
Future Approaches for Treating Chronic Myeloid Leukemia: CRISPR Therapy

 , ,
, , 
Abstract
Simple Summary
Abstract
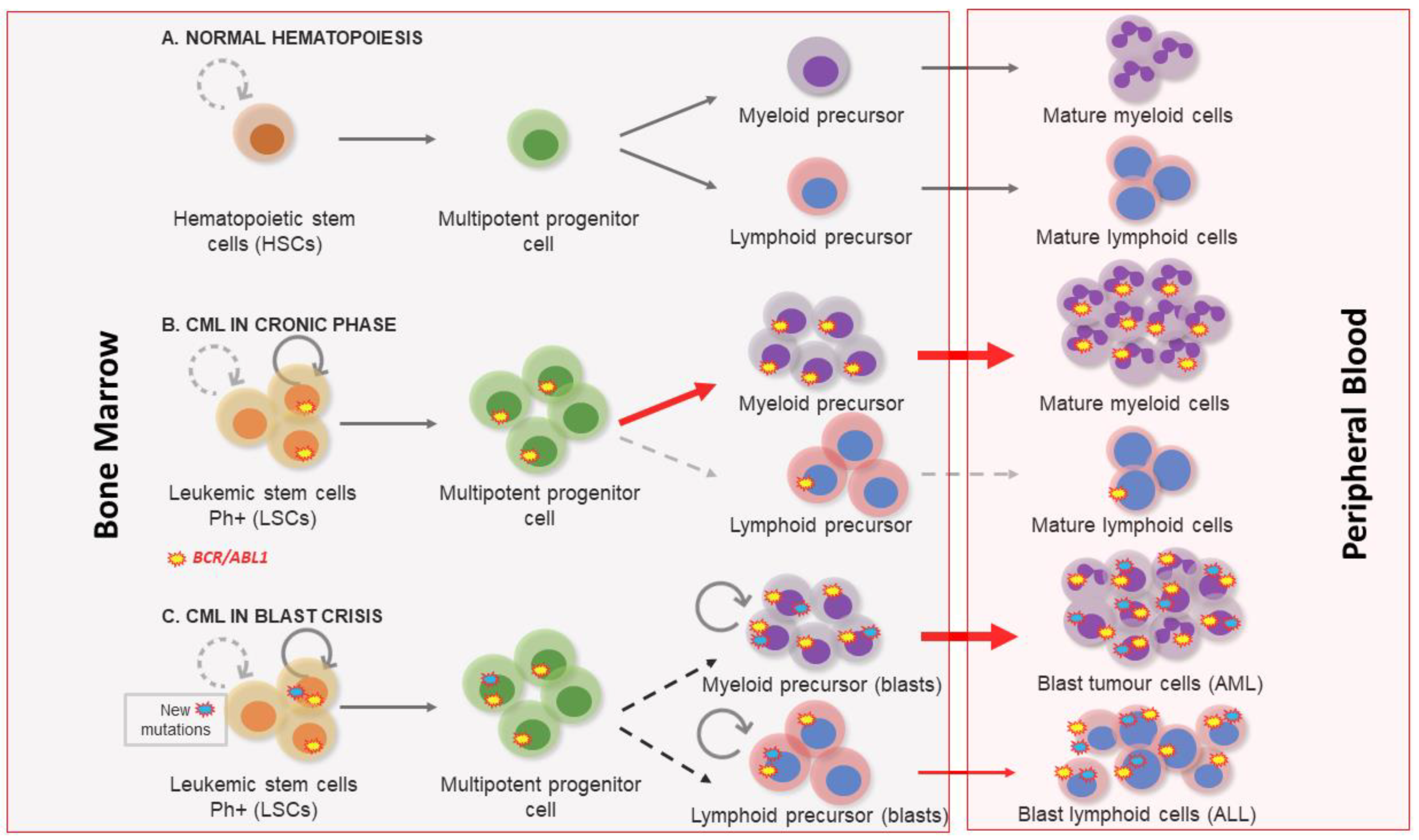
1. Clinical Features of Chronic Myeloid Leukemia
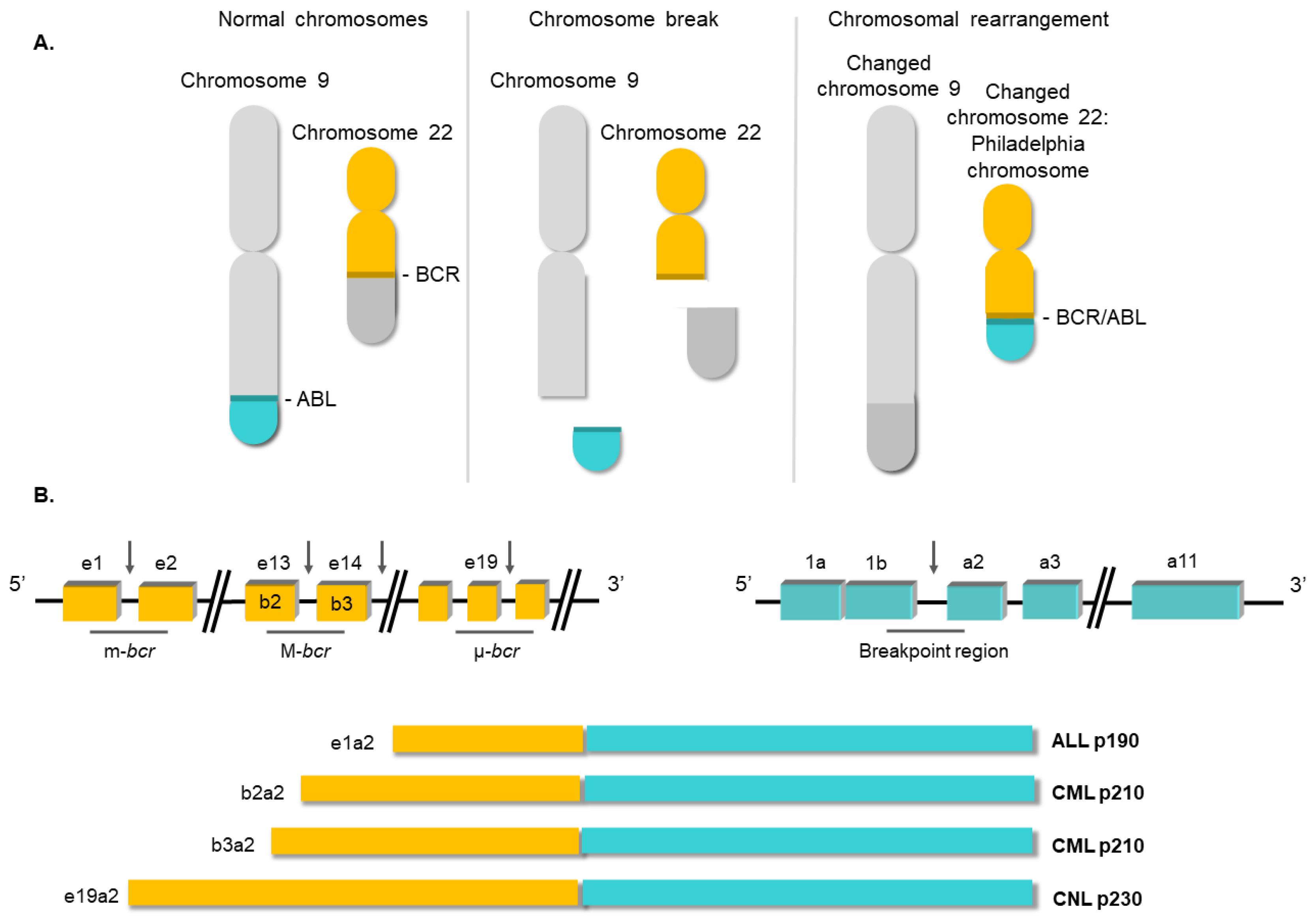
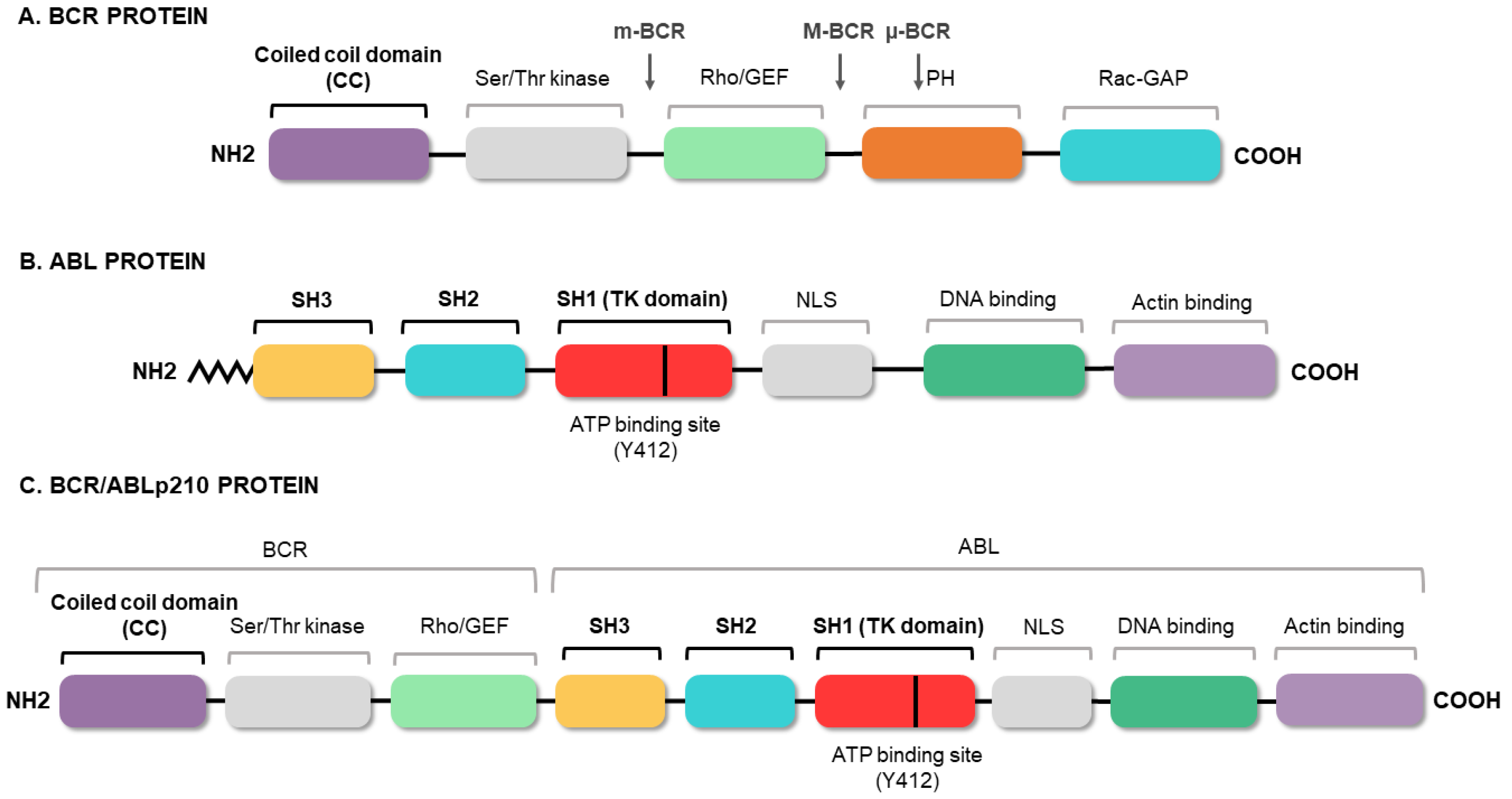
2. Molecular Biology of Chronic Myeloid Leukemia
3. Conventional Therapies for Chronic Myeloid Leukemia
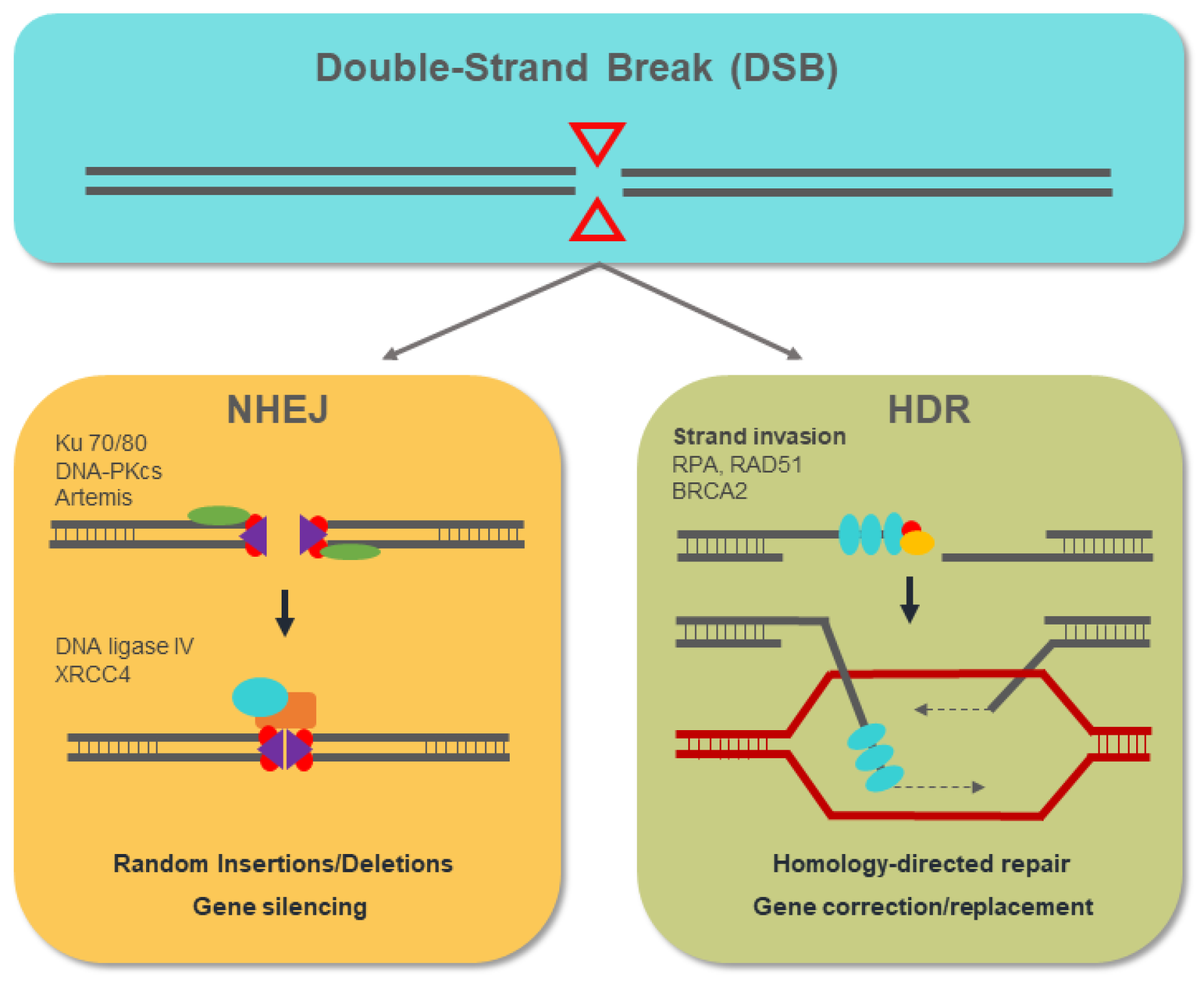
4. Genome-Editing Nucleases for Gene Therapy
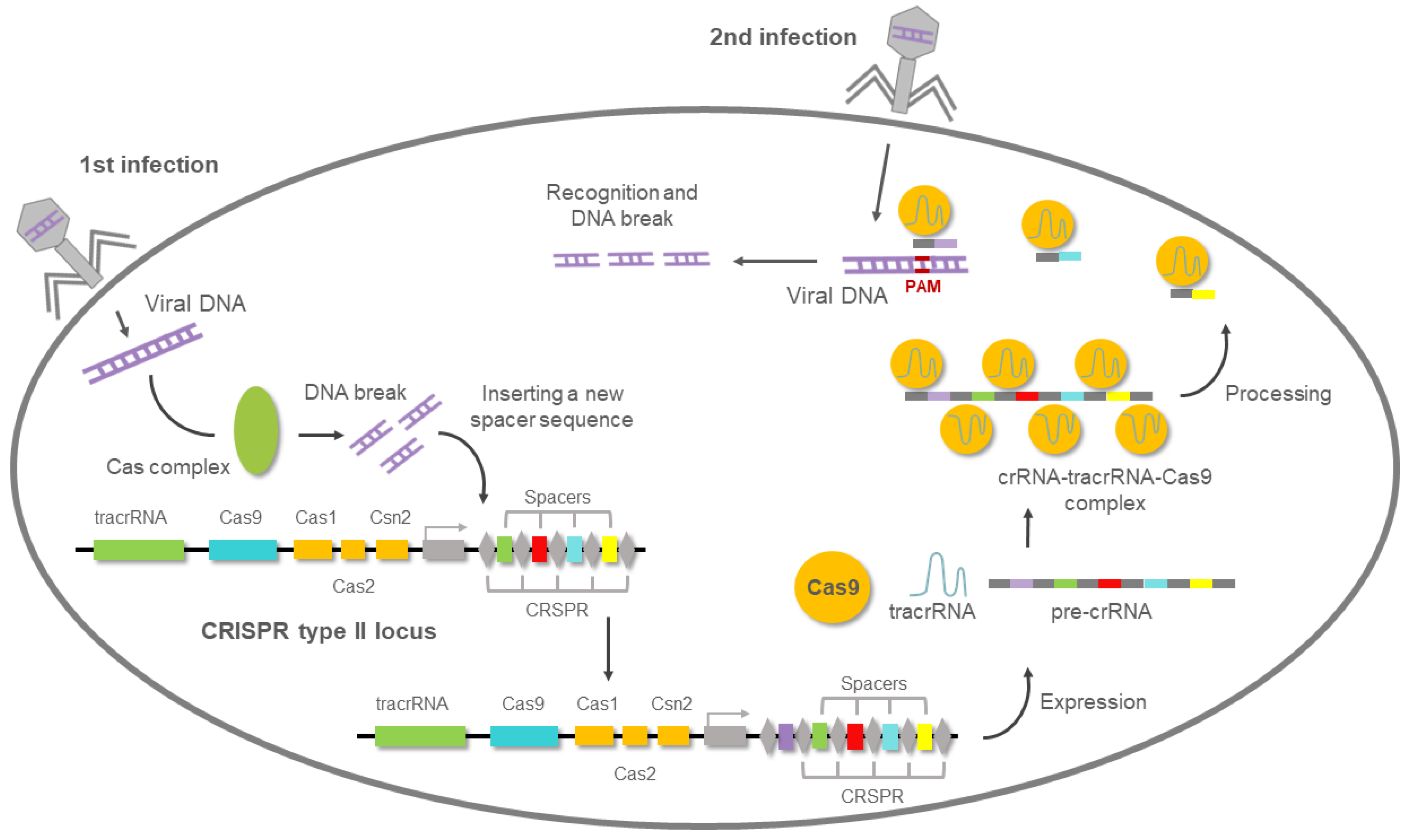
5. Overview of the CRISPR/Cas9 System
6. New CRISPR-Cas Systems and Approaches
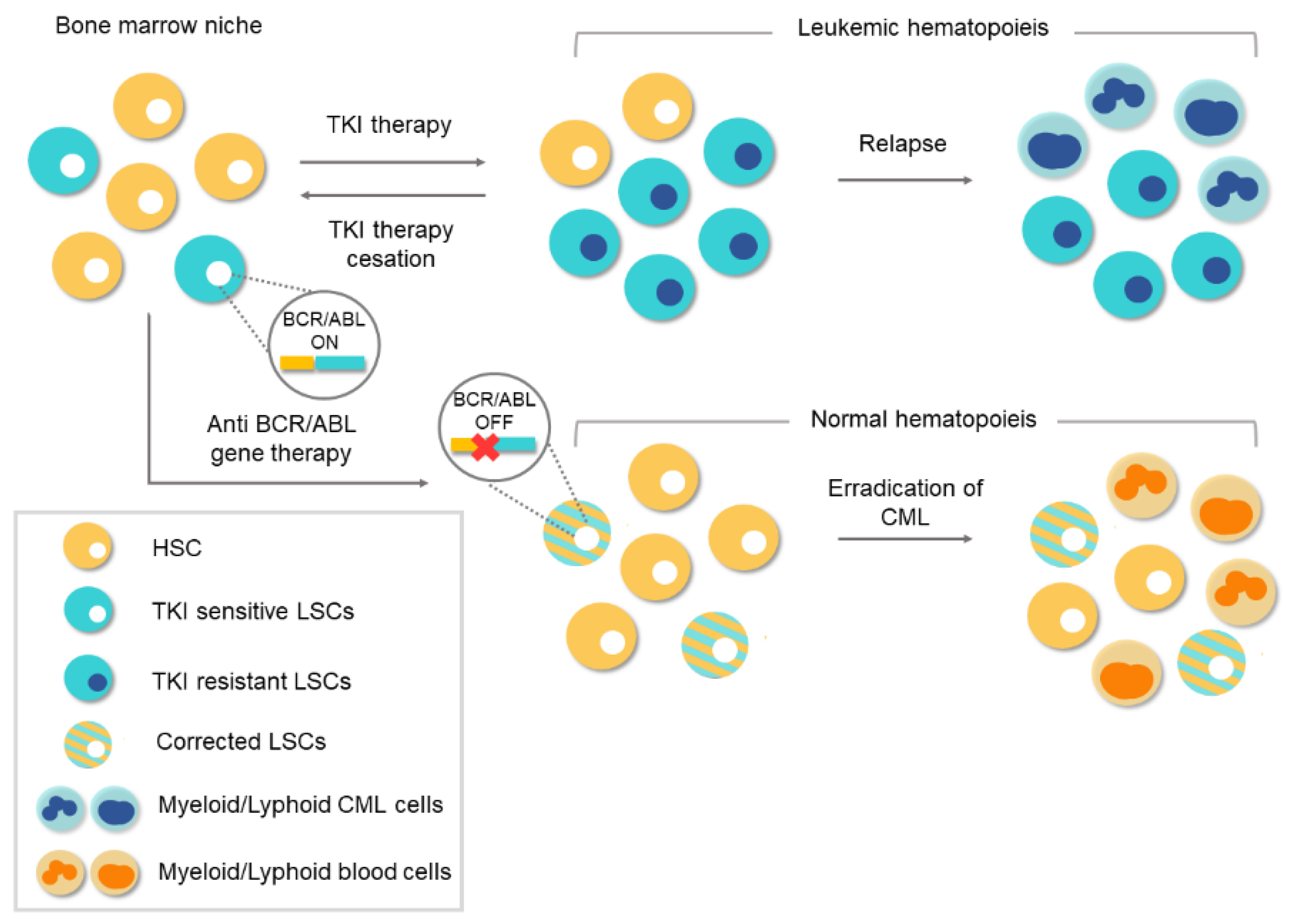
7. CRISPR Gene Therapy in CML
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quintás-Cardama, A.; Cortes, J. Chronic Myeloid Leukemia: Diagnosis and Treatment. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2006; Volume 81, pp. 973–988. [Google Scholar] [CrossRef]
- Mendizabal, A.M.; Garcia-Gonzalez, P.; Levine, P.H. Regional variations in age at diagnosis and overall survival among patients with chronic myeloid leukemia from low and middle income countries. Cancer Epidemiol. 2013, 37, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Petzer, A.; Eaves, C.; Lansdorp, P.; Ponchio, L.; Barnett, M.; Eaves, A. Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood 1996, 88, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.V.; Barnes, D.J. Chronic Myeloid Leukemia: Biology of Advanced Phase. In Myeloproliferative Disorders; Springer: Berlin/Heidelberg, Germany, 2007; pp. 37–58. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Keating, M.J.; Talpaz, M.; Walters, R.S.; Smith, T.L.; Cork, A.; McCredie, K.B.; Freireich, E.J. Chronic myelogenous leukemia in blast crisis. Am. J. Med. 1987, 83, 445–454. [Google Scholar] [CrossRef]
- Ilaria, R.L. Pathobiology of Lymphoid and Myeloid Blast Crisis and Management Issues. Hematol. Am. Soc. Hematol. Educ. Program 2005, 2005, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497–1499. [Google Scholar]
- Kantarjian, H.; O’Brien, S.; Jabbour, E.; Garcia-Manero, G.; Quintas-Cardama, A.; Shan, J.; Rios, M.B.; Ravandi, F.; Faderl, S.; Kadia, T.; et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood 2012, 119, 1981–1987. [Google Scholar] [CrossRef]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94 (Suppl. 2), S107–S121. [Google Scholar] [CrossRef]
- Bower, H.; Björkholm, M.; Dickman, P.W.; Höglund, M.; Lambert, P.C.; Andersson, T.M.-L. Life Expectancy of Patients with Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J. Clin. Oncol. 2016, 34, 2851–2857. [Google Scholar] [CrossRef]
- Deininger, M.; O’Brien, S.G.; Guilhot, F.; Goldman, D.J.M.; Hochhaus, A.; Hughes, T.P.; Radich, J.P.; Hatfield, A.K.; Mone, M.; Filian, J.; et al. International Randomized Study of Interferon Vs STI571 (IRIS) 8-Year Follow up: Sustained Survival and Low Risk for Progression or Events in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Treated with Imatinib. Blood 2009, 114, 1126. [Google Scholar] [CrossRef]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nat. Cell Biol. 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Daley, G.Q.; Mes-Masson, A.M.; Witte, O.N.; Baltimore, D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986, 233, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Score, J.; Calasanz, M.J.; Ottman, O.; Pane, F.; Yeh, R.F.; A Sobrinho-Simões, M.; Kreil, S.; Ward, D.; Hidalgo-Curtis, C.; Melo, J.V.; et al. Analysis of genomic breakpoints in p190 and p210 BCR–ABL indicate distinct mechanisms of formation. Leukemia 2010, 24, 1742–1750. [Google Scholar] [CrossRef] [PubMed]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; De Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Verstovsek, S.; Lin, H.; Kantarjian, H.; Saglio, G.; De Micheli, D.; Pane, F.; Garcia-Manero, G.; Intrieri, M.; Rotoli, B.; Salvatore, F.; et al. Neutrophilic-chronic myeloid leukemia: Low levels of p230 BCR/ABL mRNA and undetectable p230 BCR/ABL protein may predict an indolent course. Cancer 2002, 94, 2416–2425. [Google Scholar] [CrossRef] [PubMed]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Kabarowski, J.H.; Witte, O.N. Consequences of BCR–ABL Expression within the Hematopoietic Stem Cell in Chronic Myeloid Leukemia. Stem Cells 2000, 18, 399–408. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, R. Leukemia stem cells: The root of chronic myeloid leukemia. Protein Cell 2015, 6, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Janossy, G.; Roberts, M.; Greaves, M. Target Cell in Chronic Myeloid Leukæmia and Its Relationship to Acute Lymphoid Leukæmia. Lancet 1976, 308, 1058–1061. [Google Scholar] [CrossRef]
- Cohen, G.B.; Ren, R.; Baltimore, D. Modular binding domains in signal transduction proteins. Cell 1995, 80, 237–248. [Google Scholar] [CrossRef]
- Mayer, B.J.; Baltimore, D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol. Cell. Biol. 1994, 14, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, J.R.; Galasso, D.L.; Wang, J.Y. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Biol. 1993, 13, 7587–7595. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Lu, D.; Wu, Y.; Liu, J.; Arlinghaus, R.B. Bcr phosphorylated on tyrosine 177 binds Grb2. Oncogene 1997, 14, 2367–2372. [Google Scholar] [CrossRef][Green Version]
- Steelman, L.S.; A Franklin, R.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef]
- Walker, S.R.; Frank, D.A. STAT Signaling in the Pathogenesis and Treatment of Cancer. In Signaling Pathways in Cancer Pathogenesis and Therapy; Springer: New York, NY, USA, 2012; pp. 95–108. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chappell, W.; Abrams, S.L.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; Libra, M.; et al. Targeting the translational apparatus to improve leukemia therapy: Roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia 2011, 25, 1064–1079. [Google Scholar] [CrossRef][Green Version]
- Puil, L.; Liu, J.; Gish, G.; Mbamalu, G.; Bowtell, D.; Pelicci, P.; Arlinghaus, R.; Pawson, T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994, 13, 764–773. [Google Scholar] [CrossRef]
- Bedi, A.; Zehnbauer, B.; Barber, J.; Sharkis, S.; Jones, R. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood 1994, 83, 2038–2044. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.Y.; Dowding, C.R.; Riley, G.; Goldman, J.M.; Greaves, M.F. Altered adhesive interactions with marrow stroma of haematopoietic progenitor cells in chronic myeloid leukaemia. Nat. Cell Biol. 1987, 328, 342–344. [Google Scholar] [CrossRef]
- Clarkson, B.H.; Strife, A.; Wisniewski, D.; Lambek, C.; Liu, C. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 2003, 17, 1211–1262. [Google Scholar] [CrossRef]
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J.; et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993, 75, 175–185. [Google Scholar] [CrossRef]
- Minot, G.R.; Buckman, T.E.; Isaacs, R. Chronic myelogenous leukemia: Age incidence, duration, and benefit derived from irradiation. J. Am. Med. Assoc. 1924, 82, 1489–1494. [Google Scholar] [CrossRef]
- Rushing, D.; Goldman, A.; Gibbs, G.; Howe, R.; Kennedy, B.J. Hydroxyurea versus busulfan in the treatment of chronic myelogenous leukemia. Am. J. Clin. Oncol. Cancer Clin. Trials 1982, 5, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Tura, S.; Baccarani, M. α-interferon in the treatment of chronic myeloid leukemia. Blood 1995, 85, 2999–3000. [Google Scholar] [CrossRef] [PubMed]
- Lübking, A.; Dreimane, A.; Sandin, F.; Isaksson, C.; Märkevärn, B.; Brune, M.; Ljungman, P.; Lenhoff, S.; Stenke, L.; Höglund, M.; et al. Allogeneic stem cell transplantation for chronic myeloid leukemia in the TKI era: Population-based data from the Swedish CML registry. Bone Marrow Transplant. 2019, 54, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.P.; Hehlmann, R.; Zhang, M.J.; Hasford, J.; Goldman, J.M.; Heimpel, H.; Hochhaus, A.; Klein, J.P.; Kolb, H.J.; McGlave, P.B.; et al. Survival with Bone Marrow Transplantation Versus Hydroxyurea or Interferon for Chronic Myelogenous Leukemia. Blood 1998, 91, 1810–1819. [Google Scholar]
- Van Rhee, F.; Szydlo, R.; Hermans, J.; Devergie, A.; Frassoni, F.; Arcese, W.; De Witte, T.; Kolb, H.; Niederwiser, D.; Jacobsen, N.; et al. Long-term results after allogeneic bone marrow transplantation for chronic myelogenous leukemia in chronic phase: A report from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 1997, 20, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Müller, M.; Druker, B.J.; Lydon, N.B. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56, 100–104. [Google Scholar]
- Bhatia, R.; A Munthe, H.; Verfaillie, C. Tyrphostin AG957, a tyrosine kinase inhibitor with anti-BCR/ABL tyrosine kinase activity restores β1 integrin-mediated adhesion and inhibitory signaling in chronic myelogenous leukemia hematopoietic progenitors. Leukemia 1998, 12, 1708–1717. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Milojkovic, D.; Apperley, J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.; Cortes, J. Use of Second- and Third-Generation Tyrosine Kinase Inhibitors in the Treatment of Chronic Myeloid Leukemia: An Evolving Treatment Paradigm. Clin. Lymphoma Myeloma Leuk. 2015, 15, 323–334. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Montoliu, L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol. 2016, 24, 811–820. [Google Scholar] [CrossRef]
- Wassef, M.; Luscan, A.; Battistella, A.; Le Corre, S.; Li, H.; Wallace, M.; Vidaud, M.; Margueron, R. Versatile and precise gene-targeting strategies for functional studies in mammalian cell lines. Methods 2017, 121–122, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Capecchi, M.R. Altering the genome by homologous recombination. Science 1989, 244, 1288–1292. [Google Scholar] [CrossRef]
- Rudin, N.; Sugarman, E.; Haber, J.E. Genetic and physical analysis of double-strand break repair and recombination in Sac-charomyces cerevisiae. Genetics 1989, 122, 519–534. [Google Scholar] [CrossRef]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Genet. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Mojica, F.J.; Juez, G.; Rodriguez-Valera, F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol. Microbiol. 1993, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663. [Google Scholar] [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Dusko Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; A Shabalina, S.; I Wolf, Y.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; Van Der Oost, J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR—A widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.-È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef]
- Horvath, P.; Romero, D.A.; Coûté-Monvoisin, A.-C.; Richards, M.; Deveau, H.; Moineau, S.; Boyaval, P.; Fremaux, C.; Barrangou, R. Diversity, Activity, and Evolution of CRISPR Loci in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonté, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage Response to CRISPR-Encoded Resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Dickinson, D.J.; Goldstein, B. CRISPR-Based Methods for Caenorhabditis elegans Genome Engineering. Genetics 2016, 202, 885–901. [Google Scholar] [CrossRef]
- Gratz, S.J.; Rubinstein, C.D.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. CRISPR-Cas9 Genome Editing in Drosophila. Curr. Protoc. Mol. Biol. 2015, 111. [Google Scholar] [CrossRef]
- Jiang, W.Z.; Yang, B.; Weeks, D.P. Efficient CRISPR/Cas9-Mediated Gene Editing in Arabidopsis thaliana and Inheritance of Modified Genes in the T2 and T3 Generations. PLoS ONE 2014, 9, e99225. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Shen, B.; Lu, Y.; Chen, W.; Ma, J.; Bai, L.; Huang, X.; Zhang, L. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 2014, 24, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Beal, P.A. Off-Target Editing by CRISPR-Guided DNA Base Editors. Biochemistry 2019, 58, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-S.; Li, Q.-C.; Yin, C.-Q.; Xue, W.; Song, C.-Q. Advances in CRISPR/Cas-based Gene Therapy in Human Genetic Diseases. Theranostics 2020, 10, 4374–4382. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Torres-Ruiz, R.; Puig-Serra, P.; Moreno-Gaona, P.; Martin, M.C.; Moya, F.J.; Quintana-Bustamante, O.; Garcia-Silva, S.; Carcaboso, Á.M.; Petazzi, P.; et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; O Abudayyeh, E.M.D.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Park, J.J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV–Cpf1. Nat. Methods 2019, 16, 247–254. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- Garcia-Tunon, I.; Alonso-Perez, V.; Vuelta, E.; Perez-Ramos, S.; Herrero, M.; Méndez, L.; Sánchez, J.; Martín-Izquierdo, M.; Saldaña, R.; Sevilla, J.; et al. Splice donor site sgRNAs enhance CRISPR/Cas9-mediated knockout efficiency. PLoS ONE 2019, 14, e0216674. [Google Scholar] [CrossRef]
- Luo, Z.; Gao, M.; Huang, N.; Wang, X.; Yang, Z.; Yang, H.; Huang, Z.; Feng, W. Efficient disruption of bcr-abl gene by CRISPR RNA-guided FokI nucleases depresses the oncogenesis of chronic myeloid leukemia cells. J. Exp. Clin. Cancer Res. 2019, 38, 224. [Google Scholar] [CrossRef]
- Huang, N.; Huang, Z.; Gao, M.; Luo, Z.; Zhou, F.; Liu, L.; Xiao, Q.; Wang, X.; Feng, W. Induction of apoptosis in imatinib sensitive and resistant chronic myeloid leukemia cells by efficient disruption of bcr-abl oncogene with zinc finger nucleases. J. Exp. Clin. Cancer Res. 2018, 37, 62. [Google Scholar] [CrossRef] [PubMed]
- García-Tuñón, I.; Hernández-Sánchez, M.; Ordoñez, J.L.; Alonso-Pérez, V.; Álamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernández-Rivas, J.M.; Sanchez-Martin, M.A. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget 2017, 8, 26027–26040. [Google Scholar] [CrossRef]
- Vukovic, M.; Guitart, A.V.; Sepulveda, C.; Villacreces, A.; O’Duibhir, E.; Panagopoulou, T.I.; Ivens, A.; Menendez-Gonzalez, J.; Iglesias, J.M.; Allen, L.; et al. Hif-1α and Hif-2α synergize to suppress AML development but are dispensable for disease maintenance. J. Exp. Med. 2015, 212, 2223–2234. [Google Scholar] [CrossRef]
- Valletta, S.; Dolatshad, H.; Bartenstein, M.; Yip, B.H.; Bello, E.; Gordon, S.; Yu, Y.; Shaw, J.; Roy, S.; Scifo, L.; et al. ASXL1 mutation correction by CRISPR/Cas9 restores gene function in leukemia cells and increases survival in mouse xenografts. Oncotarget 2015, 6, 44061–44071. [Google Scholar] [CrossRef]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nat. Cell Biol. 2016, 539, 479. [Google Scholar] [CrossRef]
- A Safety and Efficacy Study Evaluating CTX001 in Subjects with Transfusion-Dependent β-Thalassemia—Full Text View. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03655678?view=results (accessed on 15 January 2021).
- Aryee, K.-E.; Shultz, L.D.; Brehm, M.A. Immunodeficient Mouse Model for Human Hematopoietic Stem Cell Engraftment and Immune System Development. Methods Mol. Biol. 2014, 1185, 267–278. [Google Scholar] [CrossRef]
- Peng, C.; Li, S. Chronic Myeloid Leukemia (CML) Mouse Model in Translational Research. Methods Mol. Biol. 2016, 1438, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Hsieh, Y.-Y.; Tzeng, H.-E.; Lin, C.-Y.; Hsu, K.-W.; Chiang, Y.-S.; Lin, S.-M.; Su, M.-J.; Hsieh, W.-S.; Lee, C.-H. ABL Genomic Editing Sufficiently Abolishes Oncogenesis of Human Chronic Myeloid Leukemia Cells In Vitro and In Vivo. Cancers 2020, 12, 1399. [Google Scholar] [CrossRef]
- Vuelta, E.; Luis Ordoñez, J.; Alonso-Pérez, V.; Méndez, L.; Hernández-Carabias, P.; Saldaña, R.; Sevilla, J.; Sebastian, E.; Muntión, S.; Sánchez-Guijo, F.; et al. CRISPR/Cas9 technology abolishes the BCR/ABL1 oncogene effect in chronic myeloid leukemia and restores normal hematopoiesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Lafrance, B.; Kaplan, M.; Doudna, J.A. Conformational control of DNA target cleavage by CRISPR–Cas9. Nature 2015, 527, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Hong, S.; Chen, W.; Zuo, E.; Yang, H. Advances in detecting and reducing off-target effects generated by CRISPR-mediated genome editing. J. Genet. Genom. 2019, 46, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Cell Type | Genome Editing System | Outcomes | Reference |
|---|---|---|---|---|
| Fusion sequence | Boff p210 (mouse) | CRISPR/Cas9 | Subcutaneous injection of edited single cell derived clones was unable to generate tumors in a CML xenograft model. | [96] |
| BCR exon 1 | K562 (human) and patient derived CD34+ cells | ZFNs | Intravenous tail vein injection into NOD/SCID mice of the edited K562 showed a lower tumorigenic capacity in vivo. Lower proliferative capacity in vitro was observed in edited primary cells. | [95] |
| ABL1 exon 2 | K562 (human) and patient derived CD34+ cells | CRISPR RNA-guided FokI nucleases (RFNs) | Similar results to those of their previous work. High efficiency and greater security by reducing the frequency of off-targets, compared with CRISPR/Cas9 system. | [94] |
| ABL1 exon 2 | K562 (human) and peripheral blood mononuclear cells (PBMCs) of CML patients | CRISPR/Cas9 | Virus-mediated ABL1-targeting to edit luciferase-labeled K562 into a systemic leukemia xenograft model. Bioluminescence imaging showed a significant reduction of leukemic cells in vivo. | [103] |
| Fusion sequence | K562 (human) and patient derived CD34+ cells | CRISPR/Cas9 | Specific targeting of the BCR/ABL1 fusion sequence with a pair of guides directed towards intronic sequences of each of the genes involved in the fusion that will cause a deletion in those cells that carry the translocation. | [84] |
| ABL1 exon 6 | Boffp210 (mouse), K562 (human), Lin- CML mouse model and patient-derived CD34+ | CRISPR/Cas9 | Edited HSCs from CML mouse model restored normal hematopoiesis in NOD/SCID bone marrow niche. Edited patient-derived CD34+ are capable of regenerating normal hematopoiesis in the bone marrow niche of NOD/SCID mice. | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuelta, E.; García-Tuñón, I.; Hernández-Carabias, P.; Méndez, L.; Sánchez-Martín, M. Future Approaches for Treating Chronic Myeloid Leukemia: CRISPR Therapy. Biology 2021, 10, 118. https://doi.org/10.3390/biology10020118
Vuelta E, García-Tuñón I, Hernández-Carabias P, Méndez L, Sánchez-Martín M. Future Approaches for Treating Chronic Myeloid Leukemia: CRISPR Therapy. Biology. 2021; 10(2):118. https://doi.org/10.3390/biology10020118
Chicago/Turabian StyleVuelta, Elena, Ignacio García-Tuñón, Patricia Hernández-Carabias, Lucía Méndez, and Manuel Sánchez-Martín. 2021. "Future Approaches for Treating Chronic Myeloid Leukemia: CRISPR Therapy" Biology 10, no. 2: 118. https://doi.org/10.3390/biology10020118
APA StyleVuelta, E., García-Tuñón, I., Hernández-Carabias, P., Méndez, L., & Sánchez-Martín, M. (2021). Future Approaches for Treating Chronic Myeloid Leukemia: CRISPR Therapy. Biology, 10(2), 118. https://doi.org/10.3390/biology10020118