitz, B.; Loncaric, I.; Jakupović, D.; Vetter, S.G.; Smith, S.; Posautz, A.; Hoelzl, F.; Wagner, M.; et al. Gut microbiota of the European brown hare (Lepus europaeus). Sci. Rep. 2019, 9, 2738. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. BMC Nutr. 2015, 113, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Amir, A.; Hyde, E.R.; Metcalf, J.L.; Song, S.J.; Knight, R. Tracking down the sources of experimental contamination in microbiome studies. Genome Biol. 2014, 15, 564. [Google Scholar] [CrossRef]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The madness of microbiome: Attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 2018, 84, 84. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Yi Liang, Q.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.; Panda, S.; Mengels, G.; Martinez, X.; Azpiroz, F.; Dore, J.; Guarner, F.; Manichanh, C. Processing faecal samples: A step forward for standards in microbial community analysis. BMC Microbiol. 2014, 14, 112. [Google Scholar] [CrossRef]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet. 2014, 38, 525–552. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Fouhy, F.; Sleator, R.D.; O’Driscoll, A.; Stanton, C.; Cotter, P.D.; Claesson, M.J. Comparing apples and oranges?: Next generation sequencing and its impact on microbiome analysis. PLoS ONE 2016, 11, e0148028. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Reichlin, T.; Klansek, E.; Hackländer, K. Diet selection by hares (Lepus europaeus) in arable land and its implications for habitat management. Eur. J. Wildl. Res. 2006, 52, 109–118. [Google Scholar] [CrossRef]
- Posautz, A.; Loncaric, I.; Lundin, M.; Hoffmann, D.; Lavazza, A.; Kelemen, Z.; Beiglböck, C.; Walzer, C.; Kübber-Heiss, A. Health screening of free-ranging European brown hares (Lepus europaeus) on the German North-Sea island Pellworm. Acta Vet. Scand. 2015, 57, 43. [Google Scholar] [CrossRef] [PubMed]
- Valencak, T.G.; Tataruch, F.; Ruf, T. Energy turnover in European hares is centrally limited during early, but not during peak lactation. J. Comp. Physiol. B 2009, 179, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Johnston, C.H. Lepus europaeus. In IUCN 2010. IUCN Red List of Threatened Species; IUNC: Gland, Switzerland, 2008. [Google Scholar]
- Sangiuliano, A.; Lovari, S.; Ferretti, F. Dietary partitioning between European roe deer and European brown hare. Eur. J. Wildl. Res. 2016, 62, 527–535. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Traslational Med. 2009, 1, ra14–ra16. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011, 333, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.; Statello, R.; Carnevali, L.; Mancabelli, L.; Milani, C.; Mangifesta, M.; Duranti, S.; Lugli, G.A.; Jimenez, B.; Lodge, S.; et al. How to feed the mammalian gut microbiota: Bacterial and metabolic modulation by dietary fibers. Front. Microbiol. 2017, 8, 1749. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Bahrndorff, S.; Alemu, T.; Alemneh, T.; Lund Nielsen, J. The microbiome of animals: Implications for conservation biology. Int. J. Genom. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B 2019, 286, 20182448. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Latini, A.; Bacci, G.; Teodoro, M.; Gattia, D.M.; Bevivino, A.; Trakal, L. The impact of soil-applied biochars from different vegetal feedstocks on durum wheat plant performance and rhizospheric bacterial microbiota in low metal-contaminated soil. Front. Microbiol. 2019, 10, 2694. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2014; Available online: http//www.R-project.org (accessed on 31 August 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Clarke, K.R.; Warwick, R.M. A further biodiversity index applicable to species lists: Variation in taxonomic distinctness. Mar. Ecol. Prog. Ser. 2011, 216, 265–278. [Google Scholar] [CrossRef]
- Anderson, D.R.; Link, W.A.; Johnson, D.H.; Burnham, K.P. Suggestions for presenting the results of data analyses. J. Wildl. Manag. 2001, 65, 373–378. [Google Scholar] [CrossRef]
- Anderson, M.J. Distance-based tests for homogeneity of multivariate dispersions. Biometrics 2006, 62, 245–253. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2018; Available online: http//www.R-project.org (accessed on 31 August 2020).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B. Package Vegan. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Vántus, V.B.; Kovács, M.; Zsolnai, A. The rabbit caecal microbiota: Development, composition and its role in the prevention of digestive diseases–a review on recent literature in the light of molecular genetic methods. Acta Agrar. Kaposváriensis 2014, 18, 55–65. [Google Scholar]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Kohl, K.D.; Miller, A.W.; Marvin, J.E.; Mackie, R.; Dearing, M.D. Herbivorous rodents (N eotoma spp.) harbour abundant and active foregut microbiota. Environ. Microbiol. 2014, 16, 2869–2878. [Google Scholar] [CrossRef]
- Velasco-Galilea, M.; Piles, M.; Viñas, M.; Rafel, O.; González-Rodríguez, O.; Guivernau, M.; Sánchez, J.P. Rabbit microbiota changes throughout the intestinal tract. Front. Microbiol. 2018, 9, 2144. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [PubMed]
- De Vries, F.A.T.; De Boer, E.; Van Den Bosch, M.; Baarends, W.M.; Ooms, M.; Yuan, L.; Liu, J.G.; Van Zeeland, A.A.; Heyting, C.; Pastink, A. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 2005, 19, 1376–1389. [Google Scholar] [CrossRef]
- Hong, P.Y.; Wheeler, E.; Cann, I.K.; Mackie, R.I. Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galápagos Islands using 16S rRNA-based pyrosequencing. ISME J. 2011, 5, 1461–1470. [Google Scholar] [CrossRef]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of diet on the gut microbiota: Rethinking intervention duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Posautz, A.; Kübber-Heiss, A.; Loncaric, I. Faecal flora of captive European brown hares (Lepus europaeus). Agric. Agric. Sci. Procedia 2016, 10, 358–363. [Google Scholar] [CrossRef][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padula, A.; Bambi, M.; Mengoni, C.; Greco, C.; Mucci, N.; Greco, I.; Masoni, A.; Del Duca, S.; Bacci, G.; Santini, G.; et al. Exploring the Gut Microbiome Alteration of the European Hare (Lepus europaeus) after Short-Term Diet Modifications. Biology 2021, 10, 148. https://doi.org/10.3390/biology10020148
Padula A, Bambi M, Mengoni C, Greco C, Mucci N, Greco I, Masoni A, Del Duca S, Bacci G, Santini G, et al. Exploring the Gut Microbiome Alteration of the European Hare (Lepus europaeus) after Short-Term Diet Modifications. Biology. 2021; 10(2):148. https://doi.org/10.3390/biology10020148
Chicago/Turabian StylePadula, Anna, Marina Bambi, Chiara Mengoni, Claudia Greco, Nadia Mucci, Ilaria Greco, Alberto Masoni, Sara Del Duca, Giovanni Bacci, Giacomo Santini, and et al. 2021. "Exploring the Gut Microbiome Alteration of the European Hare (Lepus europaeus) after Short-Term Diet Modifications" Biology 10, no. 2: 148. https://doi.org/10.3390/biology10020148
APA StylePadula, A., Bambi, M., Mengoni, C., Greco, C., Mucci, N., Greco, I., Masoni, A., Del Duca, S., Bacci, G., Santini, G., Fani, R., & Zaccaroni, M. (2021). Exploring the Gut Microbiome Alteration of the European Hare (Lepus europaeus) after Short-Term Diet Modifications. Biology, 10(2), 148. https://doi.org/10.3390/biology10020148